

Abstract
Purpose of review:
Diagnosis of ataxic disorders is an important clinical challenge upon which prognostication, management, patient solace, and, above all, the hope of future treatment all rely. Heritable diseases and the possibility of affected offspring carry the added burden of portending adverse health, social and financial ramifications.
Recent findings:
Cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) is an inherited multisystem ataxia compromising cerebellar, vestibular, and sensory function. It is not uncommon, but despite early attempts the genetic defect is yet to be identified. As the search for the causative gene continues, we have found it useful to further define this syndrome in terms of its likely phenotype.
Summary:
We propose staged diagnostic criteria based on the identified pathology in CANVAS. We envisage that these criteria will aid the clinician in diagnosing CANVAS and the researcher in further elucidating this complex disorder.
Cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) is a novel ataxia comprising 3 key etiologic foci of ataxia: cerebellar, vestibular, and sensory. Although this disorder implicates 3 of the 4 cardinal props of balance (only vision remains unaffected), it is slowly progressive compared to other similar ataxias, such as Friedreich ataxia or spinocerebellar ataxia type 3. The search for the causative gene(s) in CANVAS is under way, but our initial attempts indicate that the pattern of inheritance is unlikely to be straightforward.
Our experience to date suggests that as a clinical entity, CANVAS may comprise more than 1 causative gene, exhibit considerable phenotypic heterogeneity and phenocopies, and have a complex pattern of inheritance. Patients with CANVAS range from singletons to those with affected siblings in what most closely approximates a late-onset recessive pattern. Our attempts to isolate the causative gene(s) have been aided by close scrutiny of the diagnostic criteria we apply to those patients suspected of having CANVAS. The benefit of a clinical definition that is not too exclusive is that it minimizes the chance of excluding patients who may have CANVAS, particularly should there prove to be a major degree of phenotypic heterogeneity.
Conversely, if we are too inclusive with our clinical definition, we risk diluting our attempts at identifying the underlying genetic abnormality. For this reason we propose a staged classification system such that possible cases are identified while still maintaining those characteristics that we suspect are at the clinical core of the syndrome (for example, a sensory neuronopathy [ganglionopathy]). Following on from this example, the presence of mixed sensorimotor and small fiber neuropathies seen in some patients with CANVAS highlights the possibility of a variable phenotype. We anticipate that once the genetic abnormality is identified, reviewing these alternative phenotypes may be valuable in more completely defining the phenotype (or perhaps identifying more than 1 disease).
Another reason for creating a broad definition in the “clinically possible CANVAS” category is an acknowledgement of the variability in access to, and expertise in interpreting, certain investigative modalities. This pertains to MRI, vestibular function testing, and neurophysiologic assessment. Again, drawing on the case of neurophysiology, the availability of such testing may be limited, but more importantly, the experience in differentiating between a neuropathy and neuronopathy (ganglionopathy) may be limited. It has been our experience that if a neuronopathy is not considered, a neuropathy is readily diagnosed. A similar situation may occur in the peripheral sensory examination, in that one may test sensory perception in the lower limbs and cease testing at a certain anatomical point, under the impression that a sensory level has been located. However, in certain cases pursuing sensory perception further proximally along the limb (and indeed onto the torso) may reveal a patchy sensory loss, which is more consistent with a neuronopathy than a neuropathy.
There is no discernible sequence to the onset of the 3 cardinal features of CANVAS (cerebellar impairment, bilateral vestibular hypofunction, and a somatic sensory deficit), and patients may manifest only 2 of the 3 for many years before fulfilling the minimal diagnostic requirements of this syndrome. Pathologically, CANVAS is defined by a multiple cranial nerve1 and dorsal root2 neuronopathy and a consistent pattern of cerebellar atrophy.3 Hearing loss and pyramidal signs are not components of CANVAS. Patients may have unrelated hearing loss, most commonly presbycusis or noise-induced hearing loss. Coincidental bilateral vestibular loss and cerebellar impairment would preclude consideration of a diagnosis of CANVAS, particularly in the absence of a positive family history.4 An example of cases seen is that of a posterior fossa decompression for a Chiari malformation with postoperative sepsis managed with gentamicin and complicated by a bilateral peripheral vestibulopathy.
Although these proposed criteria are constructed around the 3 cardinal features of CANVAS, it is important not to neglect the other features of CANVAS that may be seen. These include dysphagia, cough, autonomic dysfunction (such as postural hypotension), somatic allodynia, and dysesthesia.5
Clinically possible canvas
Clinical evidence of bilateral vestibular hypofunction is most commonly ascertained via the assessment of clinical markers of diminished vestibulo-ocular reflex (VOR) gain (table 1).6 Modalities that may be used include the head impulse test (see figure 1), abnormal dynamic visual acuity,7 and abnormal occlusive fundoscopy (Zee test).8
Table 1.
