

Abstract
A complete understanding of the molecular mechanisms governing vesicle formation requires quantitative assays and vesicle reconstitution using purified components. We describe a simple model membrane template for studying protein-mediated membrane remodeling and vesicle formation or fission that is amenable to both quantitative biochemical analysis and real-time imaging by epifluorescence microscopy. Supported bilayers with excess membrane reservoir (SUPER) templates are compositionally well-defined unilamellar membrane systems prepared on 2–5-μm silica beads under conditions that enable incorporation of excess membrane to form a loosely fitting bilayer that can be used to study membrane remodeling and fission. This protocol describes methods for SUPER template formation and characterization, as well as for the qualitative observation and quantitative measurement of vesicle formation and fission via microscopy and a simple sedimentation assay. SUPER templates can be prepared within 60 min. Results from either sedimentation-based or microscopy-based assays can be obtained within an additional 60 min.
INTRODUCTION
Development of the method and application
The reconstitution of cellular processes under biochemically defined conditions is an important step toward the comprehension of complex biological mechanisms. Vesicle formation, which requires the concerted action of a large number of proteins, is a pivotal process in all eukaryotic cells. These proteins assemble into coats and actively remodel membranes to generate curvature, recruit and concentrate cargo proteins, and mediate membrane fission. The role of many of these proteins in vesicular transport has been identified through overexpression or knockdown/knockout approaches; however, in many cases their direct function and the temporal hierarchy by which they are recruited and act remains unclear. The faithful reconstitution of vesicle formation and membrane fission is a prerequisite for defining the role of each component of this complex vesicle formation machinery1.
Here we describe the formation of SUPER templates and detail their general utility by using them to study membrane remodeling, membrane fission and vesicle release mediated by the large GTPase dynamin. Among dynamin’s roles, the best understood is its involvement in clathrin-mediated endocytosis (CME)2,3, the major route of entry for receptor-bound molecules into the cell4. CME is initiated by adaptor proteins that capture cargo molecules at the plasma membrane and trigger clathrin assembly for the formation of a clathrin-coated pit (CCP). The subsequent recruitment of endocytic accessory proteins drives the maturation of the CCP. During the late stages of CCP formation, dynamin self-assembles into collar-like structures at the constricted neck of a pit. GTP hydrolysis by dynamin severs the membrane and triggers disassembly of the dynamin collar. Via SUPER templates, we determined that dynamin is involved in membrane curvature generation and can mediate membrane fission without the help of accessory endocytic proteins5.
Initially, SUPER templates were used to uncover the mechanism of dynamin-catalyzed membrane fission and to study isoform-specific properties of dynamin5–7. However, in general, these templates are an ideal platform for studying how proteins interact and function in a concerted fashion to remodel membranes and drive vesicle formation. For example, SUPER templates can be used to study functional interactions between the ATPase EHD8 or the Bin-amphiphysin-Rvs (BAR) domain–containing proteins endophilin and amphiphysin9, and dynamin-1 during dynamin-mediated fission. Future challenges involve developing methods to incorporate endocytic or other cargo molecules into the membrane and deciphering the role and regulation of endocytic accessory proteins in vesicle formation.
Experimental design
For the generation of SUPER templates, a membrane bilayer of defined composition is deposited on 2–5-μm silica beads so as to incorporate an excess membrane reservoir that is amenable for the reconstitution of budding events (Fig. 1a). The addition of trace amounts of fluorescent lipids enables the quantification of vesicle release in sedimentation-based assays, as well as the real-time observation of vesicle formation in microscopy-based assays.
Figure 1.
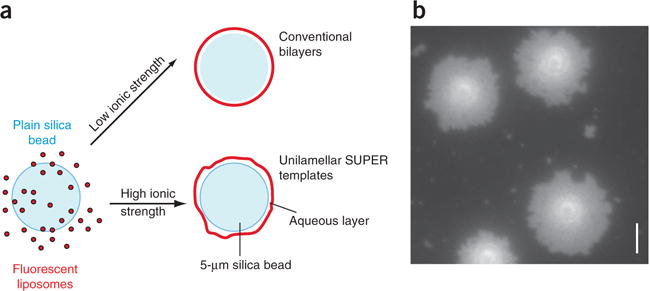
Generation of SUPER templates. (a) To generate SUPER templates, fluorescently labeled liposomes that contain negatively charged lipids are deposited onto 5-μm silica beads under high-salt conditions, so as to incorporate excess membrane reservoir. (b) The incorporation of membrane reservoir can be visualized by adding the templates onto a clean, uncoated glass coverslip in a droplet of assay buffer. The templates will bind to the glass surface, and the excess membrane will spill out to form a supported bilayer surrounding the silica bead (Supplementary Video 1). Scale bar, 5 μm.
The incorporation of excess membrane can be verified by microscopy. To this end, the templates are allowed to settle onto an uncoated clean glass coverslip in salt-containing typical assay buffers. The templates will bind to the glass surface and the excess membrane will progressively spread out on the surface of the glass around the silica bead, similar to egg whites spilling out around the yolk (Fig. 1b and Supplementary Video 1). SUPER templates can be generated with a variety of lipid compositions; however, the formation of an excess membrane reservoir is dependent on high-salt conditions during template formation and on the presence of negatively charged lipids10. If lipids with different characteristics are used, testing deposition conditions becomes crucial in order to ensure the generation of an excess membrane reservoir.
We found silicon dioxide microspheres from Corpuscular to be the most reliable source of beads, both in terms of size distribution and cleanliness. However, we recently learned that 5-μm beads can have significant lot-to-lot variation because of the added complexity of manufacturing beads >2.5 μm in diameter. Therefore, we recently switched to 2.5-μm beads. To keep the lipid-to-surface ratio constant, when preparing SUPER templates we use 4× more 2.5-μm beads than 5-μm beads. All other aspects of the protocol described below (Steps 8–15) are unchanged. Once SUPER templates have been generated, membrane fission leading to vesicle release can either be measured using a simple sedimentation assay or observed directly by fluorescence light microscopy due to the templates’ large size. The sedimentation assay is fast and quantitative, whereas the microscopy-based assay enables researchers to observe membrane-remodeling events, such as tubulation, vesicle release and individual fission events.
