

Abstract
Background
The treatment of non-localized prostate cancer involves androgen deprivation (AD) therapy which results in tumor regression. Apoptosis has been implicated in the tumor response to AD, but constitutes a small fraction of the total tumor at any time. Cellular senescence is a response to sub-lethal stress in which cells are persistently growth arrested and develop distinct morphological and biochemical characteristics. The occurrence of senescence in prostate tumor tissue after AD therapy has not previously been investigated.
Methods
Phenotypic and molecular characteristics of senescence were examined in models of androgen-sensitive prostate cancer after AD and compared with androgen-intact controls.
Results
In vitro in LNCaP cells, AD induced elevated senescence-associated β-galactosidase (SA-β-gal) staining, decreased proliferation, and increased flow cytometric side scatter while minimally affecting cell viability. The increased expression of the senescence-related proteins Glb1, the cyclin-dependent kinase inhibitor p27Kip1 and chromatin-regulating heterochromatin protein 1γ (HP1γ) were detected in LNCaP cells after AD in vitro by immunoblot and immunofluorescence microscopy. In mice bearing LuCaP xenograft tumors in vivo, surgical castration similarly increased SA-β-gal staining, increased expression of p27Kip1 and HP1γ, and decreased expression of the proliferation marker KI-67, with minimal induction of apoptosis identified by detection of cleaved caspase 3 and TUNEL. Immunohistochemical analysis of human prostate tumors removed after AD shows similar induction of Glb1, HP1γ and decreased KI-67.
Conclusions
We conclude that AD induces characteristics consistent with cellular senescence in androgen-sensitive prostate cancer cells. This finding may explain incomplete tumor regression in response to AD.
Keywords: androgen, androgen deprivation, prostate cancer, cellular senescence
Introduction
Prostate cancer growth is driven by androgen in the majority of clinical cases, and androgen deprivation (AD) is the standard treatment for advanced disease. The response of prostate cancer cells to AD and its affect on clinical outcomes have been a focus of intense investigation. Removal of androgens by surgical or chemical castration results in decreased proliferation in androgen-sensitive tumors and the presence of apoptosis in a subset of cells [1]. However, AD commonly results in incomplete tumor regression, and the development of castration-independent tumors eventually develops. The processes that regulate this transition to castration-independence and factors that affect the time to tumor recurrence are incompletely understood.
During the regression of androgen-sensitive tumors, apoptosis occurs in a subset of prostate cancer cells and growth arrest in others in vitro and in vivo [2,3]. In CWR22 xenograft tumors, apoptosis peaks 2 days after castration, then rapidly decreases [2]. The non-apoptotic bulk of the tumor persists and expresses the cyclin-dependent kinase inhibitor (CDKI) proteins p16Ink4a, p21Waf1/Cip1, and p27Kip1 [4–7]. Decreased expression of the proliferation marker KI-67 is also seen. Similarly, patient tumors after AD show histological signs of regression, decreased proliferation, but few apoptotic cells [3]. The fate and impact of non-apoptotic tumor cells after AD remains undefined.
In many ways, the phenotype of non-apoptotic, androgen-dependent prostate cancer cells after AD is similar to that displayed by senescent cells. Senescence is a distinct phenotypic response to sub-toxic stress. It is characterized by a flattened appearance in vitro, persistent growth arrest, and specific biochemical characteristics including the increased expression of secreted proteins. Senescence occurs in both non-transformed and cancer cells in response to sub-lethal insults involving DNA damage, oxidative stress, and the activities of oncogenic and stress-signaling pathways [8–10]. While there is no universal biomarker for senescence, this phenotype has been identified both in vitro and in vivo using multiple biomarkers including senescence-associated β-galactosidase (SA-β-gal) activity, CDKIs, HP1 sub-types, DNA damage and other damage-signaling events [8,11–14].
In this study, we address whether non-apoptotic cells in androgen-dependent prostate tumor models develop characteristics of cellular senescence after AD. Monitoring multiple characteristics and markers of senescence in LNCaP cells in vitro, in xenografts and patient tumors in vivo, we find that a subset of tumor cells that persist after AD develop a phenotype consistent with senescence.
