

Abstract
The aim of this study was to characterize the current molecular epidemiology of hepatitis C virus (HCV) infection and evaluate the evolutionary patterns of HCV subtypes in Beijing, China, among different subpopulations.
The whole blood samples and behavioral data were collected from a total of 10,354 subjects, including drug users (DUs), men who have sex with men (MSM), and the general population, in Beijing from 2010 to 2011. Samples were tested for HCV infection using both enzyme-linked immunosorbent assay (ELISA) and real-time PCR. All viremic subjects were then sequenced by nested PCR over core/E1 and NS5B regions. Phylogenetic and phylogeographic analysis was performed by BEAST software.
In total, 217 subjects (2.1%) were tested positive for HCV by antibody or vRNA-based testing. HCV prevalence rates for DUs, MSM, and the general population were 26.2%, 0.54%, and 0.37%, respectively. The 156 HCV RNA-positive samples were sequenced. Nine HCV genotypes, including 1a, 1b, 2a, 3a, 3b, 6a, 6n, 6u and 6v, were detected. The most prevalent subtypes were 3b (36.09%), 1b (32.54%), and 3a (16.57%). Bayesian evolutionary analysis estimated that the time of introduction of subtype 1b into Beijing was 2004 (95% CI: 1997.7, 2007.7), with subtypes 3a and 3b being introduced later in 2006. Evolutionary analyses further suggested that subtype 1b from Beijing and Shanghai were closely related, whereas subtype 3a sequences were more similar with sequences from Yunnan, Guangzhou, Hong Kong, and Jiangsu. Subtype 3b sequences were closely related to those from Yunnan, Guangdong, and Hong Kong.
Thus, the current HCV epidemic in Beijing is complex, heavily affecting DUs, and involving multiple genotypes that likely spread from different regions in China with its large migrant population.
INTRODUCTION
Hepatitis C virus (HCV) infection is a pressing global health problem.1 Global HCV prevalence is between 1% and 2% in most countries that have been studied,2 but the distribution of HCV varies considerably between subpopulations. Historically, HCV has been most frequently transmitted by percutaneous exposure to infected blood or body fluids, including transfusion of contaminated blood or blood products, nonsterile medical injection, or injection drug use; high rates of HCV seropositive reported in persons exposed to HCV through these routes.3 In China, the epidemiology of HCV infection is continually evolving. A nationwide epidemiologic survey conducted in 1992 that screened a total of 68,000 subjects across all 30 provinces, autonomous regions, and municipalities found HCV prevalence of 3.2% that was largely attributed to transfusion of contaminated blood or blood products.4
Since 1993, strict control of blood products, increased use of sterile equipment and mandatory screening for blood borne pathogens have been implemented throughout China and HCV transmission through contaminated blood products has decreased significantly.5 A national survey in 2006, screened 78,746 subjects across 31 provinces, suggested a significant reduction in HCV prevalence, with just 0.46% of tested individuals being HCV seropositive in China.6 HCV acquisition by other routes, however, has increased. Recent studies of injection drug users (IDUs) throughout China reveal available, but generally high prevalence of HCV, with between 15.6% and 98.7% testing seropositive for HCV infection.7–9 Thus, injection drug use has become the primary transmission route of HCV in China.10,11 Globally, HCV infection in men who have sex with men (MSM) is an emerging problem. Since 2000, acute HCV infections have increasingly been reported among MSM in Europe, Australia, and the United States.12–19 In China, however, recent surveys have not documented significant increases in HCV infection in urban MSM,20–22 though active surveillance continues.
The molecular epidemiology of HCV is also evolving. HCV is highly genetically diverse and is classified into 7 genotypes and 67 subtypes.23 Tracking the molecular epidemiology of HCV can be useful to elucidate transmission networks locally and globally, and it is clinically important to predict disease prognosis and response to anti-HCV therapy. Subtype 1b is the most prevalent worldwide,24 whereas other genotypes are more confined to regional epidemics. Generally, subtype 1a is most commonly seen in the United States,25 subtype 2a and 2b are predominant in North America, Europe, and Japan,26–28 subtype 3 (predominantly 3a) is the most prevalent genotype in India and Pakistan,29,30 genotype 4 is often found in the Middle East and Africa,24 subtype 5a accounts for at least 50% of the infections in South Africa,31 and genotype 6 is frequently seen in southern China and Southeast Asia.32–34 Although 6 genotypes (genotypes 1–6), 18 subtypes (subtypes1a, 1b, 1c, 2a, 2b, 2f, 3a, 3b, 4d, 5a, 6a, 6e, 6g, 6k, 6n, 6u, 6v, and 6w), and a number of unassigned variants have been detected in China, over 95% of these isolates belong to 5 major subtypes:1b, 2a, 3a, 3b, and 6a.35–41 Among them, subtype 1b has been predominant nationwide, accounting for approximately 75% of all HCV infections, followed by subtype 2a.
Recent studies of HCV epidemiology in large urban areas of China have reported complex and evolving HCV genotype patterns, suggesting the highly mobile population and many contributing epidemics. Previously, HCV genotype distribution in China was relatively homogeneous, comprised mainly of 2 genotypes, including 1b, 2a, and coinfection (1b/2a). More recently, the relative frequency of subtypes lb and 2a is slowly decreasing, and subtype 3a, 3b, and 6a prevalence are rising. In recent years, subtypes 2b, 4a, 5a, 6b, and 6n have also appeared gradually.42 Subtypes 1a, 3a, and 3b have been described among IDUs with HIV-1 coinfection in Yunnan, Guangxi, and Xinjiang,19 and subtypes 6a, 6e, 6k, 6n, and 6u are observed among IDUs from Jiangsu, Yunnan, Guangxi, and Guangdong.20–23 Although early reports from Zhang et al9 support the theory that the HCV epidemic in China originated with paid blood donors, the ongoing changes in HCV genotype distribution patterns suggest that subsequent human migration is driving the current HCV epidemic in China.43 These molecular epidemiologic trends coincide with the recent tide of human migration toward the coastal regions in China, where rapid economic growth is fueled by millions of migrants and immigrants.43
Beijing is the capital of China, the center of national politics, culture, transport, tourism, and international exchange. The city comprised of 16 districts, housing >20 million residents, and millions of migrant workers from different provinces in China and all over the world. We hypothesized that this dynamic immigration pattern and flux of people in and out of Beijing may be reflected in an increasing complex HCV epidemic in China's capital. The aim of the present study is to characterize the scope of HCV infection among DUs, MSM, and the general population in Beijing and to determine the HCV genotype distribution and evolution of these groups over time and geography. We screened over 10,000 individuals by HCV antibody and viral load testing, collected behavioral data, and performed sequencing, phylogenetic, and phylogeographic analysis of all viremic subjects. Results provide a current snapshot of the molecular epidemiology of the HCV epidemic in Beijing.