For sedimentation assays, dynamin is incubated at room temperature (20 °C) with SUPER templates in the presence of GTP. Vesicles released by dynamin-catalyzed membrane fission during incubation will remain in the supernatant (Fig. 2a) and are separated from SUPER templates by centrifugation at low speed. The amount of fluorescent lipids in the supernatant relative to the total lipids associated with the beads is taken as a measurement of membrane fission efficiency, i.e., fission efficiency = (fluorescence intensity in supernatant)/(total fluorescence intensity). To prevent the release of vesicles by shearing of the reservoir, the templates are added very carefully to the top of the reaction mixture without further mixing (Fig. 2b and Supplementary Video 2). During the course of the experiment, the templates will settle loosely at the bottom of the reaction vessel and finally be pelleted via a low-speed centrifugation.
Figure 2.
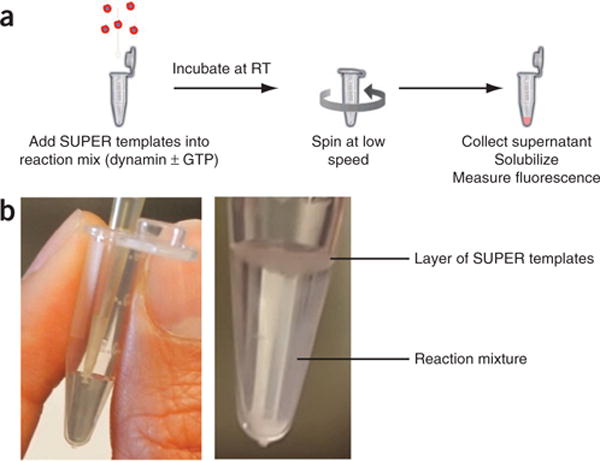
Sedimentation-based assay. (a) To quantify vesicle release, SUPER templates are incubated with dynamin-1 in the presence of nucleotides. Templates can then be easily pelleted at low speed owing to their size, and any released vesicles will remain in the supernatant. The total incorporation of membrane is determined by Triton X-100 extraction of the templates. (b) Carefully add SUPER templates to the top of the reaction mixture for a sedimentation assay without performing any additional mixing (Supplementary Video 2). RT, room temperature.
In these assays, we use the following three controls:
Template control, that is, SUPER templates alone with no added protein. This mixture is used to determine fluorescent lipid release due to mechanical shear or any remaining adsorbed liposomes.
Total control, that is, SUPER templates in 0.1% (vol/vol) Triton X-100 in 20 mM HEPES, 150 mM KCl and 1 mM MgCl2. This solubilizes the total lipids on the SUPER templates and provides information on maximum possible vesicle release.
Blank control, that is, 100 μl of 1× buffer without SUPER templates. This mixture is used to determine background signal in fluorescence measurements.
For experiments with dynamin, we also include an incubation control that contains dynamin without GTP, which is used to confirm the fact that vesicle release requires dynamin’s GTPase activity.
Because of the size of SUPER templates, microscopy-based assays enable visualization of vesicle release and membrane fission events by epifluorescence microscopy. To minimize potential photodamage to the proteins and lipids in the reaction mixture, this assay is performed in the presence of an oxygen-scavenging system. In the assay described here, SUPER templates are added to—and allowed to settle in—a drop of buffer-containing GTP that has been deposited onto a BSA-coated (or PEGylated) coverslip (Fig. 3). The subsequent addition of dynamin with a pipette leads to the formation of vesicles that can be observed as small fluorescent dots or punctae that appear next to the templates (Fig. 4 and Supplementary Video 3). We usually use an eight-well Lab-Tek chamber for this assay and spread the templates evenly in the well by pipetting (Fig. 3b,c). Once the templates have settled, the Lab-Tek chamber is transferred to a microscope stage and dynamin is carefully added to one side of the chamber with a pipette (Fig. 3d). For controls, either dynamin, GTP or both are left out of the reaction.
Figure 3.
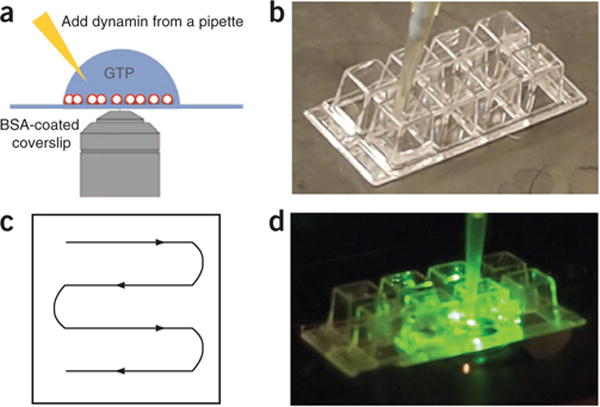
Real-time visualization of membrane fission. (a) SUPER templates are applied to a BSA-coated Lab-Tek chamber or coverslip for microscopy-based assays. Dynamin is added from a pipette to initiate the reaction. (b) SUPER templates are added to a well of a Lab-Tek chamber. (c) Spread SUPER templates evenly throughout the well by slowly moving the pipette according to the schematic while emptying the tip. (d) Once the templates are settled, transfer the well onto a microscope stage, begin to record a video and carefully add dynamin to the well.
Figure 4.
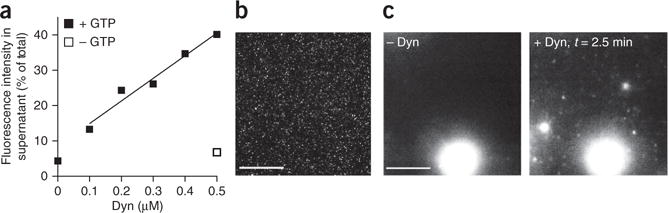
Sedimentation and microscopy assays for vesicle release. (a) Representative results from a sedimentation fission assay. SUPER templates were incubated in 20 mM HEPES, 150 mM KCl and 1 mM MgCl2 for 30 min in the presence of 1 mM GTP and with increasing amounts of dynamin (Dyn)-1. Vesicle release is dependent on dynamin-1 presence and concentration and requires GTP. (b) Fluorescence micrograph of released vesicles. (c) SUPER templates were dispersed into assay buffer containing an oxygen-scavenging system and 1 mM GTP in individual wells of an eight-well glass slide. After the beads had settled, imaging was started and Dyn-1 was added to the solution with a pipette to a final concentration of 0.5 μM. The figure shows the field of view before (left) and after (right) addition of Dyn-1. See Supplementary Video 3 for the full sequence. Scale bars, 5 μm.
