

Abstract

The recent publication of a potent and selective inhibitor of protein methyltransferase 5 (PRMT5) provides the scientific community with in vivo-active tool compound EPZ015666 (GSK3235025) to probe the underlying pharmacology of this key enzyme. Herein, we report the design and optimization strategies employed on an initial hit compound with poor in vitro clearance to yield in vivo tool compound EPZ015666 and an additional potent in vitro tool molecule EPZ015866 (GSK3203591).
Keywords: Methyltransferase, PRMT5, property based optimization, structure guided design
The mammalian protein arginine methyltransferases are a group of nine enzymes that perform NG-mono methylation-, asymmetric-, or symmetric dimethylation of arginine residues on a range of nuclear and cytoplasmic protein substrates.1 One member of this group, PRMT5, is capable of performing methylation of up to two methyl groups and is currently believed to be the predominant enzyme for symmetric dimethylation. PRMT5 may play an important role in tumorigenesis and is upregulated in several human malignancies.2−8 The mechanism behind the cell-transforming capabilities of PRMT5 has been postulated to have roles in cell death, cell-cycle progression and cell growth, and proliferation and is still under investigation.9 Whether PRMT5 drives tumorigenesis by direct signal transduction, regulating gene expression, or by some other mechanism is generally unknown, although recent studies highlight a dependency on PRMT5 as part of the spliceosomal machinery with Sm proteins, particularly for MYC-driven tumors.10
EPZ015666 has recently been characterized as a potent inhibitor and in vivo tool compound of PRMT5.11 This compound is the first inhibitor to be described with a well characterized correlation between inhibition of PRMT5 enzyme and reduction of known substrate products including symmetric-dimethylated SmD3, coupled with a corresponding effect on tumor growth inhibition. In addition, structural biology studies highlighted a unique cation−π binding mode involving the tetrahydroisoquinoline (THIQ) containing chemical series as exemplified in the EPZ015666:PRMT5:MEP50 cocrystal complex (PDB Codes: 4X60, 4X61). Herein we describe the medicinal chemistry optimization (Figure 1) in the development of tool compound EPZ015666.
Figure 1.

HTS hit compound 1 and tool compound EPZ015666.
Compound 1 was recently described11 as a hit identified from a 370 K member diversity high throughput screening (HTS) campaign, with modest inhibitory activity against PRMT5. Scheme 1 shows the synthetic route employed for the optimization of 1, as it enabled identification of SAR around the THIQ group at the penultimate step. Retaining the cyclopentylamino motif, a range of amines were used to open epoxide intermediate 5, providing the amino alcohol derivatives shown after boc-deprotection. No increase in activity was observed from this set, however, in comparison with the original hit compound (Scheme 1B). The contribution of the THIQ mediated cation−π interaction is apparent from this early data set with a number of non-THIQ containing analogues resulting in a reduced inhibitory potency. In comparison to THIQ, analogues 6a and 6b have an increased dipole moment, which is proposed to have a deleterious effect on the binding energy of the cation−π binding interaction.12
Scheme 1. Synthesis and Activity of THIQ Directed Analogues of HTS Hit Compound 1.

(A) Synthesis of analogues of compound 1. Reagents: (i) 2-(bromomethyl)oxirane, NaH, DMF, r.t., 24 h; (ii) cyclopentanamine, NaBH4, DCM, r.t. 1 h; (iii) Boc2O, DCM, Et3N, r.t., 16 h; (iv) amine, EtOH, DIPEA, microwave heating 110 °C, 20–50 min; (v) EtOAc, EtOAc/HCl, 0 °C, 4 h. (B) IC50s were determined from at least two independent experiments with IC50 values within 3-fold; see Supporting Information for details on biochemistry assay.
Additionally, analogues that may present the aryl ring in a different or less than optimal orientation with the methyl group of cofactor SAM also displayed a corresponding reduction in potency (6c and 6d).
Retaining the key THIQ-amino alcohol scaffolding motif, alternate substitutions on the alkoxy phenyl ring were initially probed with the majority of analogues in this series showing little improvement over the original HTS hit. Identification of compound 7 marked a breakthrough in potency optimization with a 4-fold increase in biochemical inhibitory potency and a <1 μM IC50 of the enzyme in cells, as revealed by reduction of SmD3me2s levels in Z-138 cells (Table 1). During the SAR expansion, the alkoxy-phenyl ring of 7 was replaced with a 2-amino pyridyl-ring yielding compound 8, which had a 2-fold increase in potency.
Table 1. Optimization of Lead Seriesa.

