

Abstract
Objective:
To investigate the association between periventricular white mater hyperintensities (PVWMH) and biomarkers of elevated cerebral β-amyloid (Aβ) in the Alzheimer's Disease Neuroimaging Initiative, a large prospective multicenter observational study.
Methods:
The burden of frontal, parietal, and occipital PVWMH on 3T fluid-attenuated inversion recovery MRI was evaluated in 698 cognitively normal participants and participants with mild cognitive impairment (MCI) using a novel semiquantitative visual rating scale. Results were correlated with CSF-Aβ, florbetapir-PET, and fluorodeoxyglucose (FDG)–PET.
Results:
Increased burden of parietal, occipital, and frontal PVWMH was associated with elevated cerebral amyloid evidenced by high florbetapir-PET signal (p < 0.01) and low CSF-Aβ (p < 0.01). In logistic regression models, including PVWMH, age, sex, APOE status, vascular risk factors, pulse pressure, vascular secondary prevention medications, education, ethnicity, and race, parietal, occipital, and frontal PVWMH burden was independently associated with high florbetapir-PET uptake (p < 0.05). In a similar logistic regression model, parietal and occipital (p < 0.05) but not frontal (p = 0.05) PVWMH were independently associated with CSF-Aβ. Weaker associations were found between parieto-occipital PVWMH and elevated CSF-tau (p < 0.05) and occipital PVWMH and elevated CSF-phospho-tau (p < 0.05). PVWMH were associated with cerebral hypometabolism on FDG-PET independent of CSF-Aβ levels (p < 0.05). Absolute and consistency of agreement intraclass correlation coefficients were, respectively, 0.83 and 0.83 for frontal, 0.78 and 0.8 for parietal, and 0.45 and 0.75 for occipital PVWMH measurements.
Conclusions:
Increased PVWMH were associated with elevated cerebral amyloid independent of potential confounders such as age, APOE genotype, and vascular risk factors. The mechanisms underlying the association between PVWMH and cerebral amyloid remain to be clarified.
Pathologic and imaging studies have shown that in Alzheimer disease (AD), cerebral small vessel disease (SVD) is coexistent in up to 80% and hastens cognitive deterioration.1 Whether this reflects parallel neuronal injury or synergistic worsening of AD pathogenic mechanisms remains unclear.2
There has been recent interest in the putative role of SVD in disruption of physiologic interstitial fluid bulk flow and paravascular mechanisms of β-amyloid (Aβ) clearance.3 If SVD is an important driver of the cortical Aβ accumulation that occurs in AD, it would be expected that injury to arteries in the cortical area would be prominent. Cerebral amyloid angiopathy (CAA) and arteriosclerosis are the main causes of cortical arterial injury and result in luminal narrowing and tortuosity.4 The periventricular area is the deepest tissue supplied by cortical arteries and therefore potentially vulnerable to pathologies that damage cortical arteries and promote distal hypoperfusion.5 The etiology of periventricular white matter hyperintensities (PVWMH) remains unclear and may be multifactorial including hypoperfusion, blood–brain barrier leakage, inflammation, degeneration, and amyloidosis.6 PVWMH have recently been shown to develop by acute infarction adjacent to older lesions with extension occurring proximally towards the cortical surface favoring cortical arterial disease and hypoperfusion as an important cause.7,8
We hypothesized that measuring PVWMH burden would provide a quantitative measure of cortical arterial disease and facilitate exploration of associations with cerebral amyloid deposition. Therefore, in the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort, we investigated the association between CSF and PET markers of elevated Aβ and PVWMH.
METHODS
Overall study design and population.
MRI and PET, clinical, neuropsychological, and APOE genotype data used in this article were downloaded from the ADNI online repository on August 1, 2015 (www.adni.loni.usc.edu). All downloaded clinical data were interpreted by a study physician (see appendix e-1 on the Neurology® Web site at Neurology.org). ADNI was launched in 2003 as a public–private partnership, led by Michael W. Weiner, MD. The primary goal was to study serial MRI, PET, clinical, neuropsychological, and other biomarker data in normal cognition (NC), mild cognitive impairment (MCI), and early AD to improve future clinical trial design (see www.adni-info.org). Patients were recruited from over 50 centers across North America. Individuals enrolled in ADNI with NC or MCI, for whom clinical, fluid-attenuated inversion recovery (FLAIR) 3T MRI, and CSF-Aβ or florbetapir-PET data were available were included in the current study (appendix e-1).
MRI acquisition and PVWMH quantification.
A standardized multisequence MRI protocol was used at participating ADNI sites.9 The FLAIR images of an initial 210 randomly selected participants with MCI were evaluated using commonly applied visual rating scales for global white matter hyperintensity burden (table e-1). Based on the results, a novel PVWMH rating scale was developed (appendix e-1). A single rater (O.O.A.-J.) trained in the described semiquantitative visual rating scale and blinded to CSF and PET data rated all cases. To assess the validity and reproducibility of the rating method, a second experienced rater (M.M.) independently evaluated 30 randomly selected cases for PVWMH blinded to previous rating and CSF and PET data.
Frontal and parietal PVWMH were quantified by measuring the length of the furthest extent of signal abnormality extending out from the frontal or parietal ventricular horn tip (figure 1, appendix e-1, figure e-1). Occipital PVWMH burden can be difficult to estimate using a single measurement line on axial MRI slices due to the complex and variable 3D geometry of the occipital ventricular extensions. We therefore developed a semiquantitative visual rating scale to estimate burden of occipital PVWMH (figure 1, appendix e-1).
Figure 1. Examples of periventricular white matter hyperintensities (PVWMH) grading and measurement.

Grade 0 (A) = no PVWMH; grade 1 (B) = PVWMH present but not extending to deep sulcal cortex; grade 2 (C) = PVWMH extending to deep sulcal cortex but not into a gyrus; grade 3 (D) = PVWMH extending into a gyrus but not reaching superficial gyral cortex in 1 gyrus; grade 4 (E) = PVWMH extending to superficial gyral cortex in 1 gyrus; grade 5 (F) = PVWMH extending to superficial gyral cortex in 2 gyri; grade 6 (G, H) = PVWMH extending to superficial gyral cortex in 3 or more gyri. Frontal periventricular hyperintensities measurement (green line in I) and parietal periventricular hyperintensities measurement (green line in J) are displayed.
Data for estimates of whole brain white mater hyperintensity (WMH) using a previously reported volumetric analysis method were downloaded from the ADNI Web site.10
PET acquisition and image analysis.