Materials and Methods
In vitro Cell Culture and Androgen-Deprivation Experiments
Androgen-dependent LNCaP prostate cancer cells were routinely cultured in complete and androgen-free medium as previously described [15]. To monitor cell growth, proliferation, viability and side scatter (SSC), LNCaP cells were cultured in triplicate 35 mm wells, stained, counted, and analyzed by flow cytometry [16]. The percentage of viable cells was assessed after propidium iodide staining. Apoptotic cells were identified by Annexin V staining [16]. For immunoblot analysis of proteins, LNCaP cells were cultured on 100-mm plates and the following antibodies used: p27Kip1 (#610242; BD Biosciences, Lexington, KY), Glb1 (#ab55176; AbCam, Cambridge, MA), α-tubulin (CP06; Calbiochem, San Diego, CA), and HRP-conjugated anti-mouse and anti-rabbit secondary antibodies (#31432, #31464; Thermo/Pierce, Rockford, IL) [16]. SA-β-gal activity and immunofluorescence microscopy was performed as described [16]. For each analysis, five photographs at high magnification were taken for each of three independent replicates per condition. The per-cell nuclear expression of HP1γ was measured in each image using Cell-Profiler 2.0 and averaged [17]. HP1γ positive and negative nuclei were counted in each image to calculate the percentage of positive cells per image. Antibodies utilized included HP1γ (#05-690; Upstate, Temecula, CA) and anti-mouse—Alexa 488 (Invitrogen, Carlsbad, CA). Nuclei were counterstained with Hoechst 33342 (Molecular Probes, Eugene, OR). Experiments were reproduced in triplicate.
Xenograft Tumors
Animal studies were performed in accordance with the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care International, and approval was obtained from the University of Wisconsin Institutional Animal Care and Use Committee. LuCaP 58 and 23.1 xenograft tumors were a gift of Robert Vessella, University of Washington. Male athymic nude mice were purchased from Harlan (Madison, WI), and xenograft tumors serially cultured as previously described [18]. Mice bearing tumors of approximately 65 mm3 were subject to either mock surgery or castrated. Tumors were harvested 3 or 10 days after castration, or 10 days after mock surgery, and either frozen in sectioning medium or formalin-fixed and paraffin-embedded (FFPE). Tissue sections were processed and stained as previously described [16,18]. Stained xenograft sections were visualized by light microscopy, photographed, and increased expression in the population was measured by counting the number of positive stained cells in a high magnification image and dividing by the total number of cells per image. Cells were counted in three images from five tumors for each condition and averaged. Primary antibodies included p27Kip1 (#610242; BD Biosciences, Lexington, KY), KI-67 (VP-K452; Vector Laboratories Inc., Burlingame, CA), cleaved caspase 3 (#9661, Cell Signaling Technology, Beverly, MA), HP1γ (#05-690 Upstate, Temecula, CA), and Glb1 (#ab55176; AbCam, Cambridge, MA).
Patient Samples
FFPE-patient tissues were obtained from the University of Wisconsin TRIPP Laboratory and Tissue Bank, according to Institutional Review Board approval and policies. Tissue samples were processed, as above, and per-cell expression was measured using the Vectra™ imaging system (Caliper Life Sciences, Hopkinton, MA), as previous reported [19]. Briefly, slides were stained using antibodies against Glb1, KI67, p27Kip1 and HP1γ with colorimetric secondary antibodies. Stained tissue sections were imaged, and stain spectra were determined to minimize background and maximize signal specificity. E-Cadherin was stained to identify epithelial cells. Staining localized to the nucleus and cytoplasm was measured using automated protocols and added together to calculate the relative expression of each marker protein per cell.
Statistical Methods
Data were analyzed, averages and standard error were calculated, and Student's t-test was performed using Microsoft Excel. Error bars in all figures represent standard error. In all data where Student's t-test was performed, *P < 0.05 and **P < 0.01.
Results
We had previously noticed that after the removal of androgen, LNCaP cells frequently demonstrate an enlarged, flattened morphology reminiscent of senescence (Fig. 1A). To further evaluate senescence characteristics after AD in vitro, we cultured androgen-dependent LNCaP cells in androgen-free conditions using charcoal-stripped serum or in replete medium. Cells were first stained for SA-β-gal activity, a primary marker of cellular senescence that appears as blue perinuclear staining [13]. The percentage of SA-β-gal staining cells increased to 40% and 60% after 6 and 9 days of AD, respectively (Fig. 1A). After being returned to complete medium for 9 additional days, SA-β-gal activity remained elevated in AD cultures and cells failed to form colonies (data not shown) suggesting a terminal arrest. In contrast, staining of the androgen-independent cell line PC3 (Supplementary Fig. 1) and Du145 (data not shown) do not significantly induce SA-β-gal expression under AD.