IC50s were determined from at least two independent experiments with IC50 values within 3-fold; see Supporting Information for details on assays.
A cocrystal structure of compound 8 with PRMT5:MEP50 (Figure 2A) revealed only the THIQ and alcohol moieties made hydrogen bond interactions with the protein. It was noted that the pyridyl-ring nitrogen was 4.1 Å from the backbone NH of Leu312, a residue involved in binding histone H4 peptide.13 Amino pyridine analogues have been used as bioisosteric replacements for amides14 due to their ability to retain hydrogen bonding interactions or providing conformational restraint. Here, this bioisostere replacement strategy was reversed, aiming to increase the conformational flexibility of the H-bond acceptor and increasing the potential of gaining a hydrogen bond with Leu312. The strategy was successful as shown by the structure of aromatic amide compound 9, which revealed both a direct hydrogen bond to the backbone of Leu312 and a water-mediated interaction with the backbone carbonyl of Ser578 (Figure 2B) and a concomitant improvement in IC50 (Table 1).
Figure 2.
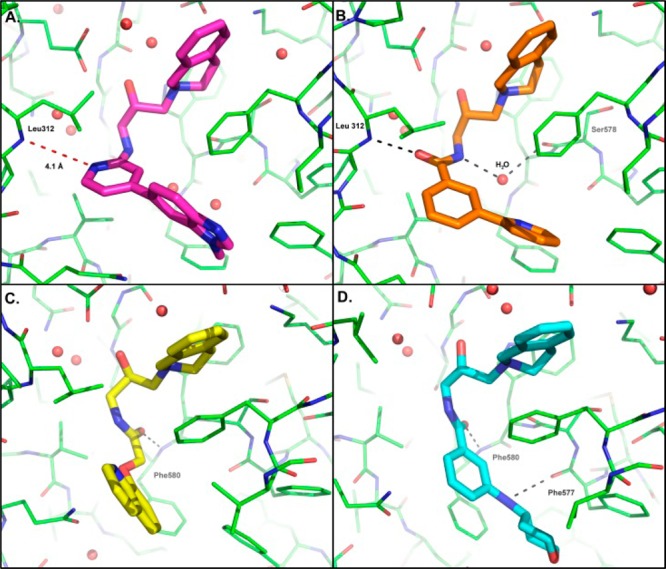
Co-crystal structures of PRMT5:MEP50 (green) with compound 8 (A, magenta), compound 9 (B, orange), compound 10 (C, yellow), and compound 15 (D, cyan). Hydrogen bonds are indicated with black dashed lines; red lines highlight distances between protein and compound. Select residues are labeled.
Aliphatic amides were also potent PRMT5 inhibitors as exemplified by compound 10. The structure revealed a new H-bonding interaction with the PRMT5 protein (Figure 2C). In contrast to 9, the H-bond interaction with 10 was observed between the amide carbonyl and the NH of Phe580. Additionally, the preferred stereoisomer for the alcohol was also reversed from that seen in 9.
As we approached the tight binding limit of the biochemical assay,11 the primary data for potency SAR increases was then driven by the cellular methylation and high throughput proliferation assays. The cellular assays11 consisted of an In-Cell Western (ICW) format assay following inhibition of symmetric dimethylation of arginine containing substrates, and a cell proliferation IC50 assay. Both assays were run with Z-138 cells, a Mantel Cell Lymphoma (MCL).
Although racemic compounds 9 and 10 both displayed a cellular methylation IC50 of ∼36 nM, compound 10 exhibited >3-fold increase in antiproliferative activity over 9. Furthermore, analysis of the individual stereoisomers of 9 (11 and 12) and 10 (13 and 14) revealed a modest chemotype-dependent potency preference for one particular isomer, with the aromatic and aliphatic amides presenting a preference for S- and R- stereochemistry, respectively. Crystallization of the racemic mixtures resulted in structures of the preferred isomer for both series.
Although significant improvements in cellular potency had been achieved, these chemical series were unstable in both human and mouse liver microsomal incubations with scaled clearance (CL) values approaching hepatic blood flow (Table 1). Compound design efforts were therefore focused on identifying less lipophilic amide analogues with reduced clogD values in an attempt to address high turnover in liver microsomes.15
This strategy initially yielded compound 15, which had a modest improvement in potency but a more significant impact on microsomal stability. Furthermore, the binding mode of this aryl amide switched to mimic that of the aliphatic amide subseries (e.g., compound 10), despite the difference in the preferred stereoisomer (Figure 2D). In doing so, the NH of the 4-amino tetrahydropyran motif was now capable of forming a hydrogen bond with the carbonyl of Phe577. Further optimization in this vector generated EPZ015666 and an extremely potent in vitro tool compound EPZ015866. EPZ015866 maintained excellent selectivity against other PMT enzymes16 observed with EPZ015666.11
To identify an appropriate in vivo tool compound, we focused on compounds with scaled HLM clearance values ≤50% hepatic blood flow (Figure 3A), believing this chemical space was more likely to provide data, which could eventually be used to identify candidate quality molecules. Minimal species-dependent SAR was observed when the same compound set were incubated with mouse LMs (Figure 3B). Further assessment of the chemical series in mouse PK studies highlighted a number of compounds with moderate to high plasma clearance and a wide range of oral bioavailability, F (Figure 3C,D). Volume of distribution values derived from these studies ranged between 0.6 and 45 L/kg with the majority of data points being <5 L/kg (Supplementary Figure 1).
Figure 3.