MATERIALS AND METHODS
Study Population
A total of 10,354 individuals were recruited from 2010 to 2011 in Beijing. Among them, 684 were DUs, 1296 were MSM, and 8,374 were general population. The blood samples of drug users (DUs) were collected from compulsory detoxification center and methadone maintenance treatment clinics in Beijing. MSM were randomly recruited at voluntary counseling and testing sites (VCT) in Beijing Chaoyang District Center for Disease Control and Prevention and self-identified as MSM. General population was randomly recruited from hospital health examination centers and reported neither DUs nor MSM. This study was approved by the Institutional Research Ethics Community, China Chaoyang CDC (NO.1206), and all subjects signed informed consent forms before blood collection. Epidemiological data were collected by trained interviewers (including age, marriage status, inhabit time in Beijing, nationality, education, and risk behaviors). Blood plasma was separated and stored at −70°C before genetic analysis.
Enzyme Immunoassay
All 10,354 plasma samples were screened for anti-HCV antibodies using ELISA kit (Wantai, China). The results of the assay were expressed quantitatively as the ratio of the optical density (OD) of the test sample to the calculated cutoff absorbance as recommended by the manufacturer. Serum samples with OD values over the cutoff value were considered to be positive, whereas those with OD values less than cutoff value were considered negative. Positive samples were confirmed with an additional ELISA assay kit (Kehua, China). Only samples testing positive by both assays were counted as positive.
RNA Extraction and Real-time PCR
Viral RNA was extracted from 200 μL of ethylene diammine tetraacetice acid anticoagulated plasma using MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche Applied Science, Manheim, Germany) according to the manufacturer's instructions. All 10,354 serum samples were screened for HCV RNA load using Real-time PCR kit (Qiagen, Germany) by Roche 480.
HCV Amplification and Sequencing
The HCV RNA underwent reverse transcription by SuperScript III Reverse Transcriptase kit (Invitrogen, USA). Regions of the HCV Core/E1 (475 bp) and NS5B (381 bp) genes were amplified by nested-PCR, using a genotyping method as previously described.44 The amplification was performed with predenaturation at 94°C for 15 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 1.5 min, followed by an additional extension at 72°C for 10 min in a 25-μL volume. The nested PCR was performed in a 50-μL volume and the cycling condition includes predenaturation at 94°C for 3 min, 35 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, extension at 72°C 1.5 min, followed with an additional extension at 72°C for 10 min. The PCR products were visualized by 1% agarose gel electrophoresis and sequenced by ABI 3730xl Automated DNA Analyzer (Applied Biosystems, Foster City, CA). Each step was carried out with negative controls.
Phylogenetic Analysis
The HCV genotype was determined after alignment of 82 sequences with reference sequences (Appendix 1) from GenBank (http://www.ncbi.nlm.nih.gov/genbank). Sequences were aligned using CLUSTAL_X and edited by BioEdit. Maximum Likelihood (ML) trees were constructed to subtype each sequence by Mega 6. The Core/E1 and NS5B sequences from the same patient were merged. Bayesian Markovchain Monte Carlo (MCMC) analyses were performed with the selected nucleotide substitution model using a Bayesian uncorrelated log-normal relaxed molecular clock method as implemented in BEAST 1.8.0. MCMC analysis was run for 30 million steps, with sampling every 1000 steps. Convergence of parameters was assessed using TRACER v1.4.45 To assess the sampling convergence of the MCMC procedures, the estimated effective sampling sizes (ESSs) were evaluated. In this study, when all of the ESSs were ≥200, sufficient sampling was considered to have been achieved. Maximum clade credibility (MCC) trees were constructed from the posterior tree distribution using Tree Annotator and displayed by FigTree 1.4.
Phylogeographic Analysis of Subtype 3b, 1b, and 3a
The main subtypes of Beijing in this study were subtype 3b, 1b, and 3a. To investigate the possible geographic origins of HCV in Beijing, HCV 3b, 1b, and 3a NS5B region nucleotide sequences with known sampling years and locations in China were collected from Genbank. Each sequence was assigned a character state reflecting its sampling location. The maximum clade credibility tree was constructed using a MCMC method implemented in the BEAST software.
Statistical Analysis
All statistical analyses were done using SPSS software (version 16.0, SPSS Inc, Chicago, IL). Data were presented as mean ± SD. Numerical data were analyzed using student t test. The distribution of genotypes among DUs and non-DUs was analyzed by bidirectional Mann-Whitney test. P value <0.05 was considered statistically significant.