Fluorine-18 florbetapir-PET was used to estimate cerebral amyloid load. A native-space MRI was segmented and parcellated to create a summary cortical gray matter region of interest (ROI) for elevated amyloid including frontal, anterior/posterior cingulate, lateral temporal, and lateral parietal cortex subregions. A reference unaffected ROI was also defined in the cerebellum. These were coregistered with florbetapir-PET data to calculate mean cortical and reference region uptakes. A cortical summary measurement for florbetapir-PET was generated by dividing the cortical by cerebellar values (sumROI-amyloid). A sumROI-amyloid cutoff of 1.11, using this method, is equivalent to the upper 95% confidence interval (CI) above the mean of a group of young normal controls and was used to define florbetapir-PET positive and negative cases in this and other studies.11
Fluroine-18 fluorodeoxyglucose PET (FDG-PET) was performed according to standardized protocols.12 Pre-defined ROIs cited in other FDG-PET studies comparing AD, MCI, and participants with normal cognition were used for this analysis, which included coordinates for the most hypometabolic regions in AD. These subregions included the angular, inferior temporal, and posterior cingulate gyri and were combined (sumROI-FDG) to investigate for FDG-PET hypometabolism as described in previous studies.13 The sumROI-FDG, sumROI-amyloid, and subregion uptake measurements were downloaded from the ADNI online repository.
CSF analysis.
CSF analysis for Aβ, tau, and phospho-tau in the ADNI study was performed at a central laboratory at the University of Pennsylvania. Never before thawed aliquots collected at participating sites were analyzed using the xMAP Luminex platform and Innogenetics (Ghent, Belgium)/Fujirebio (Malvern, PA) AlzBio3 immunoassay kits.14 A CSF-Aβ threshold of 192 pg/mL was used to define CSF-Aβ positive and negative cases. Standard CSF-tau and CSF-phospho-tau thresholds for defining AD were also used.14
Statistical analysis.
The distribution of data was assessed using the Shapiro-Wilk test. The demographic, clinical, and genetic data described in table 1 were compared between groups using 2-sample Student t test or Wilcoxon rank-sum for interval or ordinal data, respectively, and the χ2 or Fisher exact test were used for categorical data. Frontal and parietal PVWMH measurements were scalar; therefore, associations with CSF and PET biomarkers and volumetric WMH analyses were evaluated using Student t test and Pearson correlation coefficient (r) as appropriate. For ordinal scales, including occipital PVWMH grade, the Wilcoxon rank-sum and Spearman rank correlation coefficient (ρ) were used. Interrater reliability was estimated using single-measures intraclass correlation coefficients (ICC) for both absolute and consistency of agreement using a 2-way random-effects model.15 Logistic regression models were used to estimate the marginal effect of predictors on dichotomized CSF and PET biomarkers. Linear regression models were used to estimate the effect of predictors on continuous FDG-PET data. All tests were 2-sided with a significance threshold set at p < 0.05. Analyses were performed using Stata version 13.0 (College Station, TX).
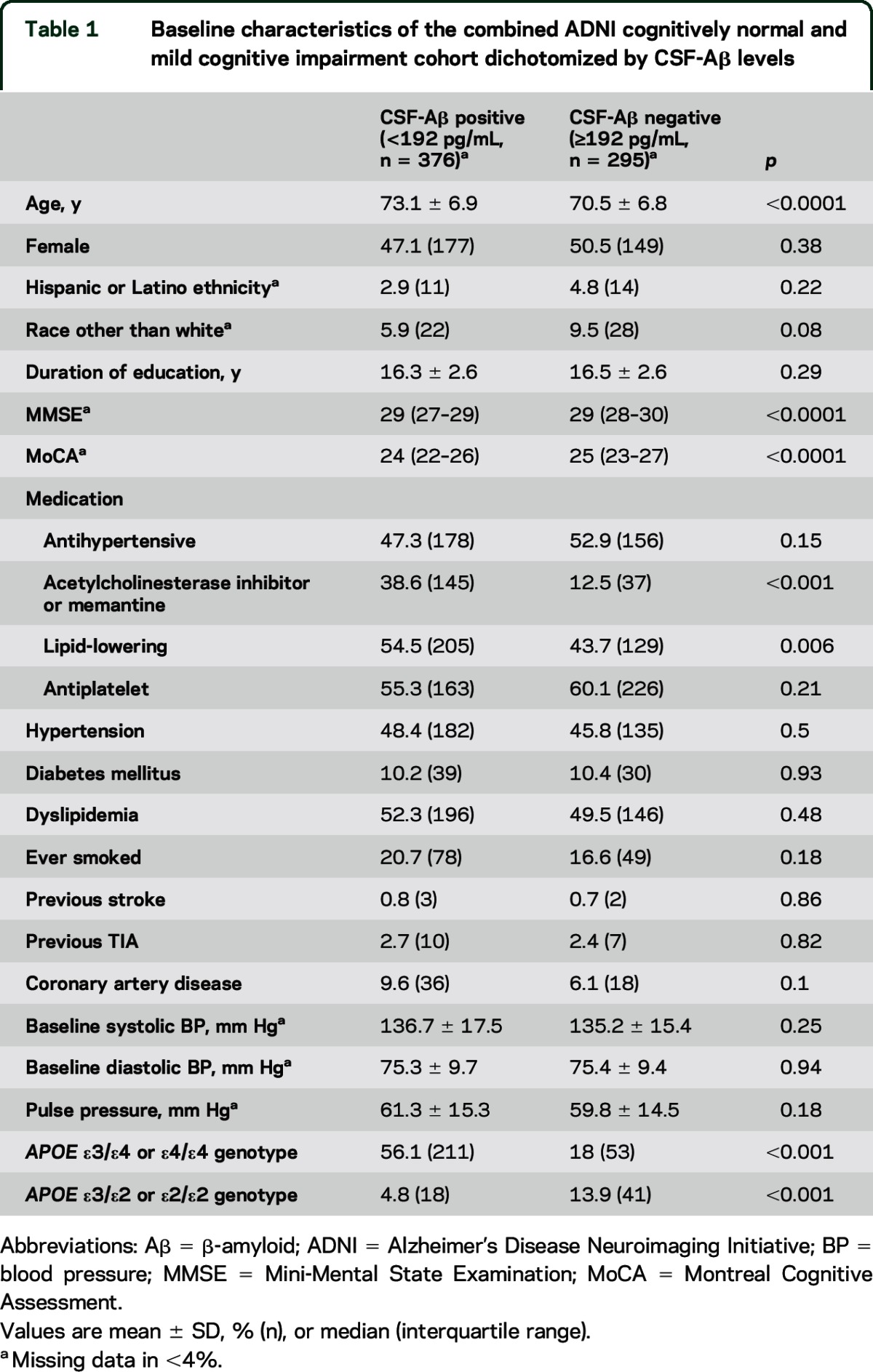
Table 1.