Plot of cLogD vs scaled clearance (CL) from human microsomes (A) and mouse liver microsomes (B). Plot of proliferation IC50 in Z-138 cells vs observed CL in mouse PK (C) and percentage oral bioavailability %F from mouse PK (D) dosed at 2 mg/kg IV and 10 mg/kg PO.
From these combined data, EPZ015666 emerged as the compound with the best combination of low plasma clearance and high oral bioavailability maintaining potent antiproliferative activity against a known mantle cell lymphoma cell line (Z-138 cells). Plasma protein binding in mouse plasma was determined to be approximately 30%. Further PK studies revealed that at 100 mg/kg in mouse, the unbound plasma concentration was equivalent to or above, the methyl mark IC90 for a period of 12 h, thus suggesting that BID dosing of EPZ015666 could effectively inhibit PRMT5 in vivo (Figure 4). Additional species PK and ADME characterization data on EPZ015666 were reported by Rioux et al.17
Figure 4.

Plasma PK profile of EPZ015666 in male CD-1 mouse following single dose administration at 10 and 100 mg/kg p.o. Methyl mark IC90 corrected for PPB shown as dotted line.
In summary, we have outlined the medicinal chemistry strategies employed in the optimization of a submicromolar HTS hit (1) to the selection of a potent in vivo tool compound to evaluate the pharmacology of PRMT5 inhibition. Early SAR exploration around the THIQ motif confirmed the contribution of the cation−π interaction with cofactor SAM and was thus retained within the series as a key binding interaction.
Additional compound design relied upon two parallel strategies; rational structure based design in identifying and optimizing hydrogen-bonding interactions, driving cellular inhibitory potency to low nanomolar IC50s, and physiochemical property driven optimization to minimize CL. These efforts identified EPZ015666, the first in vivo tool compound for PRMT5. Additionally, EPZ015866 was shown to be an extremely potent compound suitable for in vitro studies.
Acknowledgments
We thank Jian Wang and Charles Ding and the team at Wuxi App Tec for their contributions to the synthetic work.
Glossary
ABBREVIATIONS
- CL
clearance
- i.v
intravenous
- p.o.
per os
- THIQ
tetrahydroisoquinoline
- PK
pharmacokinetic
- PRMT5
arginine protein methyltransferase 5
- MEP50
methylosome protein 50
- SmD3me2s
Small ribonucleoprotein particle protein 3 symmetric dimethylation
- ICW
in-cell western
- LM
liver microsome
- MCL
Mantle Cell Lymphoma
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00380.
X-ray crystallographic data, synthetic procedures for all final compounds, and PK derived volume of distribution data (PDF)
Author Present Address
† H3 Biomedicine, Cambridge, Massachusetts 02139, United States.
Author Present Address
‡ Warp Drive Bio, 400 Technology Square, Cambridge, Massachusetts 02139, United States.
Author Present Address
§ Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States.
Author Present Address
∥ Agile Biostructure Solutions, Cambridge, Massachusetts 02139, United States.
Author Present Address
⊥ Raze Therapeutics, Cambridge, Massachusetts 02139, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors are/were employees or consultants of Epizyme, Inc. Work was funded by Epizyme and GlaxoSmithKline.
Supplementary Material
References