Owing to the high fluorescence of the beads, individual fission events are difficult to observe. Therefore, an alternative approach for visualizing fission makes use of membrane tethers generated from SUPER templates, which can serve as substrates for dynamin-mediated membrane fission. These tethers are generated by gently rolling plain silica beads that are larger than those used to make the SUPER templates over a layer of SUPER templates to capture and pull out membrane tethers (Fig. 5a). For this assay, SUPER templates are allowed to settle on a BSA-coated well of an eight-well Lab-Tek chamber (Fig. 3b,c). Next, an aliquot of the above-mentioned larger, untreated silica beads is added. Because of their size, these beads settle readily within the pipette tip (Fig. 5b). The volume of the bead pellet within the pipette tip is deposited into one corner of the well. By gently tilting (and tapping) the Lab-Tek chamber, the beads are rolled over the templates in a path that resembles a ‘W’ shape (Fig. 5c,d and Supplementary Video 4). During this procedure, the beads form clumps when rolled over SUPER templates. To prevent the disruption of already formed tethers, it is important not to tilt the chamber too many times. Once the tethers are formed, the Lab-Tek chamber is transferred to a microscope stage and dynamin is carefully added to one side of the well (Fig. 3d). Once again, incubation mixtures missing either dynamin, GTP or both serve as controls.
Figure 5.

Fission of membrane tethers. (a) SUPER templates are applied to a BSA-coated Lab-Tek chamber. After the templates settle, untreated silica beads of sizes (diameter = 20 μm) bigger than those used to produce the SUPER templates are added to the microscopy chamber in order to create tethers. Owing to their size, the 20-μm beads readily settle within the pipette tip. (b) Only the volume of the mixture that contains the settled beads is added into one corner of the well. (c) When they are first added to the wells and rolled over the templates, the larger beads clump. (d) Gently roll the clumped larger beads over the templates another three or four times by tilting the chamber in a W-shaped path. (Supplementary Video 4).
Comparison with other methods
CME in vivo requires the major coat proteins clathrin and AP2 adaptors, dynamin-mediated membrane fission and numerous endocytic accessory factors11. Clathrin-coated vesicle formation and the selective uptake of cargo molecules has been reconstituted in biological membranes, such as perforated cells12 or purified plasma membrane sheets13, by using crude cytosol preparations. However, complete reconstitution of clathrin-coated vesicle formation, including cargo capture, with purified components remains an elusive goal, owing to its complexity.
Multiple aspects of clathrin-coated vesicle formation, including coat assembly, curvature generation and fission, have been reconstituted with purified protein components using liposomes14,15 or lipid monolayers16 as substrates. In particular, liposome-based assays in combination with electron microscopy have provided insight into the structural mechanisms of membrane deformation and remodeling by dynamin17–19 and other endocytic proteins containing BAR14,20–22 or epsin N-terminal homology (ENTH)/AP180 N-terminal homology (ANTH)23 domains. However, these approaches are typically endpoint assays that require application of samples to electron microscopy grids. Time-resolved cryo-electron microscopy experiments have provided evidence that fission events in these liposome-based assay systems occur only as a result of mechanical stress during sample preparation19. Membrane sheets and tethers pulled from giant unilamellar vesicles have been used to overcome the limitations of electron microscopy and to develop dynamic, light microscopy–based assays for dynamin-mediated curvature generation and membrane fission24,25. However, these assays often require specialized equipment24,26, and the efficiency of fission events in these assays is not readily quantifiable. Thus, the development of facile and quantitative assays for vesicle formation and membrane fission from model lipid bilayers has been a limitation to the reconstitution of clathrin-coated vesicle formation.
Protein-mediated membrane fusion is readily detected by content or lipid mixing of two separately prepared liposome substrates27 and can be visualized by fluorescence microscopy as fluorescently labeled liposomes fuse with conventional supported bilayers formed on coverslips28. By contrast, it is difficult to unambiguously distinguish released vesicles from their liposome donors without resorting to electron microscopy, which is often a time-consuming approach and does not allow the easy quantification of the efficiency of vesicle release. Supported bilayers have not proven to be suitable for studying membrane fission owing to the lack of a membrane reservoir, which is essential for the reconstitution of budding events. SUPER templates can overcome this deficit and represent a quantitative approach to measure membrane fission. Furthermore, the incorporation of fluorescent lipids and/or the use of fluorescently labeled proteins enable the direct observation of protein-mediated membrane remodeling and vesicle formation by fluorescence microscopy in real time. Finally, SUPER templates are inexpensive to make and are easily produced and handled in any cell biology, molecular biology or biochemistry laboratory.
Limitations
Although SUPER templates are easy to prepare, their low-tension membrane reservoir is subject to shedding under conditions of high mechanical stress. Therefore, it is crucial to handle the templates gently with minimal pipetting and mixing in order to avoid membrane shearing. Because of this limitation, the number of samples that can be handled at a time (for example, during sedimentation assays) is limited. The integrity of the reservoir is also dependent on temperature. SUPER templates are not stable at 4 °C; therefore, they must be prepared before each experiment. This need to prepare fresh SUPER templates before use might result in an increased experiment-to-experiment variability, and thus it is important to include positive and negative controls in each experiment. Other potential sources of experimental variations are batch-to-batch differences in protein and liposome preparations.
The templates are also subject to extraction of lipids by means other than vesiculation. For example, we attempted to reconstitute CME using rat brain cytosol, but found that the protein-mediated extraction of lipids from the templates was not dependent on the addition of nucleotides or Mg2+ and occurred independently of the presence of dynamin-1 (S.N., unpublished observations). Furthermore, the extracted lipids did not seem to be present in small vesicles, as determined by fluorescence microscopy. Future approaches should use purified components or other sources of complex protein mixtures. Indeed, lipids were not extracted by purified coat proteins, although these preparations did not stimulate dynamin-1–dependent vesicle release (S.N., unpublished observations). Again, these observations reflect the necessity for proper controls when measuring the fluorescence in the supernatant. To confirm the validity of the results, we suggest imaging the released vesicles by fluorescence microscopy (Fig. 4b).
