

Abstract
Bruton’s tyrosine kinase (BTK) is a Tec family kinase with a well-defined role in the B cell receptor (BCR) pathway. It has become an attractive kinase target for selective B cell inhibition and for the treatment of B cell related diseases. We report a series of compounds based on 8-amino-imidazo[1,5-a]pyrazine that are potent reversible BTK inhibitors with excellent kinase selectivity. Selectivity is achieved through specific interactions of the ligand with the kinase hinge and driven by aminopyridine hydrogen bondings with Ser538 and Asp539, and by hydrophobic interaction of trifluoropyridine in the back pocket. These interactions are evident in the X-ray crystal structure of the lead compounds 1 and 3 in the complex with the BTK enzyme. Our lead compounds show desirable PK profiles and efficacy in the preclinical rat collagen induced arthritis model.
Keywords: BTK, rheumatoid arthritis (RA), rat CIA model, kinase selectivity, human peripheral blood mononuclear cells (hPBMC), whole blood, pharmacokinetic, X-ray crystal structure
Rheumatoid arthritis (RA) is an autoimmune disease that causes chronic inflammation of the joints, the surrounding tissues, and other organs in the body. Initiation of the disease involves the systematic dysregulation of T and B lymphocytes, which leads to a breach of self-tolerance, resulting in immune responses directed against self-antigens.1 Despite available efficacious biological agents for the treatment of this condition, not all patients tolerate or respond to those therapies, leaving open the need for novel agents with different mechanisms of action. Moreover, orally bioavailable small molecule drugs are desirable in the treatment of RA as they offer an alternative to parenteral administration of the biologic agent. The ability of small molecule therapies to effectively treat rheumatoid arthritis was demonstrated by Tofacitinib, a selective JAK inhibitor, which achieved efficacy comparable to biological agents.2,3
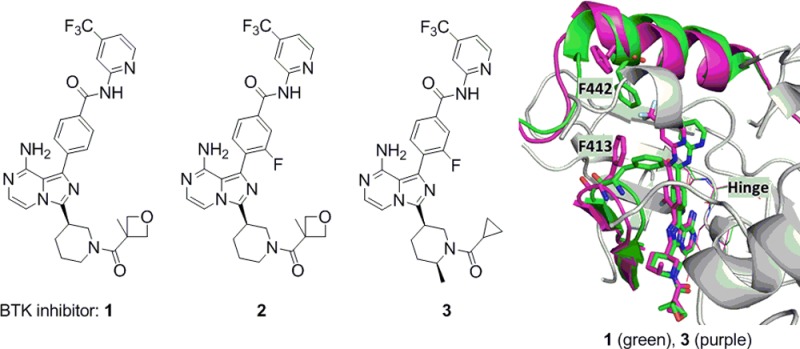
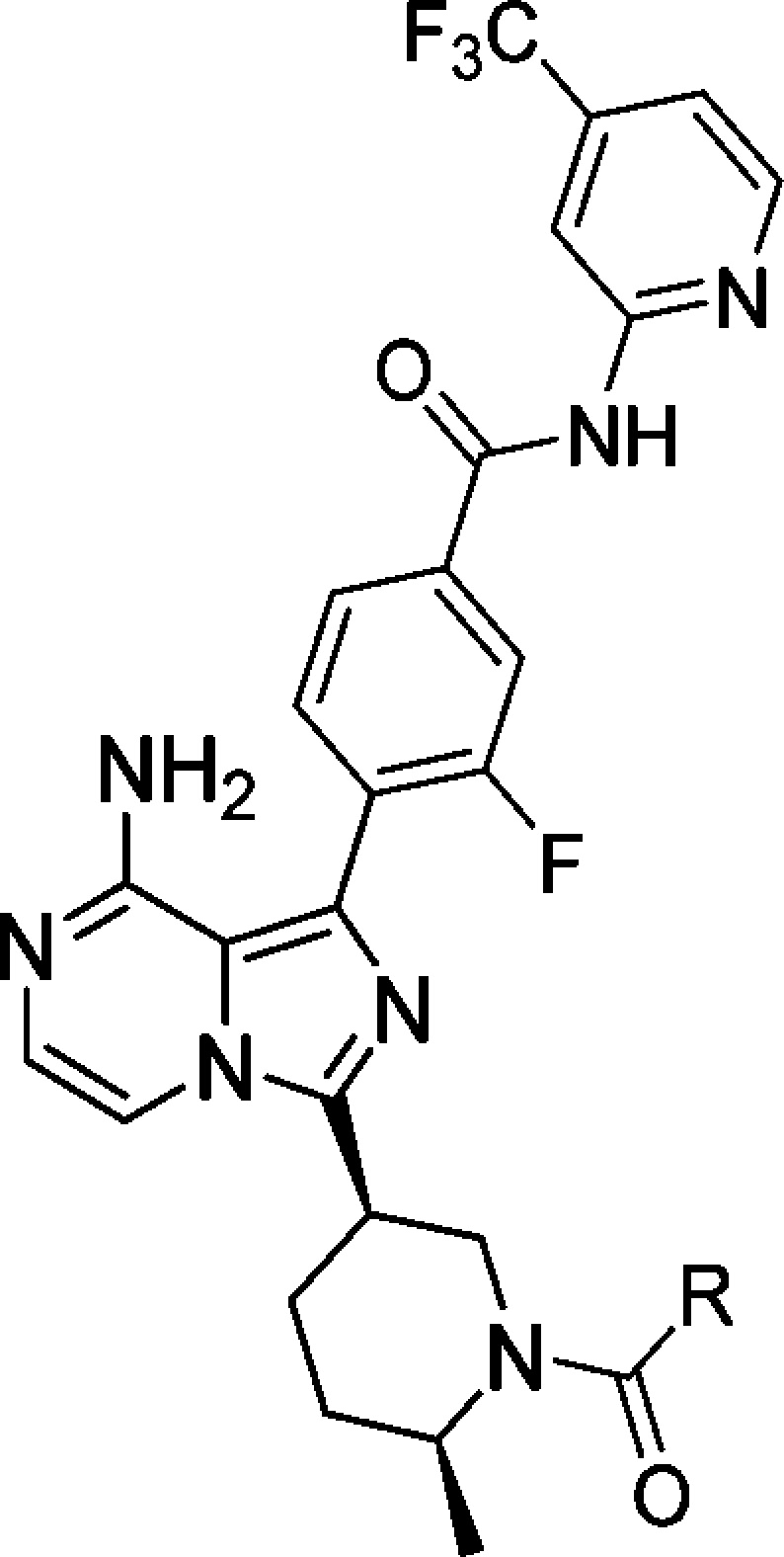
Bruton’s tyrosine kinase (BTK) is a Tec family kinase expressed in certain immune cells including B cells, mast cells, and macrophages.4,5 It plays a critical role in multiple pathways such as the B cell receptor (BCR) and Fcγ receptor (FcR) signaling cascades, where it regulates the survival, activation, proliferation, differentiation, and maturation of B cells. BTK’s role in these pathways makes it a