Fig. 1.
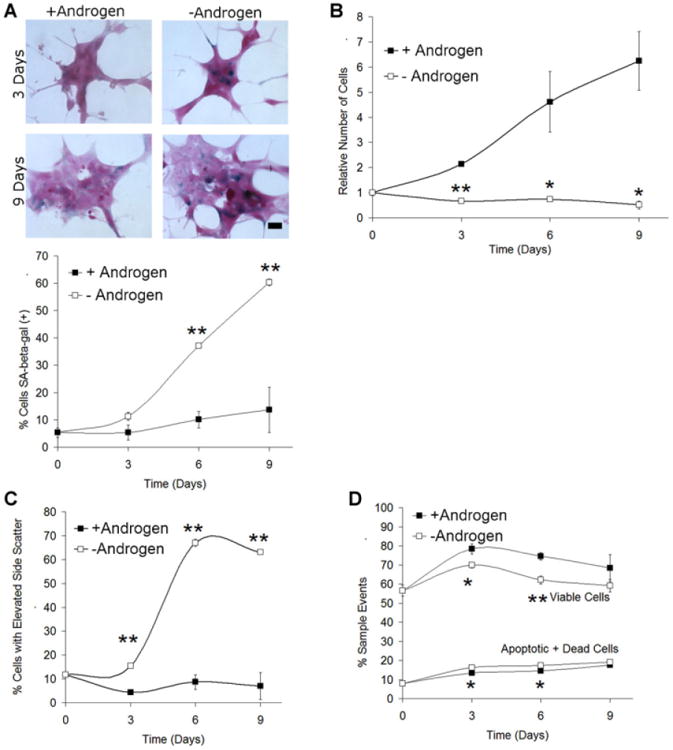
Androgen deprivation of LNCaP in vitro induces phenotypic characteristics of cellular senescence. A: LNCaP cells were cultured in androgen-free or repletmedium for up to 9 days, stained for SA-β-gal activity, and counterstained with nuclear fast red as described. Scale bars: low magnification, × 200,40 μm; inset, 20 μm. The number of cells staining intensely positive (blue) and the total number of cells per high power microscope image were counted and used to calculate the percentage of SA-β-gal positive cells used in the figure. B: LNCaP cultured in androgen-free or control medium were counted using flow cytometry. Androgen-free medium results in a decrease in the total number of viable cells. C: Flow cytometry was used to examine the measure of cellular complexity side scatter. AD increases the fraction of cells with elevated cellular complexity and size. D: AD causes a minor decrease in the viable fraction of cells, and an increase in apoptosis, as measured using Annexin V/propidium iodide staining. For all conditions and experiments, n = 3. Error bars represent Standard Error. Student's t-test: *P < 0.05 and **P < 0.01.
Other characteristics of the cultures after AD were assessed. As previously described [15], the number of cells after AD decreases by approximately 50% over 9 days with the majority of cell loss occurring in the first 3 days (Fig. 1B). DNA staining using propidium iodide and cell cycle modeling was performed. Decreased proliferation after AD was seen with an increased G1/G0 fraction and decreased S phase (Table I). A second characteristic of senescence is altered morphology. To confirm our visual assessment of cellular enlargement and complexity, we utilized flow cytometry to evaluate side scatter (SSC), a parameter commonly increased in senescence [8]. A significant increase in side scatter was seen after 3 days and continued to increase in longer term AD cultures (Fig. 1C). The majority of cells in the population remained viable, another senescence characteristic (Fig. 1D). Apoptosis was assessed by Annexin V staining and flow cytometry. Significant but smaller (3%) changes in apoptosis were seen at 3 and 6 days when compared with control (Fig. 1D). Therefore, AD induces phenotypic characteristics of cellular senescence, as well as increased SA-β-gal and decreased proliferation in LNCaP.