Finally, although the microscopy-based assays are useful for visually confirming membrane remodeling, vesicle release and fission from SUPER templates, they are ill-suited to the quantification of reaction efficiency. Thus, a major advantage of SUPER templates is that they can be used in parallel to microscopy-based real-time imaging assays in order to perform quantitative sedimentation assays that measure protein-membrane interactions or vesicle release.
MATERIALS
REAGENTS
! CAUTION All lipid stocks are stored in chloroform. Chloroform is a chemical hazard. Do not breathe gas/fumes/vapor/spray. Wear suitable protective clothing. Work in well-ventilated areas or in a fume hood.
▲ CRITICAL Lipids are typically purchased as chloroform stocks from Avanti Polar Lipids and stored at −20 °C. Take care in storing lipid stocks. Owing to their hygroscopic nature, 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) and L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) stocks are prone to precipitation if stocks are left open.
Chloroform (VWR, cat. no. JT9180-22)
1,2-Dioleoyl-sn-glycero-3-phosphocholine, chloroform stock, (DOPC; Avanti Polar Lipids, cat. no. 850375C)
1,2-Dioleoyl-sn-glycero-3-phospho-L-serine, sodium salt, chloroform stock (DOPS; Avanti Polar Lipids, cat. no. 840035C)
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl), ammonium salt, chloroform stock (RhPE; Avanti Polar Lipids, cat. no. 810150C)
L-α-Phosphatidylinositol-4,5-bisphosphate (brain, porcine, ammonium salt) (PI(4,5)P2), Brain extract, CHCl3/CH3OH/H2O (20:9:1) stock, Avanti Polar Lipids, cat. no. 840046X or synthetic PI(4,5)P2, powder, Avanti Polar Lipids, cat. no. 850155P. The synthetic PI(4,5)P2 works better in the fission assay, probably because the dioleoyl chain lengths are compatible with the rest of the lipids.
HEPES, 1 M, pH 7.5 (Jena Biosciences, cat. no. CSS-192)
Magnesium chloride, 2.5 M (MgCl2; Jena Biosciences, cat. no. CSS-212)
Potassium chloride (KCl; Fisher Scientific, cat. no. BP366-1)
DTT (RPI, cat. no. D11000)
Triton X-100 (Fisher Scientific, cat. no. BP151-500)
-
Silicon dioxide microspheres (silica beads). We have used both 5-μm beads (Corpuscular, cat. no. 140226-10) and 2.5-μm beads (Corpuscular, cat. no. 140216-10) for SUPER templates. The latter are more easily manufactured and therefore have greater lot-to-lot consistency. We use larger 20-μm beads (cat. no. 140248-10) to pull out membrane tethers.
▲ CRITICAL We found Corpuscular to be the most reliable source of beads in both size distribution and cleanliness. Please order the beads at http://www.microspheres-nanospheres.com/and refer to the ‘m-type’ of silica. Beads from other sources can be used; we suggest that they first be cleaned by a piranha wash11.
Glucose oxidase (USB, cat. no. 1614650)
Catalase (Calbiochem, cat. no. 219261)
BSA, 2 mg ml−1 (albumin standard; Thermo Scientific, cat. no. 23209). Other sources of BSA may also be suitable.
GTP, Axxora (Jena Biosciences, cat. no. JBS NU-1012) ▲ CRITICAL We have found Axxora to be the most reliable source of GTP.
Milli-Q water
Absolute ethanol
Glucose
EGTA
Glycerol
NaCl
EDTA
EQUIPMENT
Disposable borosilicate glass tubes (Fisher Scientific, cat. no. 14-961-26)
Microscope coverslips, 22 mm × 50 mm (Fisher Scientific, cat. no. 12-545-E)
Hamilton syringes, 10 μl, 25 μl and 2 × 250 μl
Water bath
Nitrogen
DNA SpeedVac 110 vacuum concentrator, Savant
Avanti mini extruder (Avanti Polar Lipids, cat. no. 610000)
Nuclepore track-etched polycarbonate membrane, 100 nm (Avanti Polar Lipids, cat. no. 610005)
Filter supports (Avanti Polar Lipids, cat. no. 610014)
Low-adhesion polycarbonate microcentrifuge tubes (USA Scientific; cat. no. 1405-2600, 0.5 ml and cat. no. 1415-2600, 1.5 ml) or siliconized microcentrifuge tubes (Sigma, cat. no. T3281 or T3531) ▲ CRITICAL We have found low-adhesion tubes from these providers to work best for SUPER template generation.
Neubauer counting chamber (VWR, cat. no. 15170-208)
Allegra 6R centrifuge (Beckman Coulter)
Costar 96-well plates (cat. no. 3925)
Synergy MX fluorescent plate reader (BioTek)
Inverted fluorescence microscope, Olympus IX71 UPlanSApo, ×100, 1.40 numerical aperture (NA) oil objective, Sutter Instruments LB-LS/OF30 Xenon arc lamp equipped with neutral density filters
Hamamatsu Orca-ER (Hamamatsu, model no. 04741-12AG)
Lab-Tek chamber, eight-well (Nunc, cat. no. N155411; Thermo Fisher, cat. no. 12565470)
Falcon tubes
Siliconized polypropylene tubes
REAGENT SETUP
▲ CRITICAL Please note that all solutions are filtered through a 0.22-μm filter.
2× assay buffer
Mix 40 mM HEPES (pH 7.5), 300 mM KCl and 2 mM MgCl2. This buffer can be stored for months at room temperature.
Microscopy assay buffer
Mix 20 mM HEPES (pH 7.5), 150 mM KCl, 1 mM MgCl2 and an oxygen-scavenging system containing 50 μg ml−1 glucose oxidase, 10 μg ml−1 catalase and 1 mM DTT. This buffer is freshly prepared and can be kept at room temperature for 1 d. Fresh glucose at a final concentration of 20 mM is added just before each experiment (2 μl of a 2 M glucose stock in water for a 200-μl sample).
Triton X-100 solutions
Prepare a 0.1% (vol/vol) and a 0.4% (vol/vol) Triton X-100 solution in Milli-Q water. These solutions can be stored for months at room temperature. Detailed protocols for transient dynamin-1 expression in insect cells5 and purification29 have been published elsewhere. Dynamin-1 can be stored at −80 °C in 20 mM HEPES, 150 mM KCl, 1 mM EGTA, 1 mM DTT and 10% (vol/vol) glycerol for months without loss of activity. Before the addition of glycerol, purified protein is pelleted at 100,000g for 10 min at 4 °C to remove any aggregated protein.