Baseline characteristics of the combined ADNI cognitively normal and mild cognitive impairment cohort dichotomized by CSF-Aβ levels
Standard protocol approvals, registrations, and patient consents.
Procedures were approved by an institutional ethical standards committee on human experimentation at each center. Written informed consent was obtained from all study participants or their guardians.
RESULTS
Baseline characteristics.
A total of 698 participants (263 NC and 435 MCI) were identified who met eligibility criteria. Results for the combined group are reported unless otherwise stated. Baseline demographic, clinical, neuropsychometric test, and genotype data are described in table 1 dichotomized according to a CSF-Aβ threshold of 192 pg/mL. Those with CSF-Aβ <192 pg/mL were older, had lower baseline Mini-Mental State Examination and Montreal Cognitive Assessment scores, were more likely to be taking cholinesterase inhibitors or memantine and lipid-lowering medication, were more likely to have an APOE ε3/ε4 or ε4/ε4 genotype, and were less likely to have an APOE ε3/ε2 or ε2/ε2 genotype. Vascular risk factors did not differ between the groups. Baseline characteristics of the cohort dichotomized according to regional PVWMH burden are described in table e-2.
Interrater reliability for rating of PVWMH.
Absolute and consistency of agreement ICCs were, respectively, 0.83 (CI 0.68–0.92) and 0.83 (0.67–0.91) for frontal, 0.78 (0.57–0.89) and 0.8 (0.62–0.9) for parietal, and 0.45 (0.1–0.78) and 0.75 (0.53–0.87) for occipital PVWMH measurements.
Association of PVWMH with florbetapir-PET.
Table 2 outlines the proportion of parietal, occipital, and frontal PVWMH by quartile or grade and their association with biomarkers of elevated amyloid. Parietal PVWMH burden was greater in florbetapir-PET-positive compared to florbetapir-PET-negative participants (mean parietal PVWMH measurement 7.5 mm [±5.3 mm] vs 5.3 mm [±4.8 mm], p < 0.0001). Parietal PVWMH correlated with increased florbetapir-PET uptake in the sumROI-amyloid (r = 0.18, p < 0.0001) and the lateral parietal subregion (r = 0.19, p < 0.0001). In a logistic regression model including parietal PVWMH, age, sex, APOE status, vascular risk factors, pulse pressure, vascular secondary prevention medications, education, ethnicity, and race, parietal PVWMH burden was independently associated with florbetapir-PET positivity (odds ratio [OR] 1.09 [95% CI 1.05–1.13], p < 0.001, see table 3). For example, based on the calculated adjusted OR for the combined NC and MCI group, individuals with a parietal PVWMH measurement of 10 mm were predicted to be twice as likely to have a florbetapir-PET scan consistent with AD compared to those with 1 mm.
Table 2.
Association between quartile/grade of frontal, parietal, and occipital PVWMH and biomarkers of elevated cerebral amyloid
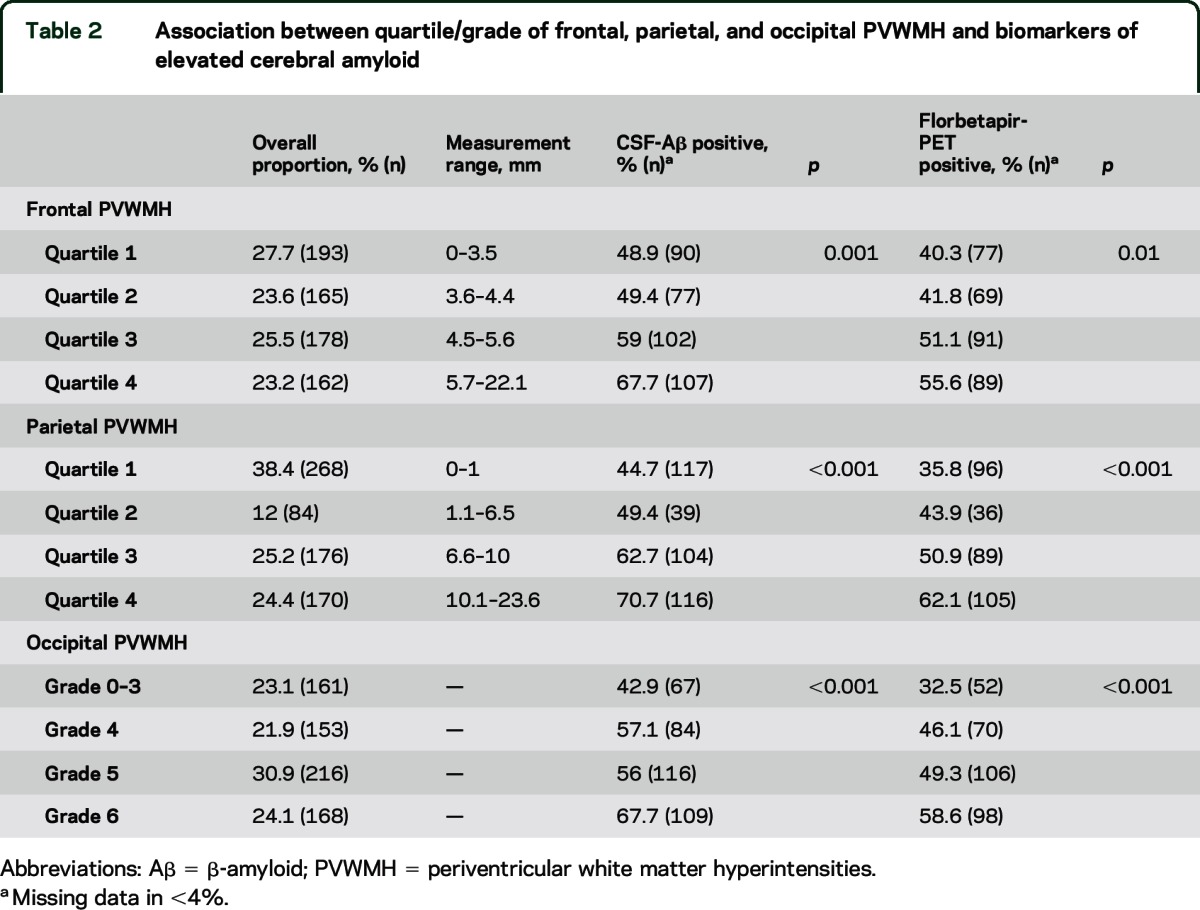
Table 3.