uniquely attractive target for the treatment of B cell related diseases. In fact, several groups have reported small molecule BTK selective inhibitors as cancer therapies or for the treatment of RA,6 with ibrutinib currently approved for mantle cell lymphoma and chronic lymphocytic leukemia.7 Ibrutinib also shows excellent efficacy in the rat collagen induced arthritis model (CIA), indicating its potential use for the treatment of RA.8 Ibrutinib is an orally administered selective BTK inhibitor, which covalently binds to the sulfhydryl group of C481 leading to irreversible inhibition of its kinase activity. A small portion (5.3%) of patients encountered relapse during ibrutinib therapy for chronic lymphocytic leukemia, majorly due to a cysteine to serine mutation at C481, which results in a reduction in binding affinity of ibrutinib to BTK.9 A potent noncovalent BTK inhibitor, which does not utilize C481 covalent binding for affinity, could still be efficacious for this portion of patients with mutation. Several research groups have reported different noncovalent binding BTK inhibitors.6 This letter describes the discovery of the reversible noncovalent binding, potent and selective BTK inhibitors based on 8-amino-imidazo[1,5-a]pyrazines, exemplified by compounds 1–3 (Figure 1).
Figure 1.

Structures of ibrutinib and compounds 1–3.
Preparation of this series of BTK inhibitors is depicted in Schemes 1 to 3. The modular nature of the synthetic route allows for quick SAR exploration around various parts of the BTK inhibitor series. Scheme 1 depicts the preparation of the boronic ester intermediate 7. 4-Bromo-3-fluorobenzoic acid 4 was converted to the intermediate benzoyl chloride by the treatment with oxalyl chloride/DMF in dichloromethane, and then further converted to 6 by coupling with 4-trifluoromethyl-pyridine-2-amine 5 (diisopropylethylamine and a catalytic amount of DMAP in acetonitrile). The aromatic bromine in 6 was then converted to boronic acid pinacol ester 7, by reacting with bis(pinacolato) diboron, catalyzed by palladium acetate with XPhos. Commercially available 4-bromobenzoic acids, with different substitutions on the ring, facilitated SAR development for different substitutions on the phenyl ring. Similarly, when different substituted pyridine-2-amines or heterocyclic amines are used for the synthesis in Scheme 1, optimization of substitutions on the pyridine ring or the heterocyclic replacements can be quickly accomplished.