Table I. Cell Cycle Modeling of LNCaP Cells+/− Androgen (P < 0.05).
| Days AD | Condition | %G | %S | %G2/M |
|---|---|---|---|---|
| 0 | 56 ±1 | 36 ±1 | 8 ± 1 | |
| 3 | +Androgen | 54 ±4 | 32 ±3 | 14 ± 1* |
| −Androgen | 74 ± 9* | 21 ± 11* | 4 ± 2* | |
| 6 | +Androgen | 79 ± 4** | 18 ± 4** | 3 ± 1** |
| −Androgen | 85 ± 6** | 11 ± 5** | 4 ± 1* | |
| 9 | +Androgen | 76 ± 2** | 16 ± 3** | 8 ± 1 |
| −Androgen | 79 ± 4** | 16 ± 3** | 4 ± 2 |
Next, we analyzed the expression of the senescence-associated proteins p27Kip1, Glb1, and HP1γ in LNCaP cells after AD in vitro. The expression of proliferation-regulating CDK inhibitors is associated with senescence and include increased p27Kip1 in prostate cancer [20]. Immunoblot analysis of protein lysates collected after AD showed increased expression of p27Kip1 over time (Fig. 2A). Expression of lysosomal β-galactosidase Glb1, a protein that is the target of SA-β-gal staining [21], also increases after AD (Fig. 2A). Proteins in the HP1 family, including HP1γ, regulate gene expression by altering chromatin structure and are consistently linked with senescence in normal and cancer cells [14]. Immunofluorescence was performed on LNCaP cells to assess nuclear accumulation of the HP1γ protein. AD showed nuclear HP1γ expression increases by approximately 1.8-fold, and the fraction of positive cells also increased (Fig. 2B and C). Samples from control cells analyzed at day 0 resembled control cells cultured in androgen-containing medium (data not shown). Therefore, the expression of senescence-associated proteins increases with AD in vitro.
Fig. 2.
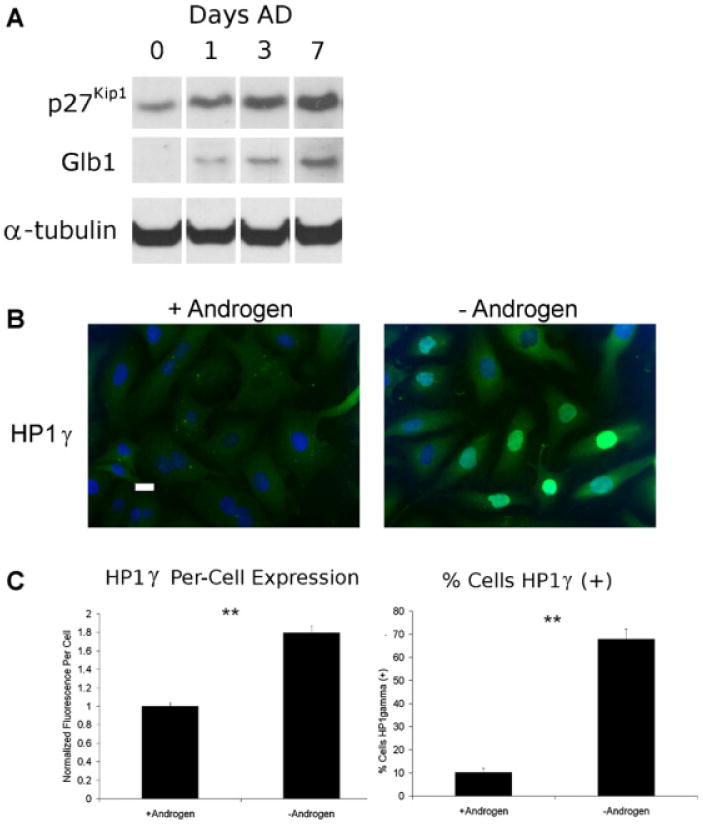
Androgen deprivation in vitro induces expression of senescence-associated proteins. Subconfluent LNCaP cells were cultured in androgen-free or supplemented medium and cells underwent fixation and immunofluorescence or had lysates harvested for western analysis. A: Increased expression of p27Kip1 and Glb1 using western blotting were observed after 1, 3, and 7 days culture in androgen-free medium. Cells collected at timepoint0 days were grown in complete medium before androgen removal. These results are representative of two experiments. B: The expression of HP1γ in LNCaP nuclei in control cells and after 9 days AD was detected by immunofluorescence microscopy. Scale bar = 20 μm. C: Cells individually increased expression of HP1γ (left) in a greater fraction of cells (right) after AD. Image fluorescence was measured using a CellProfiler 2.0. Foreach condition, n = 3. Error bars represent Standard Error. Student's t-test:*P < 0.05 and **P < 0.01.