Logistic regression models for the association of PVWMH with florbetapir-PET signal (dichotomized as >1.11 or ≤1.11) adjusting for potential confoundersa
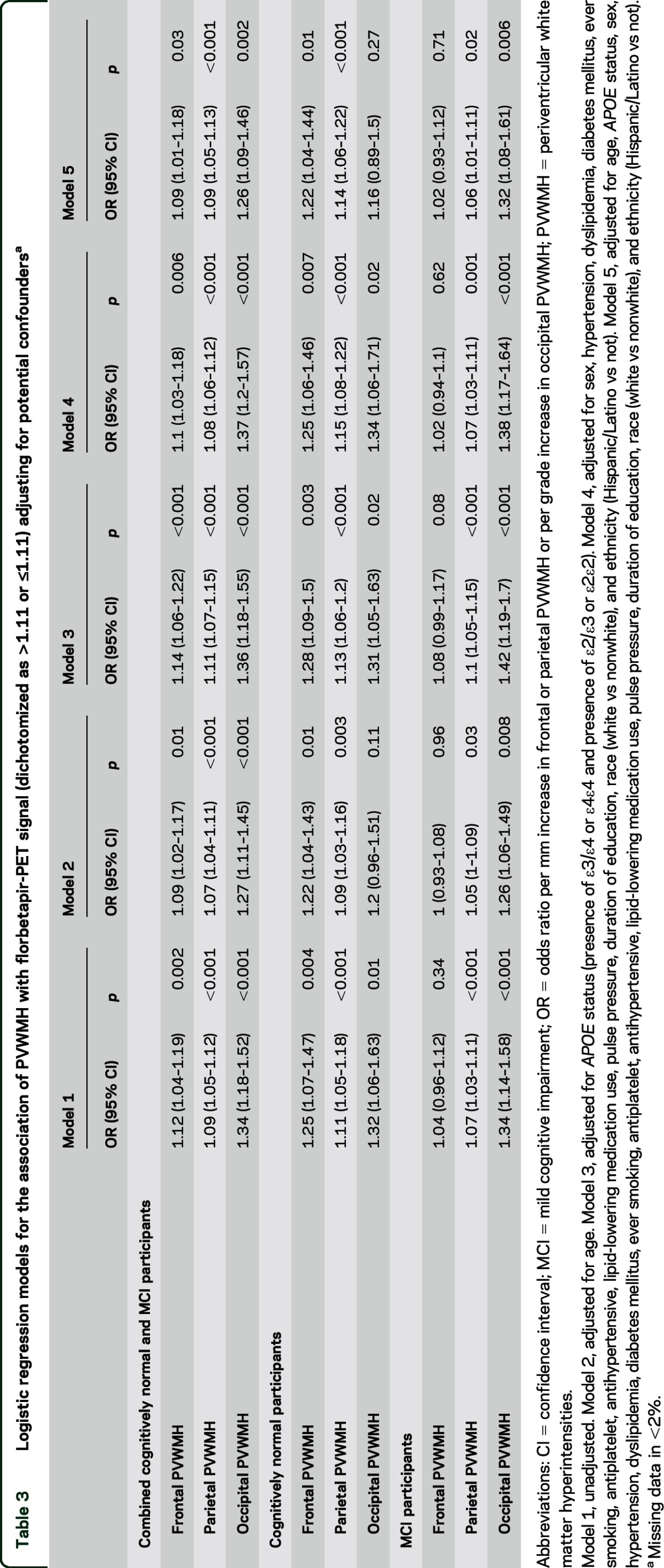
Occipital PVWMH grade was greater in florbetapir-PET-positive compared to florbetapir-PET-negative individuals (Wilcoxon rank-sum p < 0.0001). Occipital PVWMH burden correlated with increased florbetapir-PET signal in the sumROI-amyloid (ρ = 0.15, p < 0.0001). In a logistic regression model, occipital PVWMH were independently associated with florbetapir-PET positivity (OR 1.26 [1.09–1.46], p = 0.002, see table 3). For example, based on the calculated adjusted OR for the combined NC and MCI group, individuals with occipital PVWMH grade 5 were predicted to be 2.5 times as likely to have a florbetapir-PET scan consistent with AD compared to those with grade 1.
Frontal PVWMH were associated with florbetapir-PET positivity (mean frontal PVWMH measurement = 5.2 mm [±2.6 mm] vs 4.6 mm [±2.2 mm], p = 0.001). Frontal PVWMH burden correlated with florbetapir-PET signal in the sumROI-amyloid (r = 0.13, p = 0.0006) and the frontal subregion (r = 0.13, p = 0.0009). In a logistic regression model, frontal PVWMH were independently associated with florbetapir-PET positivity (OR 1.09 [1.01–1.18], p = 0.03, see table 3).
Association of PVWMH with CSF-Aβ.
Parietal PVWMH burden was greater in CSF-Aβ-positive compared to CSF-Aβ-negative individuals (mean parietal PVWMH measurement = 7.3 mm [±5.3 mm] vs 5.1 mm [±4.7 mm], p < 0.0001). Parietal PVWMH burden correlated with reduced CSF-Aβ levels (r = −0.26, p < 0.0001). In a logistic regression model, parietal PVWMH were independently associated with CSF Aβ positivity (OR 1.08 [1.04–1.13], p < 0.001, see table 4).
Table 4.