Scheme 1.

Reagents: i. (COCl)2, DMF,CH2Cl2, rt; ii. DIEA, DMAP, THF, 50 °C, 12 h, 77% two steps; iii. Bis(pinacolato) diboron, Pd(OAc)2, XPhos, KOAc, dioxane, 90 °C, 6 h, 75%.
Scheme 3.

Reagents. i. Pd(dppf)CH2Cl2, 2 M K2CO3 (aq), dioxane, 60 °C, o/n, 80%; ii. TMSI, CH2Cl2, 5 °C, 99%; iii. HATU, DIEA, DMF, 0 °C, 1 h, 70%.
The synthesis of intermediate 13 is depicted in Scheme 2. Starting from (3-chloropyrazin-2-yl)methanamine 8, coupled with (R)-N-Boc-piperidine-3-carboxylic acid 9, using HATU as coupling reagent and triethylamine as the base, provided compound 10. Cyclization of the amide in 10 to imidazole was facilitated by treatment with phosphorus oxychloride in acetonitrile at 80 °C for 5 h, followed by careful quenching with 20% aqueous ammonium solution and crushed ice to provide intermediate 11 in good yield. Bromination of intermediate 11 was carried out using N-bromosuccinimide in DMF at room temperature for 1 h, producing intermediate 12 in quantitative yield. The chloro intermediate 12 was converted to 13 in high yield with an ammonia solution, in isopropanol, at 120 °C in a sealed vessel overnight. SAR studies with different substitutions on the piperidine ring were achieved with substituted piperidine-3-carboxylic acids, which were either commercially available or readily prepared.
Scheme 2.

Reagents. i. HATU, TEA, CH2Cl2, 0 °C; ii. POCl3, CH3CN, 57% two steps; iii. NBS, DMF, rt, 95%; iv. 2 M NH3 in IPA, 120 °C, 97%.
Suzuki coupling (Scheme 3) of 7 and 13, using palladium catalyst such as Pd(DPPF)CH2Cl2 and potassium carbonate 2 M aqueous solution in dioxane at 60 °C overnight provided a good yield of product 14. In the following step, the Cbz group was successfully removed with treatment of iodotrimethylsilane in methylene chloride at low temperature in quantitative yield. The free piperidine amine was then coupled with acids such as 3-methyloxytane-3-carboxylic acid 16, using HATU, with Hunig’s base in DMF to provide the final product 2.10 SAR for optimizing amide substitutions was done using different acid to replace 16 at the final amide formation step.
All BTK inhibitors were evaluated in both a BTK enzymatic assay11 and a human PBMC functional assay.11 SAR was carried out on different areas of this series of BTK inhibitors based on 8-amino-imidazo[1,5-a]pyrazine core, using different starting materials as indicated in the aforementioned synthesis schemes. Table 1 describes the activities of compounds with different aromatic amines, which make up the central amide. 2-Pyridyl amides are more potent than other heteroaromatic amides such as pyrimidine (26, 27), pyrazine (28), pyridiazine (25), and thioazole (29, 30). 4-Position substitutions on the 2-pyridyl were best tolerated with different sizes and heteroatoms. These structural changes also contributed to tuning the off target activities and pharmacokinetic profiles of this series of compounds while maintaining excellent potencies for the BTK inhibition.
Table 1. SAR of the Amide Aromatic Amines.

The data are average of at least two repeated tests. ND: not determined.
SAR with different substitutions on the middle phenyl ring are described in Table 2. Compound 1 with no substitution has excellent BTK inhibition potency. The 3-fluoro analogue (2) is ∼10× more potent than the 2-fluoro (31) and 2-chloro (32) substitutions. However, compounds with a methoxy substitution on 2- and 3-positions (35 and 33) are equally potent. The 2,3-difluoro compound (36) has similar enzyme binding activity as 2, but was approximately 2-fold less potent in the cellular assay.
Table 2. SAR on Substitutions on the Phenyl Linker.