Proposed diagnostic criteria

Figure 1. Bilateral vestibulopathy shown on horizontal impulsive testing recorded on the video head impulse test in a patient with CANVAS.

The head rotation stimulus is shown in black and the eye movement response is shown in red. Maximum gain of the vestibulo-ocular reflex (VOR) is less than 0.2 (normal >0.68) in each direction and compensatory “catch-up” saccades are seen following the head impulse. Inset: normal bilateral horizontal VOR gain (∼1).
Given that all 3 of the cardinal features of CANVAS (cerebellar impairment, bilateral vestibulopathy, and a somatic sensory deficit) may confer ataxia in their own right (that is, cerebellar ataxia, vestibular ataxia, and a sensory ataxia), it is imperative that each component is demonstrated to be independent of the other 2. In the case of cerebellar ataxia, clinical evidence may be inferred only when a cerebellar-specific impairment is observed. These would include cerebellar oculomotor abnormalities (e.g., abnormally broken-up smooth pursuit, gaze-evoked or direction-changing nystagmus, dysmetric saccades to target, rebound nystagmus), cerebellar dysarthria, scanning speech, and truncal ataxia. Gait ataxia is a nonspecific sign and may be caused by cerebellar, vestibular, or sensory impairment.9 Similarly, a positive Romberg test may reflect cerebellar, vestibular, or spinal posterior column dysfunction.10
Nerve conduction studies should include abnormal sensory nerve action potentials in at least one upper and one lower extremity nerve to meet the minimum requirement of clinically possible CANVAS. The clinical examination for a somatosensory deficit has been found to be unreliable; in particular, intact tendon reflexes have been seen in patients with abnormal sensory nerve action potentials in the same limb.
Clinical assessment of an abnormal visually enhanced VOR (VVOR; previously referred to as the “doll's head” or “doll's eye” reflex) is demonstrated by turning a patient's head from side to side in the yaw plane at about 0.5 Hz while the patient stares at an earth-fixed target (e.g., the clinicians nose) and observing that the compensatory eye movements are saccadic rather than smooth3 (see figure 2 and the video at Neurology.org/cp).
Figure 2. Video-oculographic measurement of the visually enhanced vestibulo-ocular reflex.

Top panel: normal horizontal visually enhanced vestibulo-ocular reflex (VVOR) gain (∼1) recorded using portable rapid video-oculography equipment. Vertical axis represents velocity (degrees/s) and horizontal axis represents time (s). The head rotation stimulus is shown in red and the eye movement response is shown in black. Lower panel: A reduced VVOR gain results in salvos of catch-up saccades during slow sinusoidal head rotation (0.5–1.0 Hz) in the horizontal (yaw) plane.
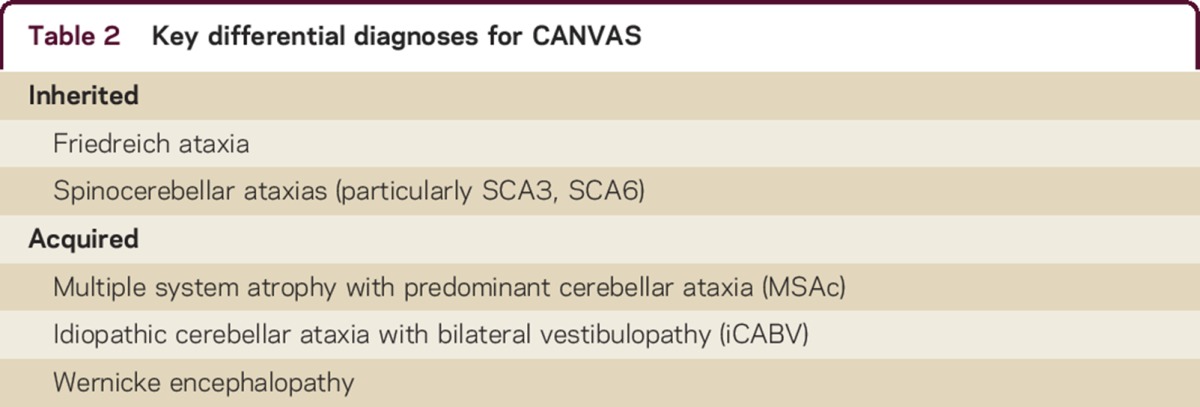
Ataxic conditions that may present similarly to CANVAS include the spinocerebellar ataxias (SCAs), particularly SCA3, which may comprise cerebellar ataxia, a bilateral peripheral vestibulopathy,11–14 and a sensory neuropathy15,16 (table 2). We recommend screening for SCA 1,2,3,6, and 7, as there is considerable variability in their presentation17 and gene tests are generally available. Adult-onset Friedreich ataxia may be composed of cerebellar impairment,18–20 a bilateral vestibulopathy,21,22 and a sensory neuronopathy,23,24 so we advise that all suspected cases of CANVAS be tested for a mutation in the frataxin gene.23
Table 2.