We next addressed whether AD similarly induces characteristics of senescence in androgen-sensitive xenograft tumors. LuCaP 58 and 23.1 xenografts were established in athymic nude mice subjected to castration or mock surgery and harvested 3 or 10 days later. Frozen tumor sections were stained for SA-β-gal activity, while protein expression in FFPE tumors was assessed by immunohistochemistry (Fig. 3A). SA-β-gal staining in frozen tumor sections increased significantly from 5% of tumor cells in intact animals to 23% and 22% of tumors 3 and 10 days after castration, respectively (Fig. 3B). A similar induction of SA-β-gal staining was seen in another androgen-sensitive line, LuCaP 23.1, after castration (data not shown). Expression of the CDK inhibitor p27Kip1 significantly increased after castration, from 22% of cells in tumors from in intact animals to 72% and 67% after castration, while HP1g expression increased from 15% of tumor cells to 50% and 85%, respectively (Fig. 3B). At the same time, expression of KI-67 decreased significantly. Increased expression of p27Kip1 and Glb1 in AD tumors was confirmed by immunoblot analysis of four pooled LuCaP 23.1 tumor lysates per condition (Supplementary Fig. 2).
Fig. 3.
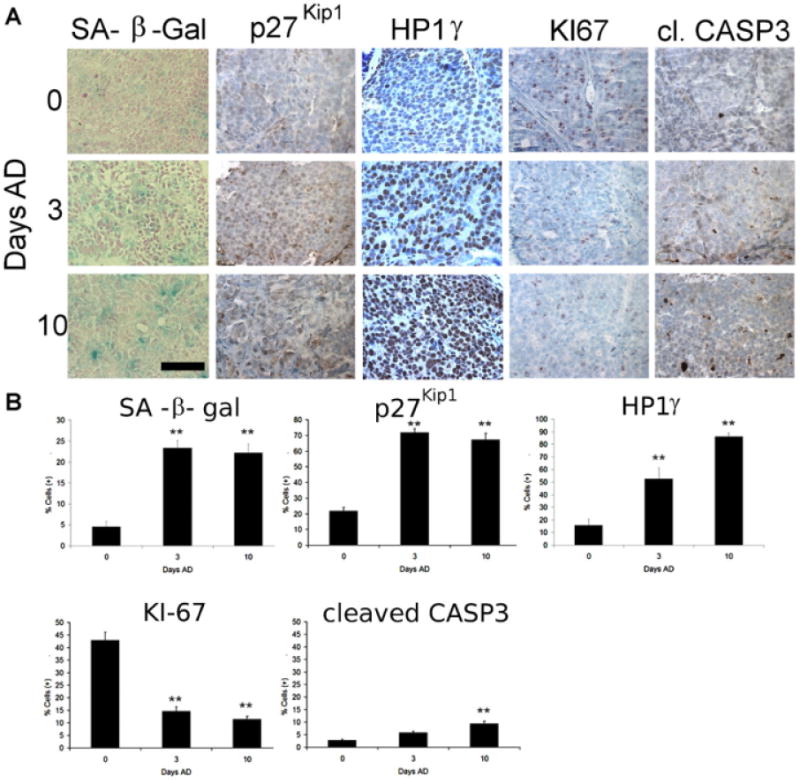
Induced expression of senescence-associated marker proteins in LuCaP 58 tumors of castrated mice. Xenograft tumors were established and when 65 mm3 mice were subject to either mock surgery or castration and harvested at 0, 3, or 10 days. A: Staining of SA-β-gal activity, p27Kip1 protein, and HP1γ expression increases in tumors after AD while KI-67 decreases. Apoptosis detected as cleaved caspase 3 increases to a minor but significant extent. Scale bar = 20 μm. B: The number of total and positive-staining cells in each high power image (three images per tumor in five tumors) were counted and used to calculate the percentage of positive-staining cells for each marker. These data are representative of the results from two independent experiments. Error bars represent Standard Error. Student's t-test: **P < 0.01.