Logistic regression models for the association of PVWMH with CSF-Aβ (dichotomized as ≥192 pg/mL, <192 pg/mL) adjusting for potential confoundersa
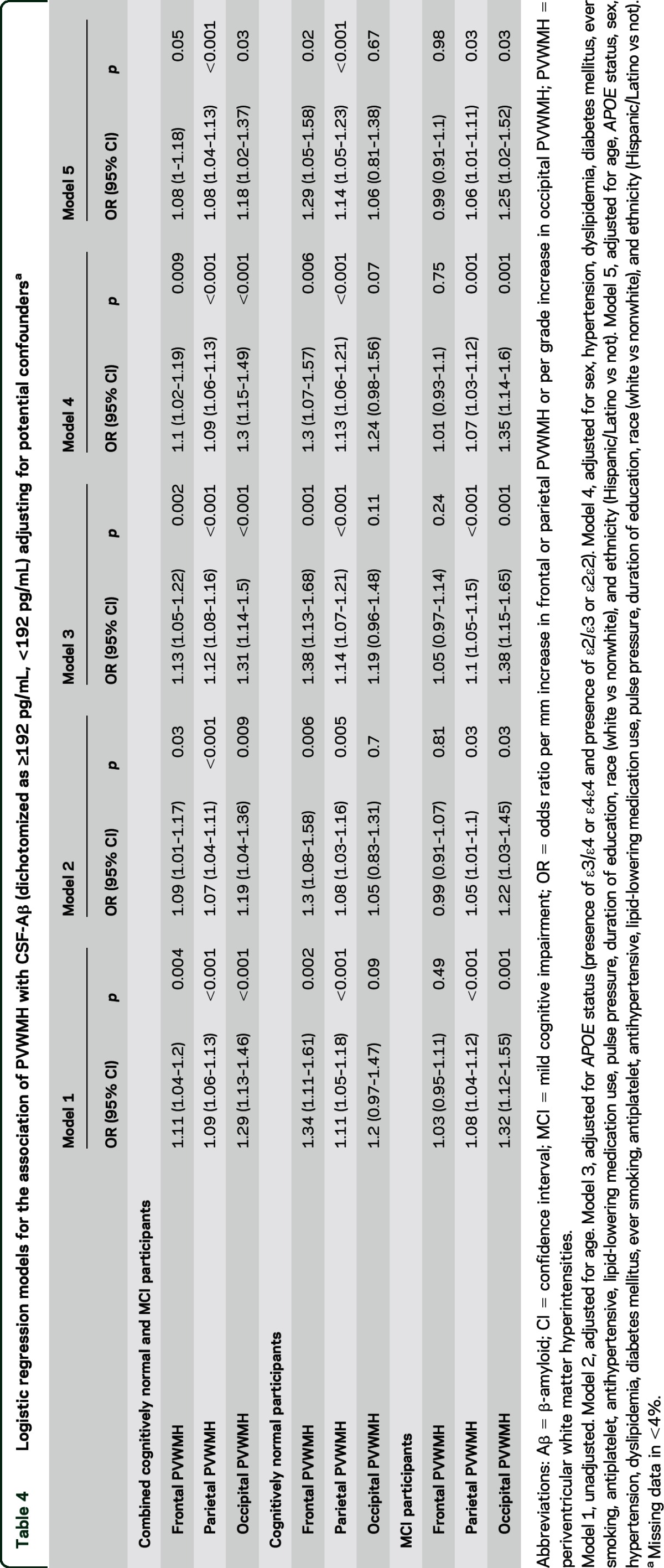
Occipital PVWMH grade was greater in CSF-Aβ-positive compared to CSF-Aβ-negative individuals (Wilcoxon rank-sum p = 0.0001). Occipital PVWMH burden correlated with reduced CSF-Aβ levels (ρ = −0.15, p = 0.0001). In a logistic regression model, occipital PVWMH were independently associated with CSF Aβ positivity (OR 1.18 [1.02–1.37], p = 0.03, see table 4).
Frontal PVWMH were associated with CSF Aβ positivity (mean frontal PVWMH measurement = 5.1 mm [±2.4 mm] vs 4.5 mm [±2.3 mm], p = 0.003). Frontal PVWMH burden correlated with reduced CSF-Aβ levels (r = −0.14, p = 0.0002). In a logistic regression model, frontal PVWMH were not independently associated with CSF Aβ positivity (OR 1.08 [1–1.18], p = 0.05; see table 4).
Association of PVWMH with CSF tau and phospho-tau.
The burden of PVWMH differed between CSF tau-positive and tau-negative cases in parietal (Student t test p = 0.04) and occipital (Wilcoxon rank-sum p = 0.008) but not frontal regions (Student t test p = 0.12). Parietal (r = 0.08, p = 0.04) and occipital (ρ = 0.14, p = 0.0004) but not frontal (r = 0.04, p = 0.26) PVWMH were correlated with increased CSF-tau levels. Parietal (p = 0.49), occipital (p = 0.3), and frontal (p = 0.82) PVWMH were not associated with CSF tau positivity in a logistic regression model including PVWMH, age, sex, APOE status, vascular risk factors, pulse pressure, vascular secondary prevention medications, education, ethnicity, and race.
Occipital PVWMH grade was associated with CSF phospho-tau positivity (Wilcoxon rank-sum p = 0.002). The burden of PVWMH did not differ between CSF phospho-tau-positive and phospho-tau-negative cases in frontal (Student t test p = 0.07) or parietal (Student t test p = 0.09) regions. Occipital PVWMH correlated with increased CSF-phospho-tau levels (ρ = 0.13, p = 0.0006). There was no correlation between CSF-phospho-tau levels and frontal (r = 0.03, p = 0.43) or parietal (r = 0.07, p = 0.08) PVWMH. In a logistic regression model including PVWMH, age, sex, APOE status, vascular risk factors, pulse pressure, vascular secondary prevention medications, education, ethnicity, and race, parietal (p = 0.84), occipital (p = 0.09), and frontal (p = 0.38) PVWMH were not associated with CSF phospho-tau positivity.
Association of PVWMH with FDG-PET.
Parietal (r = −0.17, p < 0.0001), occipital (ρ = −0.1, p = 0.006), and frontal (r = −0.12, p = 0.001) PVWMH were correlated with reduced sumROI-FDG uptake. Parietal PVWMH were also correlated with reduced FDG uptake in the parietal (angular and posterior cingulate) subregions (r = −0.18, p < 0.0001). In linear regression models including PVWMH and CSF-Aβ levels, parietal (coefficient = −0.013 [−0.023 to −0.005] per mm increase, p = 0.003), occipital (coefficient = −0.041 [−0.079 to −0.003] per grade increase, p = 0.03), and frontal (coefficient = −0.026 [−0.045 to −0.006] per mm increase, p = 0.009) PVWMH were independently associated with sumROI-FDG uptake.
Association of volumetric measures of total white matter lesion burden with PVWMH, CSF biomarkers, florbetapir-PET, and FDG-PET.