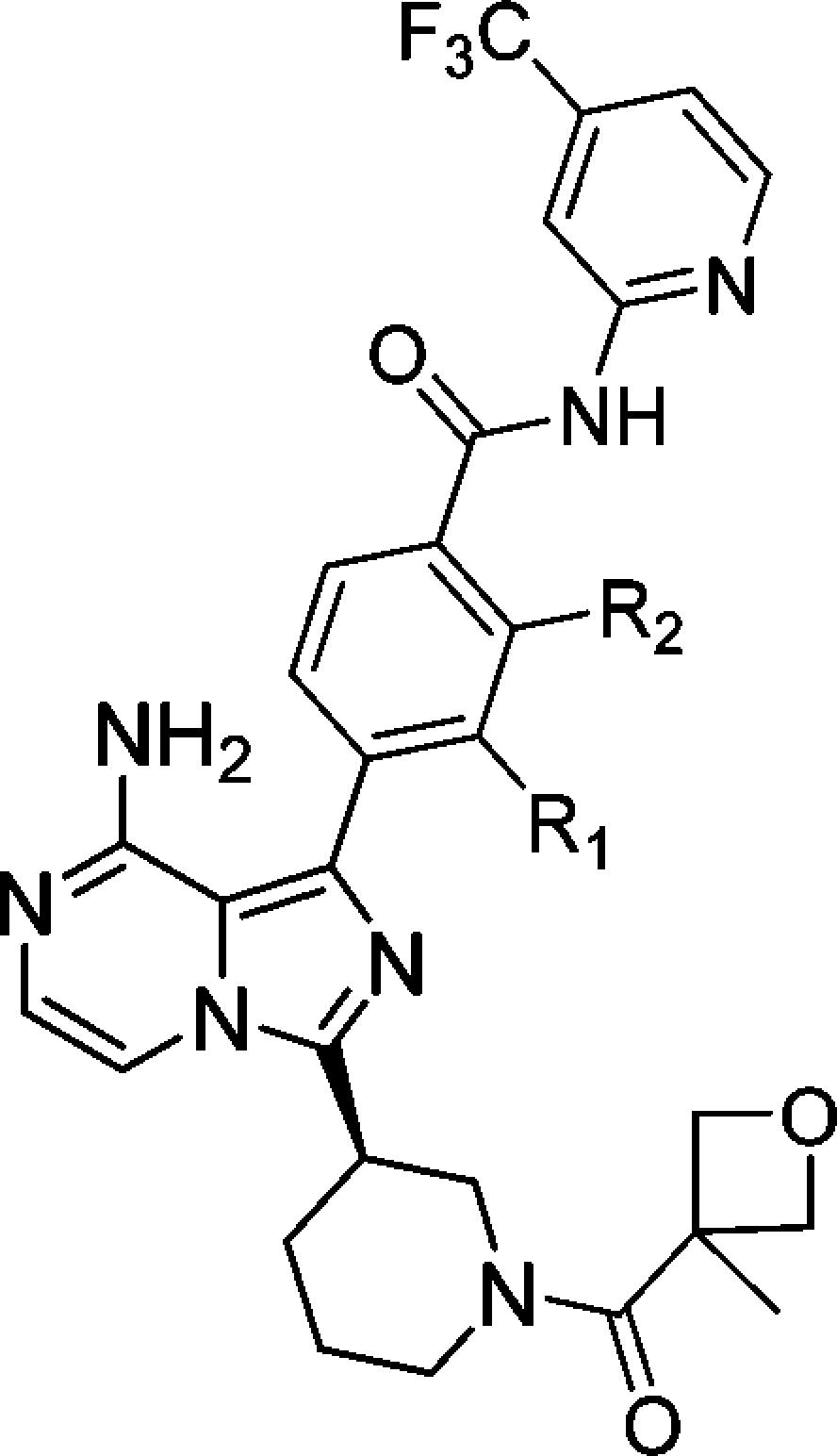
| compd no. | R1,R2 | BTK enzymatic assaya IC50 (nM) | hPBMC assaya IC50 (nM) |
|---|---|---|---|
| 1 | H, H | 0.27 | 8.0 |
| 2 | F, H | 0.32 | 7.5 |
| 31 | H, F | 2.5 | 81 |
| 32 | H, Cl | 6.3 | 181 |
| 33 | MeO, H | 1.1 | 40 |
| 34 | CF3O, H | 38 | ND |
| 35 | H, MeO | 0.98 | 48 |
| 36 | F, F | 0.41 | 18 |
The data are average of at least two repeated tests. ND: not determined.
Substitutions on the bottom piperidine ring were explored in an attempt to increase the steric hindrance around the amide, aiming to slow down the potential metabolic cleavage of the amide (Table 3). Methyl (37), difluoromethyl (38), methoxymethyl (39), and trifluoromethyl (40) were applied. Methyl substitution provides the best BTK enzymatic and cellular potency.