The increase of apoptosis, detected as cleaved caspase 3, was minor and only significant 10 days after castration (Fig. 3B). Identification of apoptosis in xenograft tumors by TUNEL staining similarly detected <5% increases in apoptotic cells in castrated samples (Supplementary Fig. 3). These data demonstrate that AD induces similar senescence-associated changes in phenotype and protein expression in LuCaP xenografts in vivo after surgical castration as those observed in vitro.
We then analyzed peripheral-zone prostate tumors from patients who received AD therapy before undergoing radical prostatectomy and compared these to samples from intact patients matched for age, tumor grade, and Gleason score (Table II). Protein expression was measured as staining intensity using the Vectra™ imaging system. This system allows automated selection of cellular subsets (epithelial vs. stromal) and quantitation of fluorescence or colorimetric staining. The average tumor volume was decreased in patients after AD therapy as expected (17.5% vs. 6.75%; one-tailed t-test: P = 0.07) (Table II). The timing of AD varied in these patient samples from 3 to 12 months. We find that the expression of Glb1 and HP1γ increased significantly, while KI-67 decreased after AD (Fig. 4). Expression of nuclear p27Kip1 was not significantly different between AD and intact groups. Large variations in p27Kip1 were noted within and between these human samples (data not shown). In all, these results demonstrate that AD induces markers of senescence in prostate cancer cells in vitro and in vivo.
Table II. Clinical Pathology Data Associated with Patient Samples.
| Age at surgery | Tumor grade | Gleason score | % Tumor (+) | Time in AD (months) | |
|---|---|---|---|---|---|
| +Androgen | 58 | T2C | 3 + 4 | 15 | |
| 66 | T2C | 4 + 3 | 35 | ||
| 64 | T3A | 4 + 3 | 5 | ||
| 66 | T3B | 4 + 5 | 15 | ||
| −Androgen | 63 | T2C | 3 + 3 | 11 | 10 |
| 55 | T2C | 3 + 3 | 7 | 6 | |
| 59 | T3B | 4 + 3 | 7 | 4 | |
| 65 | T2C | 4 + 4 | 2 | 3 |
Fig. 4.

Patient prostate tumors removed after AD therapy express senescence-associated marker proteins. Expression of senescence-associated proteins was detected in formalin-fixed tissue samples by IHC and measured using the Vectra™ imaging system. Nuclear and cytoplastic staining was measured and added together for all markers. For each condition, n = 4. Error bars represent Standard Error. Student's t-test: *P < 0.05 and **P < 0.01.
Discussion
The response of prostate tumors to androgen ablation is quite variable, with some patients having prolonged remission while others rapidly recur and progress. AD in xenograft and patient tumors is associated with decreased proliferation, but surprisingly low levels of apoptosis [1–3,18]. Apoptosis typically peaks within the first 72 hr after AD and diminishes afterward, leaving cells that are not immediately apoptotic [3]. In contrast, the hallmark increase of SA-β-gal activity and senescence morphology in cancer cells in vitro typically requires 3–6 days to develop after drug exposure [8]. The nature and fate of cancer cells that persist after AD and tumor regression remain largely undetermined.
The present data demonstrates that, in addition to apoptosis, AD induces a senescent-like phenotype in a subset of androgen-sensitive cancer cells using multiple models of androgen-sensitive prostate cancer. Without single markers that uniquely identify senescence, we adopted a strategy of assessing multiple surrogate markers associated with the senescence phenotype [22]. Hallmarks of the senescent phenotype in vitro include viability, proliferation arrest, and increased cellular complexity (SSC) (Fig. 1). The presence of a number of molecular markers of senescence were then confirmed in these cultures including increased SA-β-gal activity, p27Kip1, Glb1 and the nuclear accumulation of HP1γ, a marker of altered chromatin structure consistently linked with senescence [14]. The presence of these markers was then confirmed in a series of LuCaP tumors subjected to castration. We estimate the percentage of terminally senescent cells in these pure tumor xenografts to be as high as 30% at later timepoints.