RESULTS
Patient Characteristics
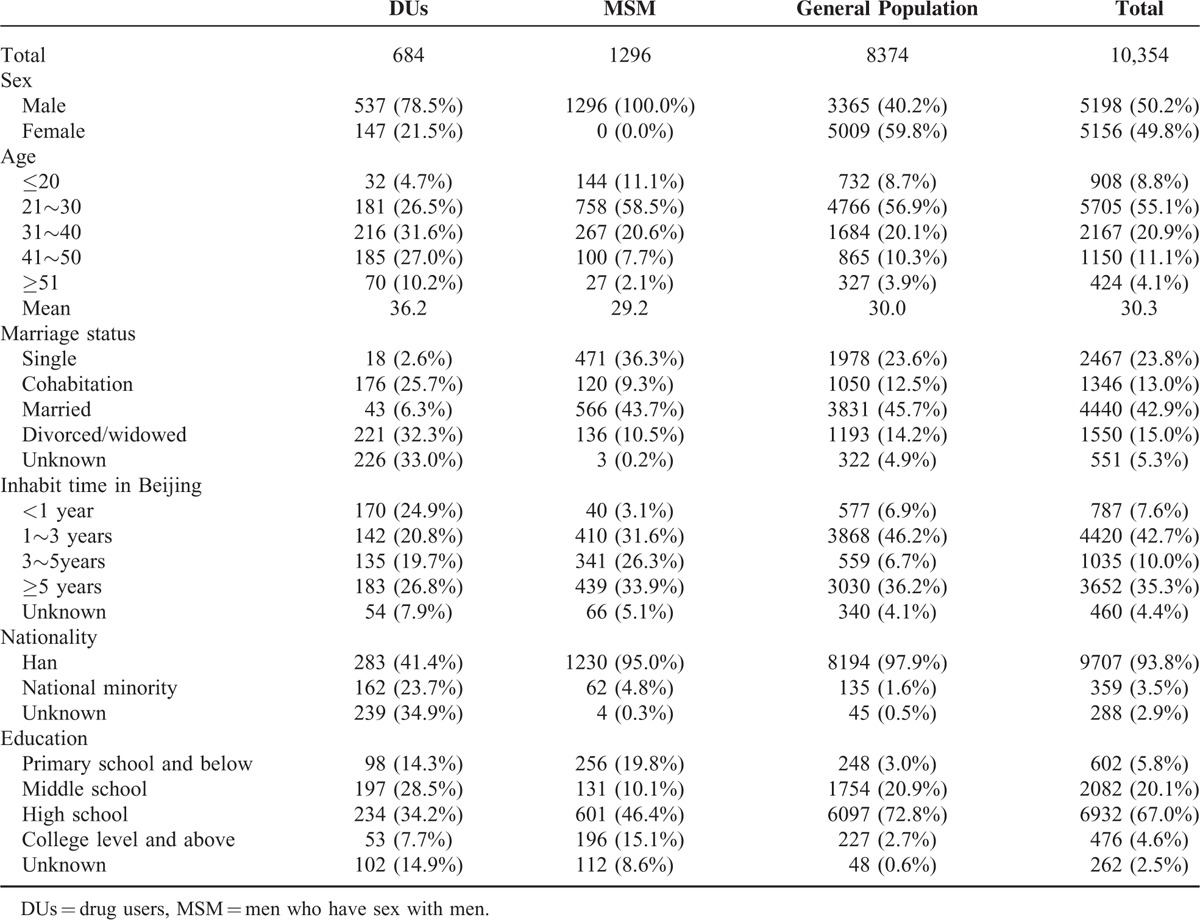
Among 10,354 participants, 684 were DUs, 1296 were MSM, and 8374 were general population. The demographics of the study population are shown in Table 1. The mean age was 30.3 years, and 63.9% of subjects were younger than 30 years. The ratio of male and female was approximately 1:1. Subjects were overwhelmingly (>90%) of Han ethnicity. 23.8% of subjects were never married, 42.9% were married, and 15% were divorced or widowed. More than 60% of the participants belonged to migrant population, having lived in Beijing for <5 years. The education level of those surveyed varied, with the majority having graduated from high school.
TABLE 1.
The Characteristic of Study Subjects
HCV Prevalence, Clearance and Acute Infection for DUs, MSM, and the General Population
Of the 10,354 participants, 181 (1.75%) were seropositive for HCV antibody by ELISA, including 144 (21.05%) DUs, 6 (0.46%) MSM, and 31 (0.37%) with neither risk factor, heretofore referred to as the general population. Screening for ongoing HCV viremia by Real-time PCR revealed 157 total positive samples (1.52%), including 143 (20.90%) DUs, 3 (0.23%) MSM, and 11 (0.13%) from the general population. Thirty-six (16.6%) of these HCV-infected individuals were seronegative and likely acutely infected. Sixty (27.65%) infected patients had serologies consistent with spontaneous viral clearance, with testing positive for HCV antibody and negative for HCV RNA. A total of 217 subjects were positive by either test, giving an overall HCV prevalence of 2.10% (217/10,354). The number of HCV infections in DUs, MSM, and the general population was 179 (26.17%), 7 (0.54%), and 31 (0.37%) (Table 2).
TABLE 2.
The Results of HCV Detection
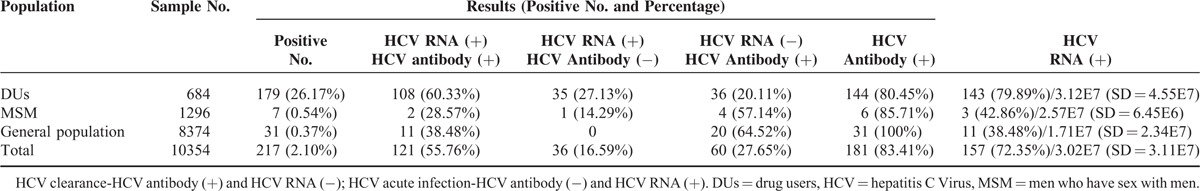
For samples testing positive for HCV RNA, quantitative viral load testing was performed. For the 156 positive samples, the median HCV viral load was 3.02E7 (SD = 3.11E7). The mean of viral load in DUs, MSM, and the general population were 3.12E7 (SD = 4.55E7), 2.57E7 (SD = 6.45E6), and 1.71E7 (SD = 2.34E7), respectively, which were statistically not different. The mean HCV viral load by genotype ranged from 1.11E6 for genotype 6 v to 5.25E7 for 2a, and did not differ statistically.
HCV Subtype Distribution
HCV genotyping was successfully performed on 156 samples (99.36%, 156/157) by amplification of subgenomic regions of Core/E1 and NS5B. A total of 9 HCV subtypes (1a, 1b, 2a, 3a, 3b, 6a, 6n, 6u, and 6v) were identified in the 156 positive samples, with genotypes 3b (36.09%), 1b (32.54%), 3a (16.57%), and 6a (8.88%) comprising >90% of circulating strains (Table 3). The frequencies of HCV subtypes in DUs, MSM, and the general population were differed. HCV subtypes 1a, 1b, 2a, 3a, 3b, 6a, and 6n were detected in DUs, whereas only subtypes 1b, 3b, and 6a were identified in MSM, and subtypes 1b, 2a, 3a, and 6v were confirmed in general population. When compared, there was a trend toward statistical difference (P = 0.003, by bidirectional Mann-Whitney) between subtype frequency in DUs compared with non-DUs (including MSM and the general population). Comparing the sequences from the 3 groups, sequences from DUs and MSM were phylogenetically interspersed within those from the general population (Figure 1 and Figure 2).
TABLE 3.