Frontal (r = 0.63, p < 0.0001), parietal (r = 0.61, p < 0.0001), and occipital (ρ = 0.33, p < 0.0001) PVWMH measurements were correlated with volumetric measurements of total white matter hyperintensity burden (volTWMH). In logistic regression models including volTWMH, age, sex, APOE status, vascular risk factors, pulse pressure, vascular secondary prevention medications, education, ethnicity, and race, volTWMH was independently associated with florbetapir-PET positivity (OR 1.04 [1.01–1.06], p = 0.003) and CSF-Aβ positivity (OR 1.05 [1.02–1.07], p = 0.002) but not CSF-tau positivity (OR 1 [0.98–1.02], p = 0.79) or CSF-phospho-tau positivity (OR 1 [0.98–1.02], p = 0.99). In a linear regression model including volTWMH and CSF-Aβ levels, volTWMH was independently associated with sumROI-FDG uptake (coefficient −0.008 [−0.013 to −0.002], p < 0.001).
DISCUSSION
In a large cohort of clinically well-characterized cognitively normal and amnestic MCI participants, we demonstrated an association between increased parietal, occipital, and frontal PVWMH burden and CSF and PET measures of elevated cerebral amyloid. This association was independent of other important predictors of cerebral amyloidosis such as age and having an APOE4 allele. The associations between PVWMH and CSF-tau and CSF-phospho-tau were weaker and topographically posterior predominant. A recent study in the ADNI cohort showed that lobar cerebral microbleeds (CMBs), a marker of cortical arterial disease, are also associated with lower levels of CSF-Aβ and high CSF-phospho-tau.16 Previously, PVWMH have been shown to be independently associated with CMBs.17 Taken together, these findings could support a hypothesis that cortical arterial disease, identified by PVWMH burden or CMBs, is associated with both increased cerebral amyloid and to a lesser extent increased tau in NC and MCI. The mechanisms underlying any such associations remain to be clarified.3,18,19
Frontal PVWMH were not associated with biomarkers of elevated amyloid in the MCI subgroup. This mirrors the lower prevalence of CAA angiopathy in frontal compared to parieto-occipital areas20 and therefore the association between PVWMH and cerebral amyloidosis could be driven by CAA-associated cortical arteriopathy.21 Supporting this, others have shown that a more prominent posterior WMH distribution is an independent predictor of pathologic evidence of CAA.22 In addition, parietal and occipital lobar CMB counts have been shown to be increased in those with greater WMH burden.23 Conversely, occipital PVWMH in the NC subgroup were less strongly associated with biomarkers of elevated amyloid. One explanation for this could be reduced burden of CAA in the NC group and therefore increased relative importance of other drivers of occipital PVWMH, such as arteriosclerosis or degeneration. Supporting this interpretation, the topography of PVWMH has been shown to correspond to areas of reduced relative perfusion on SPECT imaging irrespective of disease state (including healthy aging, MCI/AD, and CAA), suggesting that PVWMH occurrence may have a stronger relationship with hemodynamic compromise than the specific underlying pathogenic mechanism.24
PVWMH were associated with FDG-PET hypometabolism in regions of the brain known to be vulnerable to amyloid accumulation, i.e., angular, temporal, and posterior cingulate. These associations were independent of CSF-Aβ levels. Similar patterns of FDG-PET hypometabolism have previously been shown to be a risk marker for clinical progression.25 In addition, parietal WMH volume has been shown to predict incident dementia independent of hippocampal volume.26 Our findings suggest that PVWMH are a risk marker for cognitive deterioration both by amyloid-related and independent mechanisms.
Previous studies exploring the association between pathologic and in vivo biomarkers of Aβ load and WMH have shown conflicting results.27–34 Many such studies were small or used heterogeneous image acquisition protocols. In these studies, a variety of image analysis paradigms were used to define WMH burden including volumetrics and semiquantitative visual rating scales. Using multicenter standardized data acquisition protocols and the rating methods described, we were able to characterize PVWMH in the frontal, parietal, and occipital areas in a large cohort of NC and MCI patients with sufficient statistical power to control for multiple potential confounders including age and APOE genotype.
There are a number of considerations to be taken into account while interpreting our findings. First, we did not use a more complex volumetric method to quantify burden of PVWMH. Our PVWMH rating methods provide a semiquantitative estimate of disease severity. However, our measurements and findings correlated well with a well-described volumetric method of assessing total WMH burden.10 In addition, we were able to show regional variations in associations between CSF and PET markers of elevated amyloid and PVWMH. Second, we included ADNI-defined NC and MCI participants and not those with dementia. We elected to study this group as protocol eligibility criteria did not require consideration of WMH burden. In contrast, ADNI criteria for AD dementia, which include the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD, specifically exclude individuals with investigator-perceived excess of white matter lesions over age-expected norms.35,36 The inclusion of cases with a varied burden of WMH may increase the applicability of our findings to individuals seen in clinical practice. Finally, PVWMH rating in this study was performed by a blinded nonexpert rater trained in the methods described. Scores for frontal and parietal PVWMH showed excellent absolute and consistency of agreement with independent rating by an experienced vascular neurologist. This suggests that these methods could be used by clinicians and researchers of varying experience levels. Occipital PVWMH rating showed excellent consistency of agreement but only moderate absolute agreement, likely reflecting the difficulty of quantifying the less prominent MRI FLAIR hyperintensity frequently observed in lesions in this region.