Finally, we analyzed the expression of senescence-associated proteins in a set of patient tumors. Glb1 and HP1γ were increased and the proliferation marker KI-67 was decreased in patient tumors treated with AD (Table II; Fig. 4). SA-β-gal activity was not able to be assessed as these were formalin-fixed samples. A positive correlation between SA-β-gal activity and Glb1 protein levels was demonstrated in vivo and in vitro (Figs. 1 and 3). Expression of p27Kip1 was not significantly altered in patient tumors after AD. We speculate that this may be due to heterogeneity in the small number of samples examined. Alternatively p27Kip1 induction may be an early response to AD within the first 3 months of treatment that is not detected at later timepoints. A recent manuscript found that AD induces a senescence-associated secretory phenotype, in part mediated by Skp2 decreases [23]. Given the role Skp2 plays in the degradation of p27Kip1 and the induction of senescence [24,25], our results are consistent with the current understanding of this phenomenon.
Our data also demonstrate a mechanistic relationship between androgen receptor signaling and senescence induction. The induction of senescence in cancer cells is associated with exposure to specific stresses involving DNA damage, oncogenic signaling and oxidative stress [8]. Loss of androgen signaling has been demonstrated to alter genomic integrity by effecting telomere stability and DNA replication [26,27]. AD also increases cellular oxidative stress in prostate cancer cells which may be instrumental in this response [28]. Finally, AD induces the expression of CDKI protein p16Ink4a, p21Waf1/Cip1, and p27Kip1 [5–7,20,29], which have been independently shown to induce senescence in some cancer models [2,11,18,30]. The relationship of androgen receptor activity and senescence will become clearer as the regulation of senescence becomes better defined.
These results indicate that a subset of prostate cancer cells respond to AD with a senescence phenotype. Accumulating evidence suggests senescence may serve as a prognostic marker of therapeutic response. Senescence in a mouse Em-myc tumor model prolongs survival compared with apoptotic controls [31]. This may be related to increased natural killer cell activity within the hepatocarcinoma model utilized [32,33]. Senescence identified in tumor tissue may be associated with improved responses to chemotherapy and outcomes in colon cancer patients [22]. The observation that senescence is part of the prostate tumor response to AD raises questions whether this terminal phenotype may predict the duration of response to AD. The mechanisms regulating this response and the clinical relevance of this observation will be the subject of further investigation.
Supplementary Material
Acknowledgments
Grant sponsor: The John Livesey Endowment, and the Department of Defense Prostate Cancer Research Program (D.F.J. and J.A.D); Grant number: DAMD17-02-1-0163; Grant sponsor: Ruth L. Kirchstein National Research Service Award (J.A.E.); Grant number: T32 CA009681-14.
Footnotes
Additional supporting information may be found in the online version of this article.
Conflicts of interest: None.
References
- 1.Denmeade SR, Isaacs JT. Programmed cell death (apoptosis) and cancer chemotherapy. Cancer Control. 1996;3(4):303–309. doi: 10.1177/107327489600300401. [DOI] [PubMed] [Google Scholar]
- 2.Smitherman AB, Gregory CW, Mohler JL. Apoptosis levels increase after castration in the CWR22 human prostate cancer xenograft. Prostate. 2003;57(1):24–31. doi: 10.1002/pros.10271. [DOI] [PubMed] [Google Scholar]
- 3.Westin P, Stattin P, Damber JE, Bergh A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am J Pathol. 1995;146(6):1368–1375. [PMC free article] [PubMed] [Google Scholar]
- 4.Myers RB, Oelschlager DK, Coan PN, Frost AR, Weiss HL, Manne U, Pretlow TG, Grizzle WE. Changes in cyclin dependent kinase inhibitors p21 and p27 during the castration induced regression of the CWR22 model of prostatic adenocarcinoma. J Urol. 1999;161(3):945–949. [PubMed] [Google Scholar]
- 5.Agus DB, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde DW, Scher HI. Prostate cancer cell cycle regulators: Response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999;91(21):1869–1876. doi: 10.1093/jnci/91.21.1869. [DOI] [PubMed] [Google Scholar]
- 6.Loda M. p27KIP1: Androgen regulation and prognostic significance in prostate cancer. Adv Clin Path. 2000;4(4):226–232. [PubMed] [Google Scholar]