Distribution of HCV Genotypes Based on Core/E1 and NS5B Sequences in DUs, MSM, and the General population in Beijing
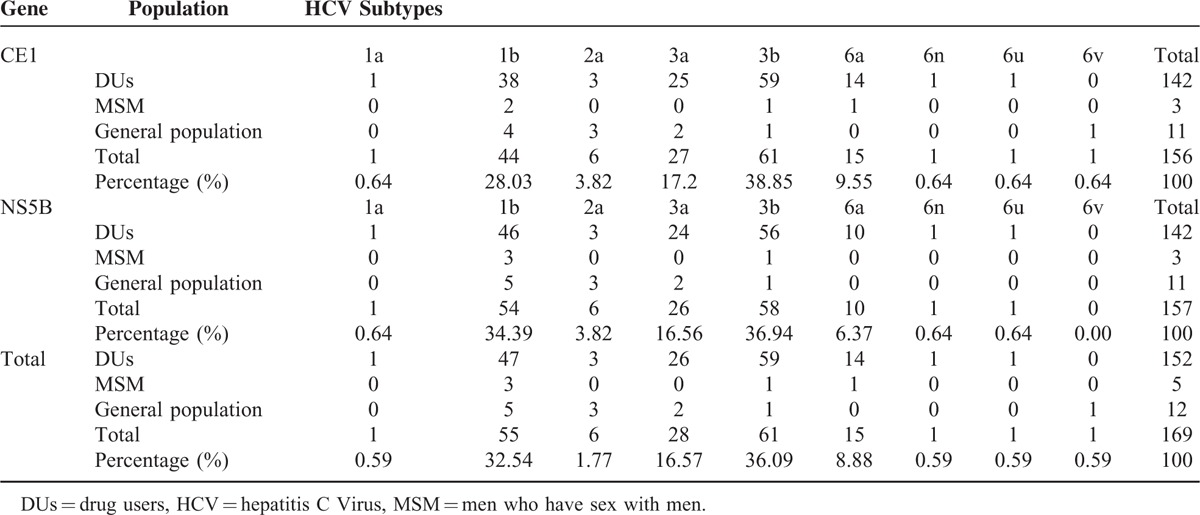
FIGURE 1.
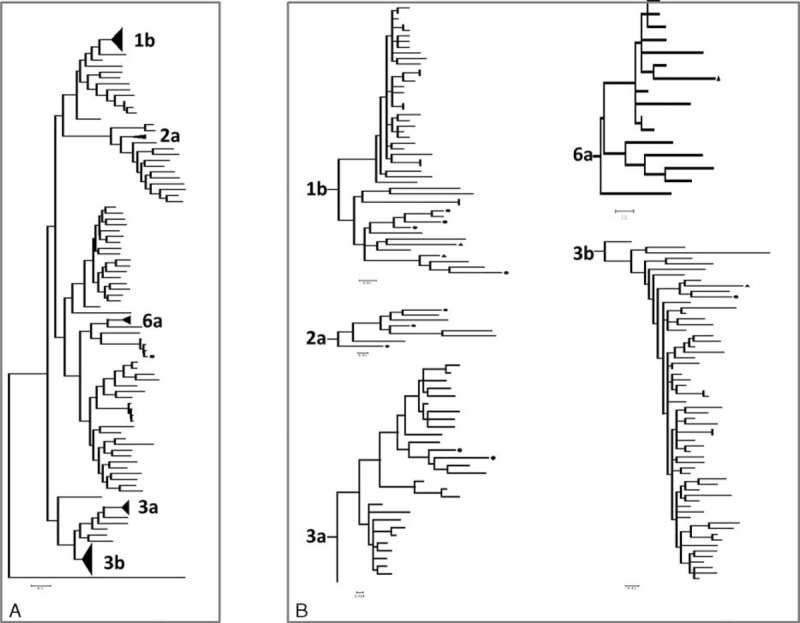
Phylogenetic relationship of HCV isolated from Beijing based on Core/E1. ML tree showed that HCV sequences from Beijing belong to 1a, 1b, 2a, 3a, 3b, 6a, 6n, 6u, and 6v genotypes. The tree was constructed with MEGA 6. The references obtained from the Genebank. Sequence of Japanese encephalitis virus strain P3 (U47032) was used as outgroup. Round dots denoted HCV strains from general population. Triangle denoted HCV strains from MSM.
FIGURE 2.

Phylogenetic relationship of HCV isolated from Beijing based on NS5B. ML tree showed that HCV sequences from Beijing belong to 1a, 1b, 2a, 3a, 3b, 6a, 6n, and 6u genotypes. The tree was constructed with MEGA 6. The references obtained from the Genebank. Sequence of Japanese encephalitis virus strain P3 (U47032) was used as outgroup. Round dots denoted HCV strains from general population. Triangle denoted HCV strains from MSM.
Figures 1 and 2 showed ML tree reconstructed by 156 sequences of Core/E1 region and 156 sequences of NS5B region. The sequences from each subtype were closely related, with generally high bootstrap value (>98%, except for NS5B 1b-79%). For subtype 1b, the phylogenies based on separate sequencing regions were highly similar. In both the Core/E1 and NS5B phylogenies, subtype 1b sequences generally formed a single major subgroup comprised of largely DU sequences; in the Core/E1 tree, all the sequences in this subgroup were from DUs, and in the NS5B tree, all but 2 sequences in the subgroup were from DUs. Sequences from subtype 3a group were divided into 3 major subgroups in both trees. The sequences were in the similar positions in both trees, with the 2 sequences from the non-DUs on the same branch. Sequences from subtype 6a fell into different phylogenetic groups in the Core/E1 and NS5B trees.
Molecular Evolutionary of HCV in Beijing
To investigate the evolutionary history of HCV in Beijing, we performed time-scaled phylogenetic analyses and phylogeographic analyses using BEAST software. Three time-scaled trees (Figure 3) and 3 phylogeographic trees (Figure 4) represented the most prevalent subtypes (1b, 3a, and 3b).
FIGURE 3.