EQUIPMENT SETUP
Imaging setup
We use a neutral density filter to regulate the intensity of our light source in order to avoid photo-induced damage of dynamin. In our imaging setup, we use a setting of 50 on a scale up to 150. This setting will vary from light source to light source and must be individually determined.
PROCEDURE
Preparation of liposomes ● TIMING 2 h
▲ CRITICAL All steps must be performed in low-adhesion tubes or, preferably, silconized tubes in order to prevent nonspecific binding of proteins and lipids.
-
1| Aliquot the appropriate amount of lipids from a chloroform stock into a disposable 12 mm × 75 mm borosilicate glass tube using a 10-μl or 25-μl Hamilton syringe, which must be washed with chloroform between uses. Prepare in this way a 200-μl lipid solution with a total lipid concentration of 1 mM. For dynamin-catalyzed fission, we use a lipid composition of DOPC:DOPS:PI(4,5)P2:RhPE = 79:15:5:1% (mol/mol), although other liposome compositions also work10. Note that the conditions for SUPER template formation may have to be optimized for other lipid compositions, and that RhPE can be replaced by other fluorescent lipid probes.
▲ CRITICAL STEP For the efficient formation of excess membrane reservoir, the liposome mixture must contain at least 9% (mol/mol) of DOPS10.
2| Dry the lipids by placing the tube in a 40 °C water bath under a gentle stream of nitrogen so that a dried lipid film is formed at the bottom of the tube.
3| Dry the lipids further under vacuum in a SpeedVac for 30 min at a high heat setting. Although trace solvents may remain after this step, given the small volumes and high temperature used, this approach has proven to be satisfactory for our purposes. To ensure complete solvent removal, drying under higher vacuum (<1,000 mTorr) would be required.
4| Add Milli-Q water to the lipid film to a final concentration of 1 mM. Let the dried lipids swell into the water layer for 30 min at 37 °C.
5| Transfer the resuspended liposomes from Step 4 into a 15-ml Falcon tube. Freeze the liposome solution in liquid nitrogen and thaw it subsequently in a 37 °C water bath. Repeat this step two more times.
6| Prepare 100-nm extruded liposomes using an Avanti mini extruder. Pass the liposome solution 21 times through a 100-nm Nuclepore polycarbonate membrane. Instructions for the use of a mini extruder can be found at the Avanti website (http://www.avantilipids.com/index.php?option=com_content&view=article&id=185&Itemid=193).
-
7| Transfer liposomes to a low-adhesion or siliconized microcentrifuge tube.
■ PAUSE POINT This is a 5× liposome stock used to prepare SUPER templates. Liposomes can be stored at 4 °C and can be used for up to 2 weeks.
Preparation of SUPER templates ● TIMING 1 h
-
8| Prepare a 1:100 (vol/vol) dilution of the bead solution and apply ~15 μl to a Neubauer counting chamber. Count the number of beads in 16 squares of the counting chamber. The total concentration of beads can be calculated as follows: c (beads per ml) = number of counted beads × 104 × 100 (dilution factor).
▲ CRITICAL STEP Silica beads are usually shipped at a concentration of 5 × 108 beads per ml. However, because of lot-to-lot variability, it is important to determine the exact density of the 5-μm plain silica bead suspension using a Neubauer counting chamber.
9| Mix the beads thoroughly by vortexing them at maximum speed. Add 5 × 106 beads to a solution containing 200 μM liposomes (20 μl of 1 mM stock), 1 M NaCl (20 μl of 5 M stock) and Milli-Q water, up to a total volume of 100 μl in low- adhesion or siliconized polypropylene tubes. Mix the solution by flicking the tube gently. Please note that we have used silica beads of 2–5 μm in size. To keep protein-to-membrane ratios similar in subsequent assays, it may be advisable to keep the surface area constant (i.e., use 4× more 2.5-μm beads than 5-μm beads, see Experimental design).
10| Incubate the beads for 30 min at room temperature with intermittent mixing, which can be achieved by flicking the tube gently.
11| Add 1 ml of Milli-Q water to wash off excess unbound liposomes.
-
12| Gently vortex the tube at a low speed (just enough to dislodge the beads; we use setting 4 on a 1–8 scale) and spin it in a swinging bucket rotor at 260g for 2 min at room temperature in a benchtop, low-speed centrifuge.
▲ CRITICAL STEP Ensure that the vortexing is gentle and done at low speed in order to avoid shear stress, which could lead to shedding of the membrane reservoir.
13| Remove 1 ml of the supernatant.
14| Repeat washing (Steps 11–13) three more times.
-
15| Remove 1 ml of the supernatant. Flick the tubes to gently resuspend the templates in the remaining 100 μl until use. This is the 10× stock mixture of SUPER templates to be used in the sedimentation assay. Templates are typically used within 2–4 h, but they must be used within 6 h. For a quality check of templates, one can estimate the total lipid in this mix by solubilizing a 10-μl aliquot of the SUPER template stock mixture in 0.1% (vol/vol) Triton X-100 and determining the amount of membrane. To this end, prepare a standard curve of liposomes, from the stock generated in Steps 1–6, ranging between 0 and 20 μM liposomes in 0.1% (vol/vol) Triton X-100; plot the fluorescence against the concentration. Lipid concentration in a 10-μl aliquot of SUPER templates is typically ~6 μM.
? TROUBLESHOOTING
16| (Optional) Perform a quality check for the amount and fluidity of the deposited reservoir by depositing SUPER templates on a clean uncoated coverslip and visualizing the templates by epifluorescence microscopy. The membrane reservoir should ‘spill out’ onto the glass surface (Supplementary Video 1). Although this step is optional, it is highly recommended for first preparations.
Measurement or visualization of vesicle release
- 17| Measure (option A) or visualize (options B and C) vesicle release according to the following procedures. As described above, the sedimentation assay (option A) enables quantitative assessment of the efficiency and kinetics of vesicle release, and the microscopy-based assays (options B and C) allow for direct real-time visualization of vesicle release from the SUPER templates (option B) or of individual membrane fission events (option C). Note that the assay described in option B can also be used to monitor the generation of membrane tubules driven by the cooperative self-assembly of dynamin-1 by adding dynamin in the absence of GTP5–7. If desired, these assays can be performed in parallel with the same preparation of SUPER templates and proteins. Typically, the sedimentation assays are performed first, followed by the microscopy-based assays, ensuring that templates are used within 6 h of their formation.