Quantifying PVWMH could improve identification of those at increased risk of significant amyloid accumulation and accelerated cognitive decline. If confirmed in an independent cohort, these scales could cost-effectively identify individuals more likely to have significant cerebral amyloidosis and more likely to benefit from intervention in clinical trials of anti-amyloid therapies. Conversely, studies using eligibility criteria such as the NINCDS-ADRDA or National Institute on Aging–Alzheimer's Association, which specifically exclude those with pronounced WMH, might reduce the number of participants who may benefit, leading to increased costs and reduced statistical power.35,36 Future studies should focus on better understanding the mechanisms underlying the association between PVWMH and elevated cerebral amyloid, which may provide novel therapeutic targets in AD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the ADNI participants and its principal investigators and study teams for their efforts in generating the data used for these analyses.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- CMB
cerebral microbleed
- FDG
fluorodeoxyglucose
- FLAIR
fluid-attenuated inversion recovery
- ICC
intraclass correlation coefficient
- MCI
mild cognitive impairment
- NC
normal cognition
- NINCDS-ADRDA
National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association
- OR
odds ratio
- PVWMH
periventricular white matter hyperintensities
- ROI
region of interest
- SVD
small vessel disease
- volTWMH
volumetric measurements of total white matter hyperintensity burden
- WMH
white matter hyperintensity
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Alzheimer's Disease Neuroimaging Initiative, Michael Weiner, Paul Aisen, Michael Weiner, Paul Aisen, Ronald Petersen, Clifford R. Jack, Jr, William Jagust, John Q. Trojanowki, Arthur W. Toga, Laurel Beckett, Robert C. Green, Andrew J. Saykin, John Morris, Enchi Liu, Robert C. Green, Tom Montine, Ronald Petersen, Paul Aisen, Anthony Gamst, Ronald G. Thomas, Michael Donohue, Sarah Walter, Devon Gessert, Tamie Sather, Laurel Beckett, Danielle Harvey, Anthony Gamst, Michael Donohue, John Kornak, Clifford R. Jack, Jr, Anders Dale, Matthew Bernstein, Joel Felmlee, Nick Fox, Paul Thompson, Norbert Schuff, Gene Alexander, Charles DeCarli, William Jagust, Dan Bandy, Robert A. Koeppe, Norm Foster, Eric M. Reiman, Kewei Chen, Chet Mathis, John Morris, Nigel J. Cairns, Lisa Taylor-Reinwald, J.Q. Trojanowki, Les Shaw, Virginia M.Y. Lee, Magdalena Korecka, Arthur W. Toga, Karen Crawford, Scott Neu, Andrew J. Saykin, Tatiana M. Foroud, Steven Potkin, Li Shen, Zaven Kachaturian, Richard Frank, Peter J. Snyder, Susan Molchan, Jeffrey Kaye, Joseph Quinn, Betty Lind, Sara Dolen, Lon S. Schneider, Sonia Pawluczyk, Bryan M. Spann, James Brewer, Helen Vanderswag, Judith L. Heidebrink, Joanne L. Lord, Ronald Petersen, Kris Johnson, Rachelle S. Doody, Javier Villanueva-Meyer, Munir Chowdhury, Yaakov Stern, Lawrence S. Honig, Karen L. Bell, John C. Morris, Beau Ances, Maria Carroll, Sue Leon, Mark A. Mintun, Stacy Schneider, Daniel Marson, Randall Griffith, David Clark, Hillel Grossman, Effie Mitsis, Aliza Romirowsky, Leyla deToledo-Morrell, Raj C. Shah, Ranjan Duara, Daniel Varon, Peggy Roberts, Marilyn Albert, Chiadi Onyike, Stephanie Kielb, Henry Rusinek, Mony J de Leon, Lidia Glodzik, Susan De Santi, P. Murali Doraiswamy, Jeffrey R. Petrella, R. Edward Coleman, Steven E. Arnold, Jason H. Karlawish, David Wolk, Charles D. Smith, Greg Jicha, Peter Hardy, Oscar L. Lopez, MaryAnn Oakley, Donna M. Simpson, Anton P. Porsteinsson, Bonnie S. Goldstein, Kim Martin, Kelly M. Makino, M. Saleem Ismail, Connie Brand, Ruth A. Mulnard, Gaby Thai, Catherine Mc-Adams-Ortiz, Kyle Womack, Dana Mathews, Mary Quiceno, Ramon Diaz-Arrastia, Richard King, Myron Weiner, Kristen Martin-Cook, Michael DeVous, Allan I. Levey, James J. Lah, Janet S. Cellar, Jeffrey M. Burns, Heather S. Anderson, Russell H. Swerdlow, Liana Apostolova, Po H. Lu, George Bartzokis, Daniel H.S. Silverman, Neill R Graff-Radford, Francine Parfitt, Heather Johnson, Martin R. Farlow, Ann Marie Hake, Brandy R. Matthews, Scott Herring, Christopher H. van Dyck, Richard E. Carson, Martha G. MacAvoy, Howard Chertkow, Howard Bergman, Chris Hosein, Sandra Black, Bojana Stefanovic, Curtis Caldwell, Ging-Yuek Robin Hsiung, Howard Feldman, Benita Mudge, Michele Assaly, Andrew Kertesz, John Rogers, Dick Trost, Charles Bernick, Donna Munic, Diana Kerwin, Marek-Marsel Mesulam, Kristina Lipowski, Chuang-Kuo Wu, Nancy Johnson, Carl Sadowsky, Walter Martinez, Teresa Villena, Raymond Scott Turner, Kathleen Johnson, Brigid Reynolds, Reisa A. Sperling, Keith A. Johnson, Gad Marshall, Meghan Frey, Jerome Yesavage, Joy L. Taylor, Barton Lane, Allyson Rosen, Jared Tinklenberg, Marwan Sabbagh, Christine Belden, Sandra Jacobson, Neil Kowall, Ronald Killiany, Andrew E. Budson, Alexander Norbash, Patricia Lynn Johnson, Thomas O. Obisesan, Saba Wolday, Salome K. Bwayo, Alan Lerner, Leon Hudson, Paula Ogrocki, Evan Fletcher, Owen Carmichael, John Olichney, Charles DeCarli, Smita Kittur, Michael Borrie, T-Y Lee, Dr Rob Bartha, Sterling Johnson, Sanjay Asthana, Cynthia M. Carlsson, Steven G. Potkin, Adrian Preda, Dana Nguyen, Pierre Tariot, Adam Fleisher, Stephanie Reeder, Vernice Bates, Horacio Capote, Michelle Rainka, Douglas W. Scharre, Maria Kataki, Earl A. Zimmerman, Dzintra Celmins, Alice D. Brown, Godfrey D. Pearlson, Karen Blank, Karen Anderson, Andrew J. Saykin, Robert B. Santulli, Eben S. Schwartz, Kaycee M. Sink, Jeff D. Williamson, Pradeep Garg, Franklin Watkins, Brian R. Ott, Henry Querfurth, Geoffrey Tremont, Stephen Salloway, Paul Malloy, Stephen Correia, Howard J. Rosen, Bruce L. Miller, Jacobo Mintzer, Crystal Flynn Longmire, Kenneth Spicer, Elizabether Finger, Irina Rachinsky, John Rogers, Andrew Kertesz, Dick Drost, Nunzio Pomara, Raymundo Hernando, Antero Sarrael, Susan K. Schultz, Laura L. Boles Ponto, Hyungsub Shim, Karen Elizabeth Smith, Norman Relkin, Gloria Chaing, Lisa Raudin, Amanda Smith, Kristin Fargher, and Balebail Ashok Raj
AUTHOR CONTRIBUTIONS
Dr. Michael Marnane contributed to design and conceptualization of the study, analysis and interpretation of the data, developing the image analysis paradigm, and drafting and revising the manuscript for intellectual content. Osama O. Al-Jawadi contributed by analyzing the data, developing the image analysis paradigm, and drafting the manuscript. Shervin Mortazavi contributed by analyzing the data and developing the image analysis paradigm. Kathleen Pogorzelec contributed by analyzing the data and developing the image analysis paradigm. Dr. Bing Wei Wang contributed by analyzing the data and developing the image analysis paradigm. Dr. Howard H. Feldman contributed by revising the manuscript for intellectual content. Dr. Ging-Yuek R. Hsiung contributed to conceptualization of the study, supervision of the analysis, and revising the manuscript for intellectual content.