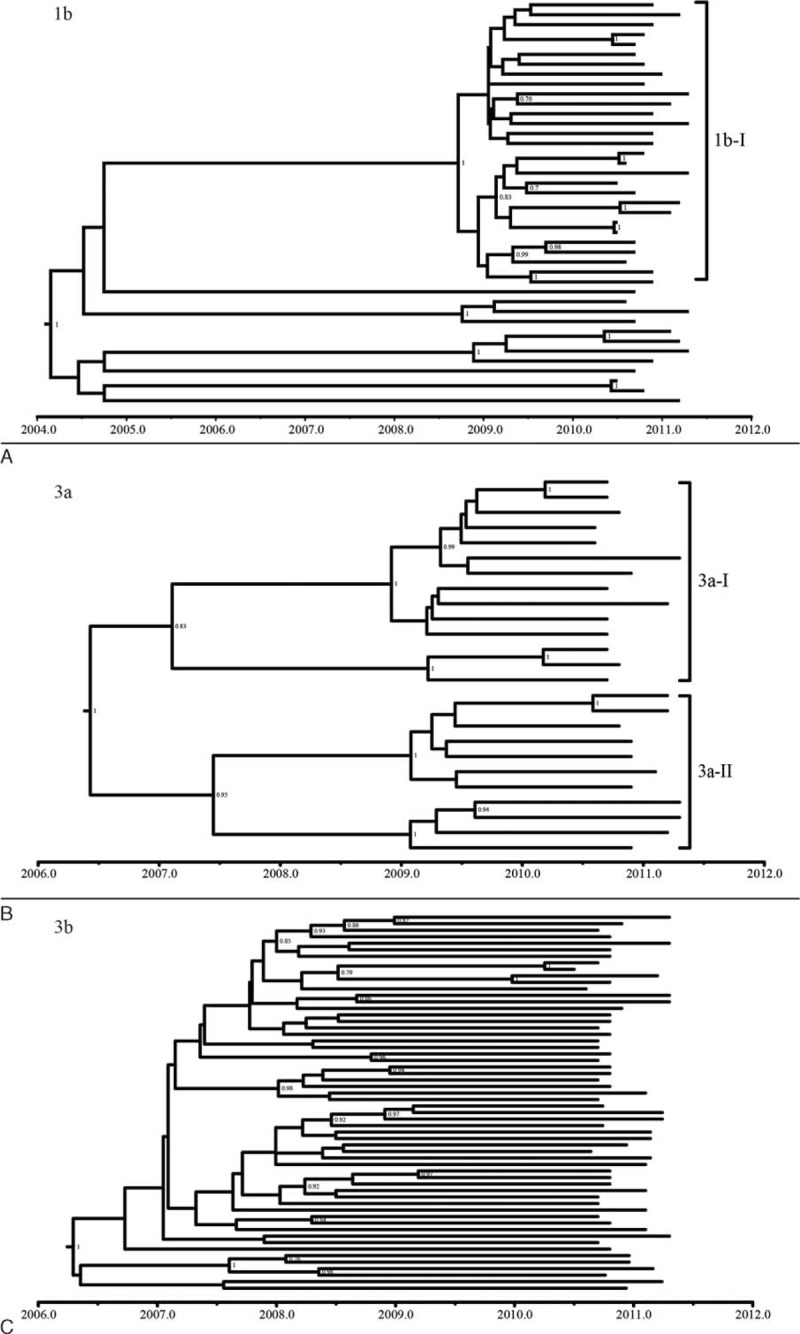
The time-scaled trees estimated with the Core/E1 and NS5B sequences of subtype 1b, 3a, and 3b.
FIGURE 4.
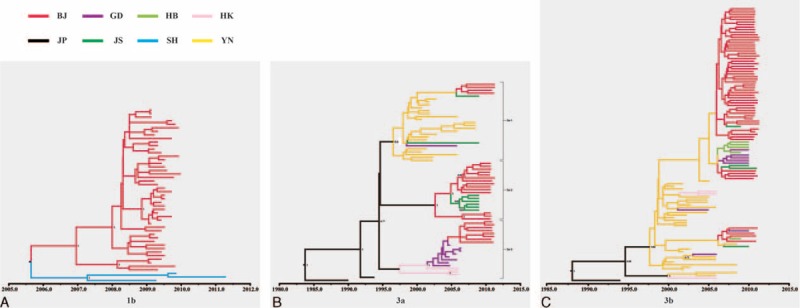
The phylogeographic trees estimated with the NS5B sequences of subtype 1b, 3a, and 3b. Branches are colored according to their geographic origins, indicated in the upper left. The sequences in these phylogeographic trees were from Beijing (BJ), Guangdong (GD), Hubei (HB), Hong Kong (HK), Japan (JP), Jiangsu (JS), Shanghai (SH), and Yunnan (YN).
Bayesian analyses inferred a time to most recent common ancestor (tMRCA) for subtype 1b of the year 2004.1 (1997.7, 2007.7), with an inferred evolutionary rate of 9.46e-3 (3.58e-3, 1.68e-2) substitution per site per year by time-scaled phylogenetic analyses. There was an obvious cluster (1b-I) in subtype 1b time-scaled tree (Figure 3A), with significant posterior probabilities of 1. All sequences in cluster 1b-I were from DUs. The phylogeographic tree reconstructed for subtype 1b suggested that most of sequences of Beijing and 3 sequences of Shanghai were related in one cluster (Figure 4A). The Beijing sequences in this cluster were from DUs, which supported the results of time-scaled analyses and ML analyses. Therefore, we used the sequences of this clade to calculate the tMRCA median and evolutionary rate of subtype 1b. The tMRCA median of the tree was 2005.7 (2005.2, 2008.3), and the evolutionary rate was 1.3e-2 (5.1e-3, 2.7e-3) substitution per site per year. The results of subtype 1b phylogeographic tree, time-scaled tree, and ML tree suggest that the HCV strains circulating among DUs and non-DUs may have different evolutionary origins. The divergence time of subtype 1b in Beijing was about 2005. HCV subtype 1b sequences circulating in Beijing among DUs and Shanghai are closely related and genetically interspersed, suggesting a common origin or frequent intermixing. The direction of migration between these cities or from a third location is unknown.