Key differential diagnoses for CANVAS
Clinically probable canvas
In this diagnostic category, the nerve conduction studies must demonstrate neurophysiologic evidence of a neuronopathy (ganglionopathy) according to our published protocol.25
Early indications of cerebellar atrophy on MRI scanning may be subtle and in our experience are often preceded by clinical evidence of cerebellar impairment. The cerebellar atrophy seen in CANVAS preferentially involves the vermis,2 and it is here that early changes may be seen.
The results of nerve conduction studies in this diagnostic category may be consistent with a sensory neuropathy or neuronopathy, a sensorimotor neuropathy, or a predominantly or purely small fiber neuropathy or neuronopathy. Motor studies are normal or show minor abnormalities (i.e., abnormalities which are proportionally less than those identified in the sensory studies).
Notably, the presence of a sensory neuronopathy can be confirmed by demonstrating low amplitude or absent sensory nerve action potentials, often with upper more than lower extremity involvement.
Clinically definite canvas
Objective evidence of an abnormal VVOR may be found on high-speed video-oculography (see figure 2), rotational chair testing, videonystagmography, or magnetic scleral search coil technology (table 1). We find the most sensitive readily available modality is high- speed video-oculography using a video head impulse system.3 Cerebellar atrophy on MRI displaying anterior and dorsal vermis atrophy (vermal lobules VI, VIIA, and VIIB) and lateral hemispheric atrophy predominantly affecting crus I (corresponding to vermal lobule VII) is best visualized in a midsagittal and parasagittal view,2 with corroborating evidence of atrophy on coronal sections (see figure 3).
Figure 3. T1-weighted MRI illustrating the characteristic pattern of cerebellar atrophy found in CANVAS.

Anterior and dorsal vermis atrophy (vermal lobules VI, VIIA, and VIIB) is seen in the midsagittal view (A), and hemispheric atrophy predominantly affecting crus I is seen in the parasagittal view (B).
Pathologically definite canvas
We have previously published the results of temporal bone histopathology1 and neurologic and spinal pathology2 (table 1) (see figure 4).
Figure 4. Autopsy demonstration of typical pathologic features.

(A) Anterior (1) and dorsal (2) cerebellar vermal atrophy. (B) Atrophic dorsal root ganglion at high magnification showing marked neuronal loss with scant residual ghost outlines (arrows), hematoxylin and eosin. (C) Cervical spinal cord cross section at low power magnification showing loss of myelinated neurons in the posterior columns (arrows), Luxol fast blue.
CONCLUSION
Identification of the pathologic gene(s) in CANVAS will lead to the development of a definitive diagnostic test and provide the hope of treatment advances. It may also alter the classification of CANVAS, be it an autosomal recessive cerebellar ataxia or an autosomal dominant SCA (albeit with markedly reduced penetrance). The “syndrome” will be clarified into a definitive disease entity, with or without subtypes or phenocopies. In the short term, the greatest benefit of these proposed diagnostic criteria may be the enhanced confidence with which clinicians will be able to inform their patients of their diagnosis, their likely prognosis, and the likelihood that subsequent generations are clinically affected.