Table 3. SAR of Substitutions on the Piperidine Ring.
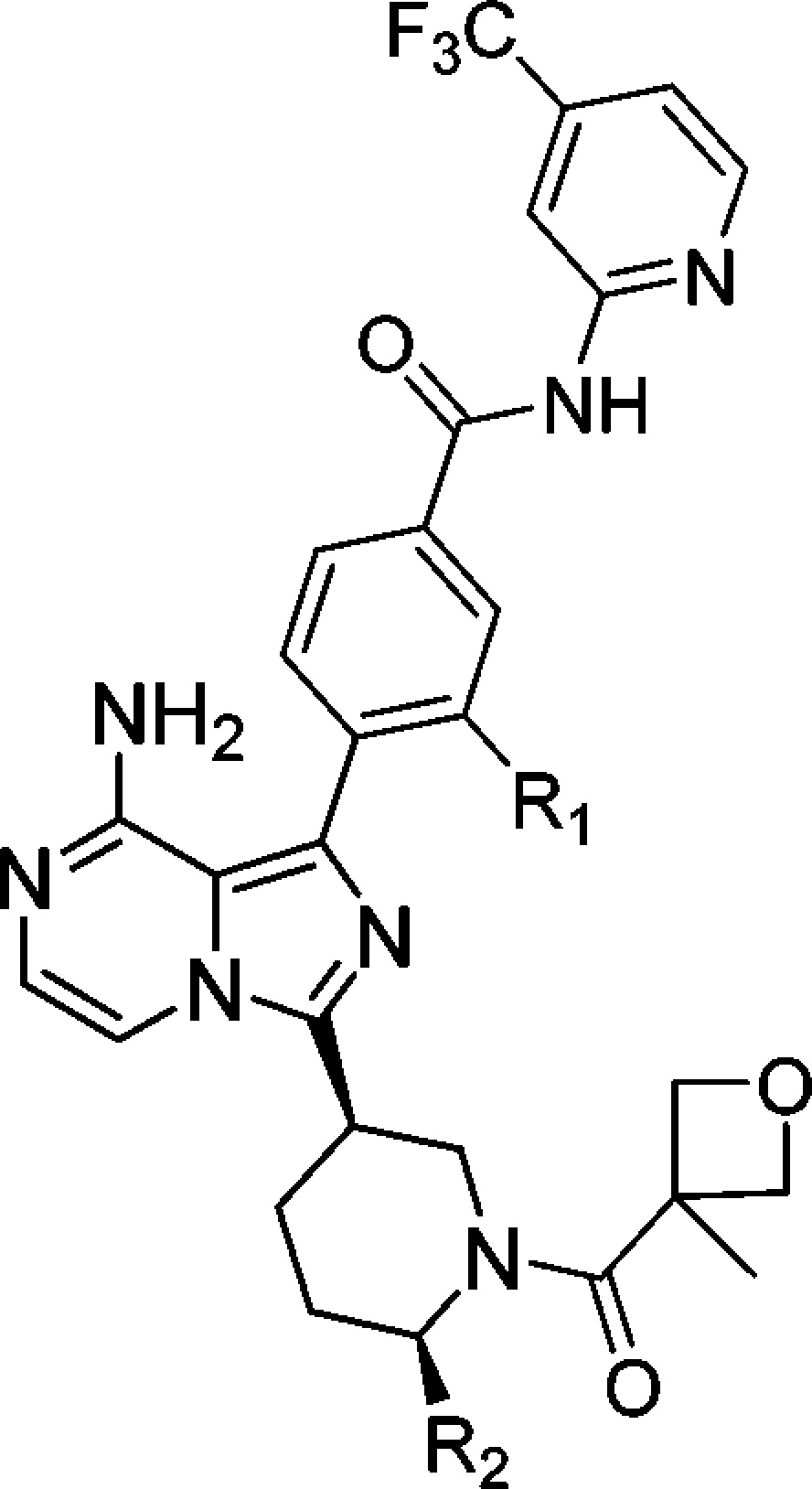
| compd | R1,R2 | BTK enzymatic assaya IC50 (nM) | hPBMC assaya IC50 (nM) |
|---|---|---|---|
| 1 | H, H | 0.27 | 8.0 |
| 37 | F, Me | 0.17 | 3.0 |
| 38 | H, CF2H | 0.86 | 20 |
| 39 | H, MeOCH2 | 0.31 | 4.0 |
| 40 | F, CF3 | 3.7 | ND |
The data are average of at least two repeated tests. ND: not determined.