- Sedimentation assay to measure vesicle release ● TIMING 45 min
- For each assay to be performed, including controls, add 50 μl of 2× assay buffer to 0.5-ml, low-adhesion or siliconized polypropylene microcentrifuge tubes—the total final volume for each tube will be 100 μl, and the final buffer concentration will be 1×.
-
Add dynamin-1 ± GTP to the assay tubes (not to the controls). Typically, we use increasing concentrations of dynamin-1 from 0.1 to 0.5 μM (five samples increasing in 0.1 μM increments) and three controls (see Experimental design), as well as a final 1 mM concentration of GTP. As many as 25 samples can be processed at a time; depending on the number of experimental variations tested, duplicates are run.▲ CRITICAL STEP Assays are started by the addition of SUPER templates, and because they settle, it is important to pipette them quickly but carefully (Fig. 2b and Supplementary Video 2) into each tube. Hence, timing is important, and it limits the number of samples that can be handled in each experiment.
- Adjust the final volume to 90 μl with Milli-Q water. Mix the solution by pipetting up and down two or three times with a 100-μl pipette.
- For the template control sample (see Experimental design), add 40 μl of Milli-Q water by pipette to the 50 μl of 2× assay buffer already present in the microcentrifuge tube (Step 17A(i)).
- (Optional) GTP can be omitted from an incubation mixture containing dynamin to serve as a control to ensure, as expected, that dynamin-mediated membrane fission depends on its GTPase activity.
- For the total control sample (see Experimental design), add 25 μl of 0.4% (vol/vol) Triton X-100 and 15 μl of Milli-Q water by pipette to the 50 μl of 2× assay buffer already present in the microcentrifuge tube (Step 17A(i)).
- For the blank control sample (see Experimental design), add 50 μl of Milli-Q water to the 50 μl of 2× assay buffer already present in the microcentrifuge tube (Step 17A(i)). Note that no templates are added to this sample.
- Resuspend the SUPER templates from Step 15 by gently flicking the tube.
- Gently add 10 μl of SUPER templates to the top of each assay solution (Fig. 2c and Supplementary Video 2) using a regular 200-μl pipette tip. Do not mix further at this stage. The reaction is initiated immediately upon the addition of the templates, which will slowly settle to the bottom of the tube during the course of the experiment.
- Incubate the mixture at room temperature for 30 min without shaking.
- Terminate the reaction by adding EDTA to a final concentration of 10 mM.
- Centrifuge the tubes at room temperature in a swinging bucket rotor at 260g for 2 min.
- Add 25 μl of 0.4% (vol/vol) Triton X-100 to a number of wells equal to the number of samples minus 1 in a 96-well plate. Remove 75 μl of supernatant from each of the assay and control tubes (except total) and add it to the 25 μl of 0.4% (vol/vol) Triton X-100. Pipette the mixture up and down slowly once or twice to ensure even mixing, but avoid the formation of bubbles. Remove 75 μl of supernatant from the total control tube (Step 17A(vi)) and add it to 25 μl of 0.1% (vol/vol) Triton X-100, pipetting up and down to ensure even mixing. Please note that the release of fluorescent vesicles into the supernatant is measured using a 96-well plate. Fluorescence of RhPE and many other fluorescent lipid probes may be partially quenched in the presence of Triton X-100. Therefore, Triton X-100 is added to each assay mixture in order to be able to compare fluorescence intensities across all samples, and thus to determine the fraction released as a reflection of fission efficiency.
- Measure fluorescence in a plate reader (for RhPE excitation = 530/25 nm bandwidth and emission = 580/25 nm bandwidth).
-
Calculate the fraction of fluorescent lipids released into the supernatant as follows. Subtract the fluorescence from the blank control sample from all values to obtain adjusted fluorescence data: % fission = 100 × adjusted sample fluorescence/adjusted fluorescence of the total control sample.? TROUBLESHOOTING
- The background release of fluorescent lipids is measured in the template control sample. This value, which should be <10% (typically ~5%) of total control, should be subtracted from ‘total % released’ to determine the specific amount of vesicle release owing to protein-mediated fission. Alternatively, one could subtract the value from another background control sample that contains dynamin but no GTP. In general, the GTP-less background is equivalent (± ~2%) to the nonspecific background, as dynamin-catalyzed release is GTP dependent.
-
(Optional) To visualize the released vesicles, carefully transfer 10 μl of the supernatant onto a clean borosilicate glass slide. The vesicles will attach to the glass surface and can then be imaged by fluorescence microscopy (Fig. 2b). This step is useful to verify that the fluorescence released corresponds to vesicles, which appear as bright spots, rather than solubilized lipids, which would appear as diffuse fluorescence.? TROUBLESHOOTING
- Real-time visualization of vesicle release from SUPER templates ● TIMING 30 min
- Wash each well of an eight-chamber Lab-Tek slide with absolute ethanol and then add 200 μl of a 2 mg ml−1 BSA solution and incubate it for 10 min. Treatment with the BSA solution ensures effective coating of the chamber and prevents the lipid from attaching to and spreading on the glass surface.
- Wash off excess BSA by rinsing the well twice with 200 μl of microscopy assay buffer.
- Add 200 μl of microscopy assay buffer containing 1 mM GTP to each well. Please note that a 100-μl volume of the buffer can be used in this step; however, we found it easier to add proteins on the microscope stage when a larger volume of buffer is present.
- Add 10 μl of a stock of SUPER templates from Step 15 with a regular 200-μl pipette tip. When performing this step, add the templates gradually throughout the entire well (Fig. 3b,c).
- Wait for the templates to settle (5 min).
- Put the Lab-Tek chamber onto the stage of an inverted fluorescence microscope. Use an ×100 oil objective to image SUPER templates. Reduce the intensity of the lamp with a neutral density filter (Equipment Setup). Focus on the templates and begin recording a video at 100-ms exposure, binning factor 2 × 2 and 1-s interval. Because of the brightness of the templates, the contrast has to be adjusted in order to see released vesicles.