Figure 3B depicts time-scaled trees reconstructed on the basis of subtype 3a sequences. The tMRCA of the tree was 2006.5 (1983.4, 2010) and the evolutionary rate was 1.26e-2 (1.04e-3, 2.71e-2) substitution per site per year. The sequences were clustered into 2 groups, groups 3a-I and 3a-II, showing significant posterior probabilities of 0.83 and 0.95, respectively. Figure 4B depicts a phylogeographic tree of the NS5B sequences of subtype 3a, which appears to have a diverse range of geographic origins. Three clusters of sequences are apparent, indicated as groups 3a-1, 3a-2, and 3a-3, with posterior probabilities of 0.6, 1, and 1, respectively. Each of the 3 groups clustered with sequences from distinct regions. Although the posterior probability of group 3a-1 was less significant, Beijing sequences cluster with primarily Yunnan in this group. Group 3a-2 was clustered with sequences from Jiangsu, and group 3a-3 was clustered with sequences from Guangdong and Hong Kong. Thus, the phylogeographic tree suggests separate migration events introducing genetically distinct subtype 3a HCV from these 3 regions. The tMRCA estimates from each of these groups further suggest different times of introduction, with the Yunnan group tMRCA of 1996, Jiangsu of 2002.5, and Guangdong/Hong Kong group of 2001.5. Compared with the time-scaled tree of 3a, we indicated a trend of subtype 3a migration from Yunnan, Guangdong, and Hong Kong to Beijing. Group 3a-2 provided a close phylogenetic relationship from Beijing isolates and Jiangsu. They may an evolutionary common ancestry.
Figure 3C depicts time-scaled trees reconstructed on the basis of subtype 3b sequences. The tMRCA of the tree was 2006.2 (2001.4, 2009.2) and the evolutionary rate was 0.25 (0.13, 0.39) substitution per site per year. The phylogeographic tree reconstructed for subtype 3b was shown in Figure 4C, with a single large group with a significant posterior probability of 0.98 compared with sequences from Yunnan, Guangdong, and Hong Kong. The vast majority of the Beijing sequences were in the large branch without significant posterior probabilities that were not phylogenetically distinct from sequences from other regions. This phenomenon was similar with the ML trees, and which indicated that subtype 3b of Beijing had specific geographical features and was different from other reference sequences from other provinces. On the basis of this tree and time-scaled tree of 3b, the migration trend appeared to have been from the south (Yunnan, Guangdong, and Hong Kong) to Beijing.
DISCUSSION
The global HCV epidemic is constantly evolving. In the present study, we characterized the molecular epidemiology of the current HCV epidemic in Beijing, China, and found evidence of an ongoing, genetically complex HCV epidemic in Beijing DUs, with limited evidence of generalization into MSMs and the general population.
Our findings revealed significant HCV prevalence in DUs, with 26% of those DUs testing positive by antibody or viral RNA for HCV infection. Testing also indicated a high incidence, with 16.59% acute HCV in DUs. The HCV prevalence modestly differed from the previous reports on DUs from national survey of registered DUs in the 15 cities of China-Gates HIV/AIDS Program in 2007, which saw higher prevalence of HCV in Beijing (32.2%, 201/624) (χ2: P = 0.016).46 In the study performed by Liu et al47 from 2005, the proportion of HCV antibody positive among DUs in Beijing was 34.36% (167/486) (χ2: P = 0.0025). According to the previous reports, HCV infection among DUs was associated with HIV status, history of incarceration, duration of injection drug use, numbers of drugs used, needle/syringe and other equipment sharing in lifetime and past 6 months, reusing needle/syringe duration of injection drug use, frequency of injection, and condom use with regular partners.8 These studies suggest that injection drug use and sharing of needle/syringe were some of the most important factors among DUs acquiring HCV. In our study, high HCV prevalence among DUs suggested that injection is major route of HCV transmission. We found a lower HCV prevalence in DUs in Beijing compared with 2005 and 2007 estimates.46,47 Thus, modestly lower estimate of HCV prevalence may be a result of a true decrease in HCV prevalence in DUs or because of sampling of cohorts with different behavioral risks, or the different sensitivities in screening tests (older ELISAs vs new ELISA/nucleic acid screening).6
Our research revealed that in Beijing, non-DUs, including MSM, maintain a very low prevalence of HCV infection. In several major metropolitan areas of the world, including Western Europe, Australia, and North America, HCV incidence has been dramatically increasing in MSM communities, especially those infected with HIV-1. Thus, the low prevalence, and lack of acute HCV, in this sizable MSM population served is notable. Continued active surveillance and HIV-1 detection of this population in China is merited.
Compared with the epidemiologic survey in 1992 (3.2%),4 the HCV prevalence rate for the general population of this study is much lower (0.37%). However, it is similar to an investigation from 2006, which showed 0.46% prevalence.6 There could be several reasons behind the decrease of HCV infection. First, to reduce the infection rate of blood transmitted diseases, such as HCV, HBV, and HIV, strict control of blood products, increased use of sterile equipment, and mandatory screening for blood borne pathogens have been implemented throughout China since 1993. Second, the detection methods are different. In this study, we used indirect ELISA; it has higher sensitivity and specificity than the second-generation UBI anti-HCV enzyme immunoassay used in the survey of 1992.6
Because we screened both for the presence of anti-HCV antibodies and HCV viral RNA, we are able to assess for spontaneous clearance of HCV.48,49 The rate of spontaneous HCV resolution has been reported to range from 10% to 60%52 and is modified by host factors such as age, sex, ethnicity, and immune response.50,51 In our study, we found a general rate of spontaneous clearance of 27.65%. The rate of HCV clearance appeared to differ among behavioral risk groups, with about 20% in DUs, 57% in MSM, and 65% in general population. It is biologically plausible that spontaneous clearance rates may be lower in the relatively immune compromised DUs who are at greater risk for reinfection than the general population. The low number of positive tests and the low pre-test probability in the latter 2 groups, however, increase the likelihood that antibody-positive, RNA-negative samples represented false-positive ELISA tests rather than true seroconversions. Thus, these differences must be taken with substantial caution.
In the present study, we found a rich diversity of circulating strains of HCV in Beijing, with a total of 8 prevalent subtypes (1a, 1b, 2a, 3a, 3b, 6a, 6n, 6u, and 6v). Four subtypes (1b, 3a, 3b, 6a) dominated, each comprising at least 15% of strains. These results differ with some older studies,43,53 in which only HCV subtypes 1b,2a, 2b or 1b, 2a, 3a were detected. Thus, our results suggest increased diversity and continued introduction of new subtypes and strains from other regions of China and the world. Our data suggest a modest difference in the distribution of the various subtypes between DUs, MSM, and the general population, but the low number of non-DUs sampled limits this analysis. The distribution of subtypes in DUs, however, does appear different than previous reports, with a decrease in prevalence of genotype 1b (the major subtype in 2007),53 and an increase in genotypes 3b and 6a. In addition to Beijing, subtypes 3b and 6a were recently found to be highly prevalent in other urban areas in the south of China, such as Guangxi, Yunnan, Wuhan, and Suzhou.34,36,37,54The geographical and genetic diversity of HCV is constantly evolving as a result of the modern transmission and increasing global travel to the national and international destination of Beijing, resulting in a complex molecular epidemiology of HCV subtypes in Beijing.