Variation of the piperidine amide was quickly explored by the library synthesis approach of amide couplings. Functional groups in different sizes were tolerated, showing excellent potency in the enzyme assay. Thus, given the wide tolerance for change, SAR in this area was applied to adjust the physical chemical properties and off-target activities for this series. Table 4 lists 12 different amides on the piperidine (37, 3, 41–50), with great BTK enzyme inhibition potency and cellular activity. Based on the overall profile of this series, compounds 1, 2, and 3 were selected for further profiling because of their excellent enzymatic and cellular potency.
Table 4. SAR of Amide Substitutions on Piperidine.

The data are average of at least two repeated tests.
The kinase selectivity profiles for compounds 1, 2, and 3 were accessed over the closely related Tec and Src family kinases. Additionally, the activity profiles of compounds 1, 2, and 3 were investigated by testing them at three concentrations (1, 0.1, and 0.01 μM) against a commercially available panel of 265 human kinases.11 Ten point compound concentration response curves were generated (at half-log intervals from 1 μM) for 17 kinases that demonstrated activity in the initial test. A summary of data for this kinase screen set is shown in Table 5. Fold selectivity over BTK enzyme is in parentheses. The selectivity ratios against these kinases, most from the Tec and Src family, are greater than 50-fold, except for compound 3, which was 34-fold for BMX and 38-fold for LCK.
Table 5. Selectivity of 1, 2, and 3 over SRC Family Kinases.
| kinase | compd 1 IC50 (nM) (fold selectivity) | compd 2 IC50 (nM) (fold selectivity) | compd 3 IC50 (nM) (fold selectivity) |
|---|---|---|---|
| BTK | 0.27 | 0.32 | 0.31 |
| BLK | 84 (311) | 42 (131) | 20 (65) |
| BMX | 28 (104) | 20 (62) | 11 (34) |
| CSK | ND | 253 (791) | 201 (648) |
| ERBB4 | 118 (437) | 81 (253) | 65 (203) |
| FGR | ND | 55 (172) | 37 (116) |
| FRK | 361 (1337) | 187 (584) | 111 (347) |
| FYN | ND | 322 (1006) | 198 (619) |
| ITK | ND | 921 (2878) | 1200 (3750) |
| LCK | 36 (133) | 22 (69) | 12 (38) |
| LYNB | 399 (1478) | 184 (575) | 131 (423) |
| PTK6 | 66 (244) | 63 (197) | 60 (194) |
| SRC | ND | 116 (362) | 89 (287) |
| SRMS | 32 (119) | 24 (75) | 23 (74) |
| TEC | 167 (619) | 69 (216) | 63 (203) |
| TXK | 49 (181) | 32 (100) | 33 (106) |
| YES1 | ND | 43 (134) | 21 (68) |
Cocrystals of compounds 1 and 3 with BTK were obtained.12 The key interactions of compound 1 with BTK are shown in Figure 2A. The 8-amino and 7-nitrogen on the imidazo[1,5-a]pyrazine each make one hydrogen bond with the BTK hinge region, and the primary amine is also within H-bonding distance to the gatekeeper T474 side-chain alcohol, while the 2-nitrogen forms a hydrogen bond with a water molecule. The amide carbonyl, on the piperidine ring, is oriented for a H-bond with the amide nitrogen of peptide G480 and C-481. A key interaction of the 2-amino-pyridine is a bidentate hydrogen bond interaction with S538 side chain hydroxy and the amide nitrogen of D539. The trifluoromethyl and the pyridine ring are in a hydrophobic pocket with great flexibility, which could be unique to the BTK enzyme. The inhibitor takes advantage of the wide access to the back pocket because of the smaller threonine gatekeeper. Overlay of the X-ray crystal structures of 1 and 3 reveals significant differences in binding mode around the trifluoromethylpyridine. There are no hydrogen bond interactions of 3 at the aminopyridine with the S538 and D539. The trifluoromethylpyridine ring extends deeper into the back pocket with the side chain of F442 and F413 moving away to accommodate a different binding mode in the complex with 3. This binding into the deep back pocket compensates the loss of interaction of the aminopyridine bidentate hydrogen bond. In the mean time, significant away movement of the loop close to the axial methyl substitution on the piperidine ring for compound 3 is noticed, which allows compound 3 to slide deeper into the pocket. The flexibility of the BTK enzyme in this region allows quite broad SAR with substitutions of various sizes (at the pyridine 4-position) to fit in the back pocket as we described before, allowing the optimization of potency and kinase selectivity. All these interactions provide this series of BTK inhibitors with their potency and selectivity.