STUDY FUNDING
Dr. Hsiung acknowledges funding support from the Ralph Fisher Professorship in Alzheimer's Research (Alzheimer Society of British Columbia, Canada) and the CIHR Clinical Genetics Investigatorship award. Dr. Marnane's salary was funded by an unrestricted research fellowship award from the Heart and Stroke Foundation of Canada. Data collection and sharing for this project was funded by ADNI (National Institutes of Health grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California.
DISCLOSURE
M. Marnane, O. Al-Jawadi, S. Mortazavi, K. Pogorzelec, and B. Wang report no disclosures relevant to the manuscript. H. Feldman reports having been the UBC site principal investigator for ADNI through 2008. He is currently UBC site co-Investigator for ADNI. He has no other disclosures related to this study. G. Hsiung reports being the UBC site principal investigator for ADNI since 2008. He has no other disclosures related to this study. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Thal DR, Attems J, Ewers M. Spreading of amyloid, tau, and microvascular pathology in Alzheimer's disease: findings from neuropathological and neuroimaging studies. J Alzheimers Dis 2014;42:S421–S429. [DOI] [PubMed] [Google Scholar]
- 2.Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement 2015;11:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of Impaired drainage of interstitial fluid. Brain Pathol 2014;25:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment: a critical update. Front Aging Neurosci 2013;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR Am J Neuroradiol 1990;11:431–439. [PMC free article] [PubMed] [Google Scholar]
- 6.Gouw AA, Seewann A, van der Flier WM, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 2011;82:126–135. [DOI] [PubMed] [Google Scholar]
- 7.Conklin J, Silver FL, Mikulis DJ, Mandell DM. Are acute infarcts the cause of leukoaraiosis? Brain mapping for 16 consecutive weeks. Ann Neurol 2014;76:899–904. [DOI] [PubMed] [Google Scholar]
- 8.Duering M, Csanadi E, Gesierich B, et al. Incident lacunes preferentially localize to the edge of white matter hyperintensities: insights into the pathophysiology of cerebral small vessel disease. Brain 2013;136:2717–2726. [DOI] [PubMed] [Google Scholar]
- 9.Jack CR, Jr, Bernstein MA, Borowski BJ, et al. Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement 2010;6:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeCarli C, Fletcher E, Ramey V, Harvey D, Jagust WJ. Anatomical mapping of white matter hyperintensities (WMH): exploring the relationships between periventricular WMH, deep WMH, and total WMH burden. Stroke 2005;36:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol 2013;74:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alzheimer Disease Neuroimaging Initiative investigators. ADNI-go PET Technical Procedures Manual AV-45 & FDG. Available at: http://www.adni-info.org/Scientists/doc/ADNIGO_PET_Tech_Manual_01142011.pdf. Accessed May 14, 2015. [Google Scholar]
- 13.Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 2011;32:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallgren KA. Computing Inter-rater reliability for observational data: an overview and tutorial. Tutor Quant Methods Psychol 2012;8:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang GC, Cruz Hernandez JC, Kantarci K, Jack CR, Jr, Weiner MW; Alzheimer's Disease Neuroimaging Initiative. Cerebral microbleeds, CSF p-tau, and cognitive decline: significance of anatomic distribution. AJNR Am J Neuroradiol 2015;36:1635–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada S, Saiki M, Satow T, et al. Periventricular and deep white matter leukoaraiosis have a closer association with cerebral microbleeds than age. Eur J Neurol 2012;19:98–104. [DOI] [PubMed] [Google Scholar]
- 18.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008;18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iliff JJ, Wang M, Zeppenfeld DM, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 2013;33:18190–18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain: II: the distribution of amyloid vascular changes. Stroke 1983;14:924–928. [DOI] [PubMed] [Google Scholar]
- 21.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014;83:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JH, Seo SW, Kim C, et al. Pathogenesis of cerebral microbleeds: in vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann Neurol 2013;73:584–593. [DOI] [PubMed] [Google Scholar]
- 24.Holland CM, Smith EE, Csapo I, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 2008;39:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewers M, Brendel M, Rizk-Jackson A, et al. Reduced FDG-PET brain metabolism and executive function predict clinical progression in elderly healthy subjects. Neuroimage Clin 2014;4:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brickman AM, Provenzano FA, Muraskin J, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol 2012;69:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polvikoski TM, van Straaten EC, Barkhof F, et al. Frontal lobe white matter hyperintensities and neurofibrillary pathology in the oldest old. Neurology 2010;75:2071–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitwell JL, Lowe VJ, Duffy JR, et al. Elevated occipital beta-amyloid deposition is associated with widespread cognitive impairment in logopenic progressive aphasia. J Neurol Neurosurg Psychiatry 2013;84:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rutten-Jacobs LC, de Leeuw FE, Geurts-van Bon L, et al. White matter lesions are not related to beta-amyloid deposition in an autopsy-based study. Curr Gerontol Geriatr Res 2011;2011:826862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenset V, Hofoss D, Johnsen L, et al. White matter lesion load increases the risk of low CSF Aβ42 in apolipoprotein E-ɛ4 carriers attending a memory clinic. J Neuroimaging 2011;21:e78–82. [DOI] [PubMed] [Google Scholar]
- 31.Kester MI, Goos JD, Teunissen CE, et al. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol 2014;71:855–862. [DOI] [PubMed] [Google Scholar]
- 32.Haight TJ, Landau SM, Carmichael O, et al. Dissociable effects of Alzheimer disease and white matter hyperintensities on brain metabolism. JAMA Neurol 2013;70:1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benedictus MR, Goos JD, Binnewijzend MA, et al. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer's disease. Neurobiol Aging 2013;34:2488–2494. [DOI] [PubMed] [Google Scholar]
- 34.Guzman VA, Carmichael OT, Schwarz C, et al. White matter hyperintensities and amyloid are independently associated with entorhinal cortex volume among individuals with mild cognitive impairment. Alzheimers Dement 2013;9:S124–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.