Supplementary Material
Footnotes
Supplemental data at Neurology.org/cp
AUTHOR CONTRIBUTIONS
David J. Szmulewicz: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data, study supervision. Leslie Roberts: drafting/revising the manuscript, study supervision. Catriona A. McLean: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data. Hamish G. MacDougall: study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis. G. Michael Halmagyi: drafting/revising the manuscript, study supervision. Elsdon Storey: drafting/revising the manuscript, study supervision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURES
D.J. Szmulewicz, L. Roberts, and C.A. McLean report no disclosures. H.G. MacDougall receives research support from National Health and Medical Research Council, Defense Science and Technology Organization, National Aeronautics and Space Administration, The Garnett Passe and Rodney Williams Memorial Foundation, Albert Bruppacher-Stiftung and OPOS-Stiftung Foundation, and OPOS-Foundation. G.M. Halmagyi serves on the scientific advisory board of the Brain Foundation of Australia; has received funding for travel from GN Otometrics; serves on the editorial boards of Acta Otolaryngologica, Otology, Neurotology, Audiology, Neuro-otology, and the Italian Journal of Otolaryngology; has served as a consultant to GN Otometrics; and receives research support from National Health and Medical Research Council. E. Storey has served on a scientific advisory board for Bethlehem-Griffiths Foundation, serves on the editorial board of Cerebellum & Ataxias, and receives research support from National Health and Medical Research Council and the NIH. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Szmulewicz DJ, Merchant SN, Halmagyi GM. Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome: a histopathologic case report. Otol Neurotol 2011;30:e63–e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szmulewicz DJ, McLean CA, Rodriguez ML, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 2014;22:1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szmulewicz DJ, Waterston JA, Halmagyi GM. Sensory neuropathy as part of the cerebellar ataxia neuropathy vestibular areflexia syndrome. Neurology 2011;76:1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szmulewicz DJ, Waterston JA, MacDougall HG, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 2011;1233:139–147. [DOI] [PubMed] [Google Scholar]
- 5.The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996;46:1470. [DOI] [PubMed] [Google Scholar]
- 6.Halmagyi GM, Curthoys IS. A clinical sign of canal paresis. Arch Neurol 1988;45:737–739. [DOI] [PubMed] [Google Scholar]
- 7.Tian J-R, Shubayev I, Demer JL. Dynamic visual acuity during transient and sinusoidal yaw rotation in normal and unilaterally vestibulopathic humans. Exp Brain Res 2001;137:12–25. [DOI] [PubMed] [Google Scholar]
- 8.Zee DS. Ophthalmoscopy in examination of patients with vestibular disorders. Ann Neurol 1978;3:373–374. [DOI] [PubMed] [Google Scholar]
- 9.Salzman B. Gait and balance disorders in older adults. Am Fam Physician 2010;82:61–68. [PubMed] [Google Scholar]
- 10.Rogers JH. Romberg and his test. J Laryngol Otol 1980;94:1401–1404. [DOI] [PubMed] [Google Scholar]
- 11.Buttner N, Geschwind D, Jen JC, Perlman S, Pulst SM, Baloh RW. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol 1998;55:1353–1357. [DOI] [PubMed] [Google Scholar]
- 12.Takegoshi H, Murofushi T. Vestibular evoked myogenic potentials in patients with spinocerebellar degeneration. Acta Otolaryngol 2000;120:821–824. [DOI] [PubMed] [Google Scholar]
- 13.Bürk K, Fetter M, Abele M, et al. Autosomal dominant cerebellar ataxia type I: oculomotor abnormalities in families with SCA1, SCA2, and SCA3. J Neurol 1999;24:789–797. [DOI] [PubMed] [Google Scholar]
- 14.Gordon CR, Joffe V, Vainstein G, Gadoth N. Vestibulo-ocular arreflexia in families with spinocerebellar ataxia type 3 (Machado-Joseph disease). J Neurol Neurosurg Psychiatry 2003;74:1403–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schöls L, Amoiridis G, Büttner T, Przuntek H, Epplen JT, Riess O. Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol 1997;42:924–932. [DOI] [PubMed] [Google Scholar]
- 16.Klockgether T, Schols L, Abele M, et al. Age related axonal neuropathy in spinocerebellar ataxia type 3/Machado-Joseph disease (SCA3/MJD). J Neurol Neurosurg Psychiatry 1999;66:222–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rüb U, Schöls L, Paulson H, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxia type 1, 2, 3, 6 and 7. Prog Neurobiol 2013;104:38–66. [DOI] [PubMed] [Google Scholar]
- 18.Klockgether T, Petersen D, Grodd W, Dichgans J. Early onset cerebellar ataxia with retained tendon reflexes. Clinical, electrophysiological and MRI observations in comparison with Friedreich's ataxia. Brain 1991;114:1559–1573. [DOI] [PubMed] [Google Scholar]
- 19.Koeppen AH, Morral JA, Davis AN, et al. The dorsal root ganglion in Friedreich's ataxia. Acta Neuropathol 2009;118:763–776. [DOI] [PubMed] [Google Scholar]
- 20.Koeppen AH, Davis AN, Morral JA. The cerebellar component of Friedreich's ataxia. Acta Neuropathol 2011;122:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furman JM, Perlman S, Baloh RW. Eye movements in Friedreich's ataxia. Arch Neurol 1983;40:343–346. [DOI] [PubMed] [Google Scholar]
- 22.Fahey MC, Cremer PD, Aw ST, et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain 2008;131:1035–1045. [DOI] [PubMed] [Google Scholar]
- 23.Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol 2009;256(suppl 1):3–8. [DOI] [PubMed] [Google Scholar]
- 24.Koeppen AH. Friedreich's ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 2011;303:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szmulewicz DJ, Seiderer L, Halmagyi GM, Storey E, Roberts L. Neurophysiological evidence for generalized sensory neuronopathy in cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome. Muscle Nerve 2015;51:600–603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.