Subtype 1b is predominant nationwide, accounting for approximately 75% of all HCV infections.43 In this study, however, subtype 1b had the highest proportion among non-DUs, but not among DUs. In the time-scaled tree, phylogeographic tree, and ML tree reconstructed on the basis of subtype 1b, sequences from non-DUs and most sequences from DUs segregated into different groups. Likely because of independent evolution, varied selective pressures, and differences in living and environmental conditions and transmission routes, these data suggest that different subtype 1b strains may have been selected in different geographic regions or different population subsets. Some strains may have been predominantly circulated among non-DUs. Other strains may be transmitted primarily via the DU network.
The phylogeographic tree reconstructed for subtype 1b included sequences from Jiangsu, Guangdong, Taiwan, Shanghai, Henan, Liaoning, Inner Mongolia, Yunnan, and Hong Kong. We found that most Beijing sequences (46/54) and 3 sequences of Shanghai were in one cluster. With the exception of one sequence from an MSM, the other Beijing sequences in this cluster were from DUs. Thus, subtype 1b sequences from Beijing DUs and Shanghai may have a shared migration trend and/or they may have the same evolutionary origins. This phenomenon also suggests that the sequences in this cluster may only exist in some regions of China and not be widely prevalent through all regions of China.
The phylogeographic results support the theory that the HCV epidemic in Beijing has been contributed to by introduction of HCV from multiple locations over time. The phylogeographic tree for subtype 3a showed 3 groups. All the groups featured sequences sampled from Beijing and other provinces, which showed that Beijing strains were related to isolates from Yunnan, Guangdong, and Hong Kong. This pattern supports the hypothesis that subtype 3a could have been introduced into Beijing from these 3 distinct regions, all of which have major trade ties to Beijing. Yunnan province is located in the far southern part of China bordering Laos, Vietnam, and Myanmar, and is a center for drug trafficking to the other parts of the world; drug trafficking routes could pass from this region to Beijing. The Guangdong province is located in the southern part of China, neighboring Guangxi. Historically, it has been an internal and international crossroads for cultural and commercial trade. Finally, Zhenjiang, a city in Jiangsu province, is located in the Yangtze River Delta and is a vital transportation station linking Shanghai with other regions of China. Previously, the coexistence of multiple HCV genotypes has been described in these provinces.35,40,54 These regions’ close communication with Beijing economically and culturally likely allowed for the importation of their distinct HCV strains into the Beijing epidemic.
The phylogeographic tree for subtype 3b showed only one statistically well supported group. On the basis of this tree, the migration trend appeared to have been from the south (Yunnan, Guangdong, and Hong Kong) to Beijing. Most of Beijing sequences (54/58) were in one subgroup without significant posterior probability. All these sequences were from DUs. In this subgroup, 45 Beijing sequences were in one cluster. In the time-scaled tree, there was no major group with significant posterior probability. There was no distinctive cluster existing in the Core/E1 and NS5B ML trees. These results suggested that Beijing HCV subtype 3b might come from the south of China, but it is possible to form its’ geographical genetic characteristic during the viral evolution.
More than half participants in this study were migrant or a “floating population.” The migration of population partly resulted in the transmission of infectious diseases. Thus, the HCV epidemic in Beijing maybe transmitted from other provinces of China. In a recent meta-analysis, Su et al42 found that subtypes 3a, 3b, and 6a had been significantly increased in conjunction with the relative decrease of subtypes lb and 2a. These results are in accordance with our data, which suggest that the dramatic population flow seen in migrants may drive much of HCV spread, especially among DUs.
In this study, samples were also tested for HBV and HIV infection using both ELISA and real-time PCR (data not shown). The results showed that there exist patients coinfected with HIV/HCV, HIV/HBV, HBV/HCV, and even HIV/HBV/HCV. However, the number of cases with coinfection is small, including 12 HBV/HCV, 3 HBV/HIV, 9 HCV/HIV, and 1 HBV/HCV/HIV coinfection patients. Meanwhile, the coinfection mainly existed in DUs and MSM groups, which supports epidemiologic reports of high rates of coinfection in MSM.55–56
In summary, we report the current molecular epidemiology of HCV in Beijing. We found that the HCV epidemic is primarily driven by drug use with a complex distribution of HCV subtypes, suggesting multiple introductions of distinct HCV strains from around China. With its large migrant population, Beijing is at the center of a highly complex and rapidly evolving HCV epidemic in China.
Limitation of the Study
Our study has several limitations. Although we benefit from analysis of both behavioral and sequence data, we are reliant on study subjects’ self-report for the behavioral data. When asking about such potentially stigmatizing behaviors as sexual activity and illegal drug use, we must recognize there may be error. Second, we sampled very few HCV infections from non-DUs. This limits our ability to appropriately compare with DUs. Finally, for internal control, we sequenced 2 subgenomic regions, Core/E1 and the NS5B. Discrepancies between genotypes in the same subject are not only likely the result of bulk sequencing in a population with a likely high frequency of mixed infection, but also possibly because of intrapatient recombination and/or experimental error. Third, we did not collect the data of treatment and clinic for HCV. Finally, because phylogeographic analysis reference sequences are limited to published studies, we may have missed some information about HCV migration routes.
Acknowledgements
None.