- 7.Waltregny D, Leav I, Signoretti S, Soung P, Lin D, Merk F, Adams JY, Bhattacharya N, Cirenei N, Loda M. Androgen-driven prostate epithelial cell proliferation and differentiation in vivo involve the regulation of p27. Mol Endocrinol. 2001;15(5):765–782. doi: 10.1210/mend.15.5.0640. [DOI] [PubMed] [Google Scholar]
- 8.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102(20):1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63(11):2705–2715. [PubMed] [Google Scholar]
- 10.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11(11):S27–S31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 11.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8(24):4112–4118. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawless C, Wang C, Jurk D, Merz A, Zglinicki TV, Passos JF. Quantitative assessment of markers for cell senescence. Exp Gerontol. 2010 doi: 10.1016/j.exger.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle. 2007;6(7):784–789. doi: 10.4161/cc.6.7.4079. [DOI] [PubMed] [Google Scholar]
- 15.Ripple MO, Henry WF, Rago RP, Wilding G. Prooxidant-antioxidant shift induced by androgen treatment of human prostate carcinoma cells. J Natl Cancer Inst. 1997;89(1):40–48. doi: 10.1093/jnci/89.1.40. [DOI] [PubMed] [Google Scholar]
- 16.Ewald JA, Desotelle JA, Almassi N, Jarrard DF. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br J Cancer. 2008;98(7):1244–1249. doi: 10.1038/sj.bjc.6604288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: Free, versatile software for automated biological image analysis. Biotechniques. 2007;42(1):71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 18.Bladou F, Vessella RL, Buhler KR, Ellis WJ, True LD, Lange PH. Cell proliferation and apoptosis during prostatic tumor xenograft involution and regrowth after castration. Int J Cancer. 1996;67(6):785–790. doi: 10.1002/(SICI)1097-0215(19960917)67:6<785::AID-IJC6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 19.Huang W, Kanehira K, Drew S, Pier T. Oncocytoma can be differentiated from its renal cell carcinoma mimics by a panel of markers: An automated tissue microarray study. Appl Immunohistochem Mol Morphol. 2009;17(1):12–17. doi: 10.1097/PAI.0b013e318173e79d. [DOI] [PubMed] [Google Scholar]
- 20.Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35(3):317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 21.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 22.Haugstetter AM, Loddenkemper C, Lenze D, Grone J, Stand-fuss C, Petersen I, Dorken B, Schmitt CA. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br J Cancer. 2010;103(4):505–509. doi: 10.1038/sj.bjc.6605784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pernicova Z, Slabakova E, Kharaishvili G, Bouchal J, Kral M, Kunicka Z, Machala M, Kozubik A, Soucek K. Andro-gen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia. 2011;13(6):526–536. doi: 10.1593/neo.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464(7287):374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewald J, Jarrard D. Decreased Skp2 expression is necessary but not sufficient for therapy-induced senescence in prostate cancer. Trans Oncol. 2012;5(4) doi: 10.1593/tlo.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iczkowski KA, Huang W, Mazzucchelli R, Pantazis CG, Stevens GR, Montironi R. Androgen ablation therapy for prostate carcinoma suppresses the immunoreactive telomerase sub-unit hTERT. Cancer. 2004;100(2):294–299. doi: 10.1002/cncr.20002. [DOI] [PubMed] [Google Scholar]
- 27.Litvinov IV, Vander Griend DJ, Antony L, Dalrymple S, De Marzo AM, Drake CG, Isaacs JT. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci USA. 2006;103(41):15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiota M, Yokomizo A, Naito S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic Biol Med. 2011;51(7):1320–1328. doi: 10.1016/j.freeradbiomed.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Gregory CW, Johnson RT, Jr, Presnell SC, Mohler JL, French FS. Androgen receptor regulation of G1 cyclin and cyclin-dependent kinase function in the CWR22 human prostate cancer xenograft. J Androl. 2001;22(4):537–548. [PubMed] [Google Scholar]
- 30.Majumder PK, Grisanzio C, O'Connell F, Barry M, Brito JM, Xu Q, Guney I, Berger R, Herman P, Bikoff R, Fedele G, Baek WK, Wang S, Ellwood-Yen K, Wu H, Sawyers CL, Signoretti S, Hahn WC, Loda M, Sellers WR. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–155. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 32.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M. Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res. 2006;66(13):6503–6511. doi: 10.1158/0008-5472.CAN-05-4671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.