Figure 2.

(A) X-ray crystal structure of 1 in the active site of BTK enzyme with hydrogen bond interactions to the key residues. (B) The overlay of X-ray crystal structures of 1 (green) and 3 (purple) in the active site of BTK enzyme showing the flexibility of the enzyme for the hydrophobic pocket around the trifluoromethylpyridine with significant movement of F442 and F413.
Compounds 1, 2, and 3 were evaluated in a human whole blood assay that assessed the effect of compounds on the upregulation of CD69 on B cells in response to BCR-cross-linking by an anti-CD79b antibody.11 Compounds 1, 2, and 3 all demonstrated excellent activity in the human whole blood assay with IC50 of 120 (n = 2), 113 (n = 11), and 94 (n = 11) nM, respectively.
In rat pharmacokinetic studies, compounds 1, 2, and 3 exhibited reasonable exposure, clearance, half-life, and bioavailabilities (Table 6). Additionally, compounds 2 and 3 were evaluated in dog pharmacokinetic studies, and compound 3 showed higher drug levels with a longer half-life compared to compound 2.
Table 6. Pharmacokinetic Parameters of Compounds 1, 2, and 3.
| compd |
1 |
2 |
3 |
|||
|---|---|---|---|---|---|---|
| rat | IV dose | PO dose | IV dose | PO dose | IV dose | PO dose |
| dose (mg/kg) | 4 | 10 | 2 | 5 | 2 | 5 |
| Cl (mL/min/kg) | 6.3 | 24 | 18 | |||
| Vss (L/kg) | 0.59 | 1.7 | 2.6 | |||
| AUCN (μM·h) | 5.1 | 1.1 | 1.6 | |||
| T1/2 (h) | 1.2 | 1.4 | 1.8 | |||
| Cmax (μM) | 4.8 | 1.0 | 0.40 | |||
| Tmax (h) | 2.2 | 0.5 | 1.0 | |||
| F (%) | 84 | 77 | 22 | |||
| dog | IV dose | PO dose | IV dose | PO dose | IV dose | PO dose |
|---|---|---|---|---|---|---|
| dose (mg/kg) | 1 | 2 | 1 | 2 | ||
| Cl (mL/min/kg) | 11 | 1.5 | ||||
| Vss (L/kg) | 2.0 | 0.65 | ||||
| AUCN (μM·h) | 3.0 | 20 | ||||
| T1/2 (h) | 4.1 | 6.5 | ||||
| Cmax (μM) | 0.78 | 3.9 | ||||
| Tmax (h) | 1.0 | 2.0 | ||||
| F (%) | 60 | 82 |
Compound 3 was tested for efficacy in the rat model of collagen-induced arthritis (CIA).11 In a prophylactic treatment protocol, compound 3 demonstrated a dose-dependent (1, 3, 10, and 30 mg/kg, PO, QD) decrease in paw thickness, as indicated by significant reduction (p < 0.0001) of paw thickness through day 30 consequent to CIA induction (Figure 3).
Figure 3.
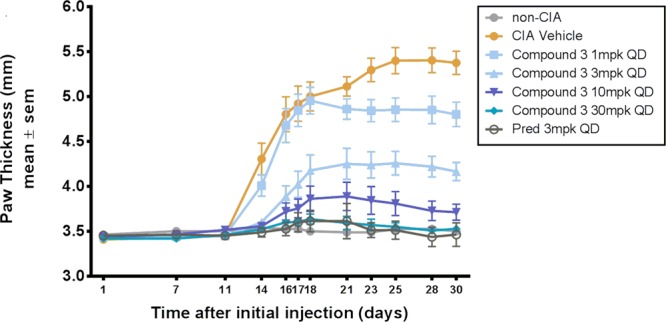
Dose-dependent changes in paw thickness in a rat CIA model at different doses of 3.
In summary, we described the identification of reversible BTK inhibitors based on the 8-aminoimidazo[1,5-a]pyrazine core. The inhibitors are characterized by high enzymatic potency, selectivity over other kinases, and acceptable oral bioavailability. SAR studies of the different parts of this series of inhibitors led to the identification of several lead compounds such as 1, 2, and 3. Further characterization by X-ray crystal structures revealed specific interactions between the inhibitors and the BTK enzyme, which likely account for the observed BTK inhibition activity and selectivity over other kinases. Compound 3 combines an optimization of the BTK enzyme binding, cellular functional activity, human whole blood assay activity, kinase selectivity, and in vivo pharmacokinetic profile. This orally available noncovalent BTK inhibitor 3 displays efficacy in reducing paw thickness in a rat model of collagen-induced arthritis. Further investigation of the SAR of this series of BTK inhibitors will be the subject of another report.