- Carefully add an aliquot of dynamin-1 with a 20-μl pipette (typically to reach a final concentration of 0.5 μM) without disturbing the microscope’s focus; ideally, pipette the dynamin-1 solution at a distance from the field of view (Fig. 3d).
-
Observe the appearance of small vesicles released from the SUPER templates after the protein diffuses into the field of view.? TROUBLESHOOTING
- Real-time visualization of membrane fission events on pulled tethers ● TIMING 30 min
- Add 200 μl of a 2 mg ml−1 BSA solution to each well of an eight-chamber Lab-Tek slide and incubate it for 10 min to coat the glass surface.
- Wash off excess BSA by rinsing the well twice with 200 μl of microscopy assay buffer.
- To generate tethers, add 10 μl of a stock of SUPER templates from Step 15. When performing this step, add the templates gradually throughout the entire well (Fig. 3b,c).
- Wait for the templates to settle (5 min).
- Collect 10 μl of a purchased bead mixture of larger, untreated silica beads (diameter = 20 μm) using a 20-μl pipette. Hold the pipette upright and wait for the beads to settle within the pipette tip (Fig. 5a). Add only the volume containing the beads (1–2 μl) into the solution at one corner of the well (Fig. 5b).
- Wait for another few minutes until the beads settle in the slide chamber.
-
To produce the tethers, tilt and rock the chamber while gently tapping the side and causing the untreated silica beads to roll over the SUPER templates to capture their membrane and pull out the tethers. After tilting the chamber one to three times, you should see the larger beads forming an aggregate, which then is rolled over the layer of SUPER templates for another two or three times to direct their movement in a ‘W’-shaped path (Fig. 5c and Supplementary Video 4).▲ CRITICAL STEP Tilting the chamber too often can disrupt tethers that have been formed already. Performing this step successfully takes some practice, but eventually it becomes reproducible.
- To confirm the formation of tethers, put the Lab-Tek chamber onto a microscope stage and look for freestanding tethers, which will appear as fluorescent lines not attached to the glass surface.
- Reduce the intensity of the lamp with a neutral density filter (Equipment Setup) and focus on a freestanding tether at a freshly imaged area of the chamber. Because of the brightness of the templates, the contrast must be adjusted in order to see the tethers. Ideally, try to find a field of view that includes few or no templates, which obscure dimmer tethers.
- Begin recording a video at a 100-ms exposure, a binning factor of 2 × 2 and a 1-s interval time.
- Carefully add an aliquot of dynamin-1 with a 20-μl pipette (typically to reach a final concentration of 0.5 μM) without disturbing the microscope focus; ideally, pipette the dynamin-1 solution at a distance from the field of view (Fig. 3d).
-
Fission of membrane tethers can be observed after protein diffuses into the field of view.? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 15 | Beads aggregate during the washing process | Batch-to-batch variation of the surface properties of the silica beads, leading to differences in membrane incorporation | Vary the concentration of NaCl used during the formation of SUPER templates (Step 9) between 200 mM and 1M to determine optimum conditions by measuring membrane spillage (Step 16) |
| 17A(xv) and 17A(xvii) | No fluorescence is detected in the supernatant or no vesicles are observed | SUPER templates lack reservoirs, possibly because of problems with lipids and/or excessive shearing of templates during preparation. | Run a quality check on templates by adding them onto a clean glass coverslip and watching their excess reservoirs spread out on the glass surface. If there is no membrane spillage, then repeat Steps 8–15 |
| Dynamin preparation is inactive | Check dynamin’s assembly-stimulated GTPase activity on liposomes as described29 | ||
| 17B(viii) | No vesicle release is observed | Contrast is not adjusted | Increase the contrast of the microscope |
| SUPER templates spill membrane reservoir onto coverslip because the coverslip is not coated properly | Focus the microscope objective on the glass surface to detect spills. Improve coating of the coverslip by increasing the BSA concentration or extending the incubation time | ||
| 17B(viii) and 17C(xii) | No fission is observed | Dynamin is photo-inactivated | Adjust the neutral filter of the microscope to a lower setting. Make sure to add an oxygen-scavenging agent to the buffer |
| Templates flow out of the field of view | Protein is added to the slide well too quickly | Pipette the protein solution carefully. Alternatively, you may use a microinjection device with a constant low pressure to add the protein solution to the reaction mixture | |
| 17C(xii) | No tethers are observed | 20-μm beads have not captured the membrane and did not pull tethers | Carefully tilt one more time until tethers are observed |
| Rocking of the 20-μm beads has disrupted the tethers that already formed | Add fresh SUPER templates to a new well and repeat Step 17C(vii), tilting the chamber only three or four times | ||
| Tethers stick to the glass surface | The large silica beads were rocked too many times over the SUPER templates | Add fresh SUPER templates to a new well and repeat Step 17C(vii), tilting the chamber only three or four times |
● TIMING
Steps 1–7, preparation of liposomes: 2 h
Steps 8–16, preparation of SUPER templates: 1 h
Step 17A, sedimentation assay to measure vesicle release: 45 min
Step 17B, real-time visualization of vesicle release from SUPER templates: 30 min
Step 17C, real-time visualization of membrane fission events on pulled tethers: 30 min
ANTICIPATED RESULTS
An example of a sedimentation fission assay is shown in Figure 4a. As can be seen, fission depends on the presence of GTP in the reaction mixture. Fission efficiency should also be dependent on dynamin-1 concentration in the 0.1–0.5-μM range. Typical fission efficiency for 0.5 μM dynamin-1 is such that, after subtracting background fluorescence, the fluorescence intensity measured in the supernatant should be ~25–30% of the total fluorescence (Fig. 4a).
Given RhPE fluorescence, the released vesicles can also be directly visualized by fluorescence microscopy. Figure 4b shows a fluorescence micrograph of released vesicles. Figure 4c shows the release of vesicles over time from SUPER templates monitored by fluorescence microscopy. The first image was recorded before the addition of dynamin to the reaction mixture, and the second image was recorded 2.5 min after this addition (Supplementary Video 3).
SUPER templates also have sufficiently large membrane reservoirs so that membrane tethers can be obtained. Figure 6 shows the fission of such preformed membrane tethers as mediated by dynamin-1 (Supplementary Video 5). Individual fission events that sever the tubes are detected by time-lapse fluorescence microscopic images recorded before and after the addition of dynamin-1 to the reaction mixture.