- Kaniskan H. U.; Konze K. D.; Jin J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–629. 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E. C.; Zheng S.; Munro S.; Liu G.; Carr S. M.; Moehlenbrink J.; Lu Y. C.; Stimson L.; Khan O.; Konietzny R.; McGouran J.; Coutts A. S.; Kessler B.; Kerr D. J.; Thangue N. B. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012, 31, 1785–97. 10.1038/emboj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J.; Karkhanis V.; Tae S.; Yan F.; Smith P.; Ayers L. W.; Agostinelli C.; Pileri S.; Denis G. V.; Baiocchi R. A.; Sif S. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) silencing. J. Biol. Chem. 2013, 288, 35534–47. 10.1074/jbc.M113.510669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Baiocchi R. A.; Byrd J. C.; Grever M. R.; Jacob S. T.; Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–69. 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Vishwanath S. N.; Erdjument-Bromage H.; Tempst P.; Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–45. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers M. A.; Fay M. M.; Factor R. E.; Welm A. L.; Ullman K. S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 2011, 71, 5579–87. 10.1158/0008-5472.CAN-11-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Pal S.; Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28, 6262–77. 10.1128/MCB.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T.-Y. W.; Juan C.-C.; Hisa J.-Y.; Su L.-J.; Lee Y.-C. G.; Chou H.-Y.; Chen J.-M. M.; Wu Y.-C.; Chiu S.-C.; Hsu C.-P.; Liu K.-L.; Yu C.-T. R. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Science 2012, 103, 1640–1650. 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis V.; Hu Y. J.; Baiocchi R. A.; Imbalzano A. N.; Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–41. 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh C. M.; Bezzi M.; Low D. H.; Ang W. X.; Teo S. X.; Gay F. P.; Al-Haddawi M.; Tan S. Y.; Osato M.; Sabo A.; Amati B.; Wee K. B.; Guccione E. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- Chan-Penebre E.; Kuplast K. G.; Majer C. R.; Boriack-Sjodin P. A.; Wigle T. J.; Johnston L. D.; Rioux N.; Munchhof M. J.; Jin L.; Jacques S. L.; West K. A.; Lingaraj T.; Stickland K.; Ribich S. A.; Raimondi A.; Scott M. P.; Waters N. J.; Pollock R. M.; Smith J. J.; Barbash O.; Pappalardi M.; Ho T. F.; Nurse K.; Oza K. P.; Gallagher K. T.; Kruger R.; Moyer M. P.; Copeland R. A.; Chesworth R.; Duncan K. W. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–7. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. The cation-pi interaction. Acc. Chem. Res. 2013, 46, 885–93. 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.; Chen L.; Civiello R.; Pin S. S.; Xu C.; Kostich W.; Kelley M.; Conway C. M.; Macor J. E.; Dubowchik G. M. Calcitonin gene-related peptide (CGRP) receptor antagonists: Pyridine as a replacement for a core amide group. Bioorg. Med. Chem. Lett. 2012, 22, 2917–2921. 10.1016/j.bmcl.2012.02.065. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- The PMT enzymes screened were PRMT1, PRMT3, PRMT4, PRMT6, PRMT7, PRMT8, DOT1L, SMYD2, SMYD3, SETBD1, SETD7, EZH1, EZH2, SETD2, NSD1, WHSC1L1, WHSC1, SUV39H1, EHMT1, and EHMT2.
- Rioux N.; Duncan K. W.; Lantz R. J.; Miao X.; Chan-Penebre E.; Moyer M. P.; Munchhof M. J.; Copeland R. A.; Chesworth R.; Waters N. J. Species differences in metabolism of EPZ015666, an oxetane-containing protein arginine methyltransferase-5 (PRMT5) inhibitor. Xenobiotica 2015, 10.3109/00498254.2015.1072253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.