Acknowledgments
We thank Mr. Wilfredo Pinto at Merck Research Laboratories for measuring the high-resolution mass and Dr. Lu Yang for taking 1HNMR and 13CNMR for key compounds.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00463.
Synthetic methods, characterization of key compounds, and biology assay protocols (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Burmester G. R.; Feist E.; Dorner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 77. 10.1038/nrrheum.2013.168. [DOI] [PubMed] [Google Scholar]
- Flanagan M. E.; Blumenkopf T. A.; Brissette W. H.; Brown M. F.; Casavant J. M.; Shang-Poa C.; Doty J. L.; Elliott E. A.; Fisher M. B.; Hines M.; Kent C.; Kudlacz E. M.; Lillie B. M.; Magnuson K. S.; McCurdy S. P.; Munchhof M. J.; Perry B. D.; Sawyer P. S.; Strelevitz T. J.; Subramanyam C.; Sun J.; Whipple D. A.; Changelian P. S. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem. 2010, 53, 8468–8484. 10.1021/jm1004286. [DOI] [PubMed] [Google Scholar]
- Garber K. Pfizer’s JAK inhibitor sails through phase 3 in rheumatoid arthritis. Nat. Biotechnol. 2011, 29, 467–468. 10.1038/nbt0611-467. [DOI] [PubMed] [Google Scholar]
- Vetrie D.; Vorechovsky I.; Sideras P.; Holland J.; Davies A.; Flinter F.; Hammarstrom L.; Kinnon C.; Levinsky R.; Bobrow M.; Smith C. I. E.; Bentley D. R. The gene involved in X-linked agammaglobulinaemia is a member of the src family of proteintyrosine kinases. Nature 1993, 361, 226–233. 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- Tsukada S.; Saffran D. C.; Rawlings D. J.; Parolini O.; Allen R. C.; Klisak I.; Sparkes R. S.; Kubagawa H.; Mohandas T.; Quan S.; Belmont J. W.; Cooper M. D.; Conley M. E.; Witte O. N. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. 10.1016/0092-8674(93)90667-F. [DOI] [PubMed] [Google Scholar]
- For reviews:; a Hendriks R. W.; Yuvaraj S. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14 (4), 219–232. 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]; b Whang J. A.; Chang B. Y. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discovery Today 2014, 19 (8), 1200–1204. 10.1016/j.drudis.2014.03.028. [DOI] [PubMed] [Google Scholar]; c Lou Y.; Owens T. D.; Kuglstatter A.; Kondru R. K.; Goldstein D. M. Bruton’s Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evalution for Inflammatory Diseases and B Cell Malignancies. J. Med. Chem. 2012, 55, 4539–4450. 10.1021/jm300035p. [DOI] [PubMed] [Google Scholar]
- Cameron F.; Sanford M. Ibrutinib: First Global Approval. Drugs 2014, 74 (2), 263–271. 10.1007/s40265-014-0178-8. [DOI] [PubMed] [Google Scholar]
- Chang B. Y.; Huang M. M.; Francesco M.; Chen J.; Sokolove J.; Magadala P.; Robinson W. H.; Buggy J. J. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Research & Therapy 2011, 13 (4), R115. 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach J. A.; Furman R. R.; Liu T.-M.; Ozer H. G.; Zapatka M.; Ruppert A. S.; Xue L.; Li D. H.; Steggerda S. M.; Versele M.; Dave S. S.; Zhang J.; Yilmaz A. S.; Jaglowski S. M.; Blum K. A.; Lozanski A.; Lozanski G.; James D. F.; Barrientos J. C.; Lichter P.; Stilgenbauer S.; Buggy J. J.; Chang B. Y.; Johnson A. J.; Byrd J. C. Resistance Mechanisms for Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The detailed synthetic procedures and characterization of the key compounds can be found in the Supporting Information.
- The protocol for the following assays are described in the Supporting Information: BTK enzymatic binding assay; BTK human PMBC functional assay; kinase panel selectivity assay; BTK human whole blood assay; the rat model of collagen-induced arthritis.
- X-ray crystal methods will be published elsewhere. The coordinations of the X-ray crystal structures for compounds 1 and 3 have been published on protein data bank with codes 5FBN and 5FBO, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.