Figure 6.
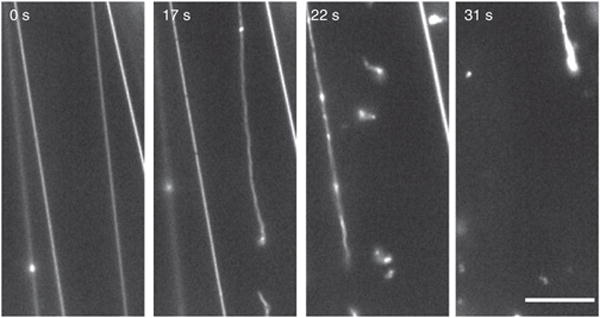
Time course of membrane tether fission. Tethers were drawn from SUPER templates as described in Figure 5 and text. After imaging was started, dynamin-1 was added to the solution (t = 0) with a pipette to a final concentration of 0.5 μM. See Supplementary Video 5 for the full sequence. Scale bar, 5 μm.
Supplementary Material
Acknowledgments
This work was funded by the US National Institutes of Health grants R01-GM42455 and R01-MH61345 to S.L.S. and fellowships from the Leukemia and Lymphoma Society to T.J.P. and from the German Research Foundation (DFG) and the American Heart Association to S.N. We thank Y.-W. Liu for her help in filming the procedures and M. Dadachov at Corpuscular for helpful discussions regarding silica bead preparation.
Footnotes
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS T.J.P. developed the method and provided comments on the manuscript. S.N. developed the detailed protocol and troubleshooting steps and wrote the manuscript with S.L.S.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Liu AP, Fletcher DA. Biology under construction: in vitro reconstitution of cellular function. Nat Rev Mol Cell Biol. 2009;10:644–650. doi: 10.1038/nrm2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mettlen M, Pucadyil T, Ramachandran R, Schmid SL. Dissecting dynamin’s role in clathrin-mediated endocytosis. Biochem Soc Trans. 2009;37:1022–1026. doi: 10.1042/BST0371022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmid SL, Frolov VA. Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol. 2011;27:79–105. doi: 10.1146/annurev-cellbio-100109-104016. [DOI] [PubMed] [Google Scholar]
- 4.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran R, et al. Membrane insertion of the pleckstrin homology domain variable loop 1 is critical for dynamin-catalyzed vesicle scission. Mol Biol Cell. 2009;20:4630–4639. doi: 10.1091/mbc.E09-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pucadyil TJ, Schmid SL. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell. 2008;135:1263–1275. doi: 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu YW, et al. Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. Proc Natl Acad Sci USA. 2011;108:E234–E242. doi: 10.1073/pnas.1102710108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakobsson J, et al. Regulation of synaptic vesicle budding and dynamin function by an EHD ATPase. J Neurosci. 2011;31:13972–13980. doi: 10.1523/JNEUROSCI.1289-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann S, Schmid S. Regulation of dynamin-mediated fission by endocytic accessory proteins. Mol Biol Cell. 2011;22:4705. Abstract no. 1030. [Google Scholar]
- 10.Pucadyil TJ, Schmid SL. Supported bilayers with excess membrane reservoir: a template for reconstituting membrane budding and fission. Biophys J. 2010;99:517–525. doi: 10.1016/j.bpj.2010.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid EM, McMahon HT. Integrating molecular and network biology to decode endocytosis. Nature. 2007;448:883–888. doi: 10.1038/nature06031. [DOI] [PubMed] [Google Scholar]
- 12.Schmid SL, Smythe E. Stage-specific assays for coated pit formation and coated vesicle budding. vitro J Cell Biol. 1991;114:869–880. doi: 10.1083/jcb.114.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miwako I, Schmid SL. Clathrin-coated vesicle formation from isolated plasma membranes. Methods Enzymol. 2005;404:503–511. doi: 10.1016/S0076-6879(05)04044-9. [DOI] [PubMed] [Google Scholar]
- 14.Takei K, et al. Generation of coated intermediates of clathrin-mediated endocytosis on protein-free liposomes. Cell. 1998;94:131–141. doi: 10.1016/s0092-8674(00)81228-3. [DOI] [PubMed] [Google Scholar]
- 15.Dannhauser PN, Ungewickell EJ. Reconstitution of clathrin-coated bud and vesicle formation with minimal components. Nat Cell Biol. 2012;14:634–639. doi: 10.1038/ncb2478. [DOI] [PubMed] [Google Scholar]
- 16.Ford MG, et al. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 17.Sweitzer S, Hinshaw J. Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell. 1998;93:1021–1029. doi: 10.1016/s0092-8674(00)81207-6. [DOI] [PubMed] [Google Scholar]
- 18.Takei K, Slepnev VI, De Camilli P. Interactions of dynamin and amphiphysin with liposomes. Methods Enzymol. 2001;329:478–486. doi: 10.1016/s0076-6879(01)29109-5. [DOI] [PubMed] [Google Scholar]
- 19.Danino D, Moon KH, Hinshaw JE. Rapid constriction of lipid bilayers by the mechanochemical enzyme dynamin. J Struct Biol. 2004;147:259–267. doi: 10.1016/j.jsb.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Gallop JL, et al. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 22.Pylypenko O, Lundmark R, Rasmuson E, Carlsson SR, Rak A. The PX-BAR membrane-remodeling unit of sorting nexin 9. EMBO J. 2007;26:4788–4800. doi: 10.1038/sj.emboj.7601889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ford MG, et al. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 24.Roux A, et al. Membrane curvature controls dynamin polymerization. Proc Natl Acad Sci USA. 2010;107:4141–4146. doi: 10.1073/pnas.0913734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roux A, Uyhazi K, Frost A, De Camilli P. GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature. 2006;441:528–531. doi: 10.1038/nature04718. [DOI] [PubMed] [Google Scholar]
- 26.Bashkirov PV, et al. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell. 2008;135:1276–1286. doi: 10.1016/j.cell.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 28.Karatekin E, et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci USA. 2010;107:3517–3521. doi: 10.1073/pnas.0914723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonard M, Song BD, Ramachandran R, Schmid SL. Robust colorimetric assays for dynamin’s basal and stimulated GTPase activities. Methods Enzymol. 2005;404:490–503. doi: 10.1016/S0076-6879(05)04043-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.