Appendix
The reference sequences used in phylogenetic analysis for C/E1 region obtained from the Genebank: M62321 (1a, USA), AF139594 (1b, USA), L02836 (1b, China-Hebei), M58335 (1b, Japan), D90208 (1b, Indonesia), D14853 (1c, Indonesia), EF115994 (1d, Canada), AY768168 (1e, Canada), L38350 (1f, Belgium), AM910652 (1g, Spain), EF115770 (1h, Canada), AY754623 (1i, Canada), AY434128 (1j, Canada), AY434122 (1k, Canada), AB047639 (2a, Japan), D00944 (2a, Indonesia), AB030907 (2b, Japan), D10988 (2b, Indonesia), D50409 (2c, Japan), L39294 (2d, Belgium), D48746 (2e, Jakarta), DQ15561 (2i, Vietnam), HM777430 (2j, Venezuela), AB031663 (2k, Japan), AY754635 (2m, Canada), FN66428 (2q, Spain), HM777422 (2r, Venezuela), D17763 (3a, Japan), D28917 (3a, Japan), D49374 (3b, Japan), D16612 (3c, Nepal), D16620 (3d, Nepal), D16614 (3f, Nepal), EF115835 (3h, Canada), EF115869 (3i, Canada), Y11604 (4a, Egypt), FJ462435 (4b, Canada), FJ462436 (4c, Canada), FJ462437 (4d, Canada), EF462435 (4e, Canada), EF589160 (4f, France), FJ462432 (4g, Canada), EF115892 (4h, Canada), FJ462438 (4k, Canada), FJ839870 (4l, Canada), FJ462433 (4m, Canada), FJ462441 (4n, Canada), FJ462440 (4o, Canada), FJ462431 (4p, Canada), FJ462434 (4q, Canada), FJ462439 (4r, Canada), FJ839869 (4t, Canada), EF684323 (4u, Egypt), Y13184 (5a, South Africa), Y12083 (6a, Hong Kong), D84262 (6b, Thailand), EF424629 (6c, Thailand), D84263 (6d, Vietnam), DQ314805 (6e,China-Guangxi), DQ835760 (6f, Thailand), D63822 (6g, Jakarta), D84265 (6h, Vietnam), DQ835762 (6i, Thailand), DQ835761 (6j, Thailand), DQ278893 (6k, China-Kunming), EF424628 (6l, USA), DQ835763 (6m, Thailand), DQ278894 (6n, China-Kunming), EF424627 (6o, Canada), EF424626 (6p, Canada), EF424625 (6q, Canada), EU408328 (6r, Canada), EU408329 (6s, Canada), EF632069 (6t, Vietnam), EF408030 (6u, China-Yunnan), EF408031 (6u, China-Yunnan), EF408032 (6u, China-Yunnan), EU798761 (6v, China-Kunming), EU798760 (6v, China-Kunming), FJ435090 (6v, China-Kunming), EU643834 (6w, Taiwan).
The reference sequences used in phylogenetic analysis for NS5B region were obtained from the Genebank: M62321 (1a, USA), AF139594 (1b, USA), L02836 (1b, China-Hebei), M58335 (1b, japan), D90208 (1b, Indonesia), D14853 (1c, Indonesia), EF115994 (1d, Canada), AY894553 (1e, Canada), AM910652 (1g, Spain), EF115992 (1h, Canada), AY754623 (1i, Canada), AY434129 (1j, Canada), AY434123 (1k, Canada), AB047639 (2a, Japan), D00944 (2a, Indonesia), AB030907 (2b, Japan), D10988 (2b, Indonesia), D50409 (2c, Japan), EF116052 (2d, Canada), D49761 (2e, Jakarta), D48769 (2f, Jakarta), DQ699770 (2i, Morocco), AY894529 (2j, Canada), GU054442 (2k, France), GU054450 (2l, France), AY754634 (2m, Canada), EF116059 (2r, Canada), D17763 (3a, Japan), D28917 (3a, Japan), D49374 (3b, Japan), D16613 (3c, Nepal), D16621 (3d, Nepal), D16615 (3f, Nepal), EF116075 (3h, Canada), EF116092 (3i, Canada), DQ202324 (3l, Iran), Y11604 (4a, Egypt), FJ462435 (4b, Canada), FJ462436 (4c, Canada), FJ462437 (4d, Canada), EF116121 (4e, Canada), EF589160 (4f, France), FJ462432 (4g, Canada), EU054465 (4h, France), FJ462438 (4k, Canada), FJ839870 (4l, Canada), FJ462433 (4m, Canada), FJ462441 (4n, Canada), FJ462440 (4o, Canada), FJ462431 (4p, Canada), FJ462434 (4q, Canada), FJ462439 (4r, Canada), FJ839869 (4t, Canada), EF694398 (4u, Egypt), Y13184 (5a, South Africa), Y12083 (6a, Hong Kong), D84262 (6b, Thailand), EF424629 (6c, Thailand), D84263 (6d, Vietnam), DQ314805 (6e,China-Guangxi), DQ835760 (6f, Thailand), D63822 (6g, Jakarta), D84265 (6h, Vietnam), DQ835762 (6i, Thailand), DQ835761 (6j, Thailand), DQ278893 (6k, China-Kunming), EF424628 (6l, USA), DQ835763 (6m, Thailand), DQ278894 (6n, China-Kunming), EF424627 (6o, Canada), EF424626 (6p, Canada), EF424625 (6q, Canada), EU408328 (6r, Canada), EU408329 (6s, Canada), EF632069 (6t, Vietnam), EF408030 (6u, China-Yunnan), EF408031 (6u, China-Yunnan), EF408032 (6u, China-Yunnan), EU798761 (6v, China-Kunming), EU798760 (6v, China-Kunming), FJ435090 (6v, China-Kunming), EU643834 (6w, Taiwan).
Footnotes
Abbreviations: DUs = drug users, ESSs = estimated effective sampling sizes, HCV = Hepatitis C Virus, IDUs = injection drug users, MCC = Maximum clade credibility, MCMC = Bayesian Markovchain Monte Carlo, ML = Maximum Likelihood, MSM = men who have sex with men, OD = the ratio of the optical density, tMRCA = time to most recent common ancestor.
YJ and XZ contributed equally to this work
This work was supported by grants from the State Key Laboratory of Infectious Disease Prevention and Control (2011SKLID102), National Natural Science Foundation of China (81361120407, 81172733, 8141101211), the 11th Five-Year National Science & Technology Major Project (2009ZX10004–903–006), the 12th Five-Year National Science & Technology Major Project (2012ZX10004–702, 2012ZX10004904–002–002), and the Excellent Talents Foundation of Beijing (2013D008001000005).
The authors report no conflicts of interest.
REFERENCES
- 1.World Health Organization. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J Viral Hepat 1999; 6:35–47. [PubMed] [Google Scholar]
- 2.Wasley A, Alter MJ. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis 2000; 20:1–16. [DOI] [PubMed] [Google Scholar]
- 3.Thomas DL, Hepatitis C. Epidemiologic quandaries. Clin Liver Dis 2001; 5:955–968. [DOI] [PubMed] [Google Scholar]
- 4.Xia GL, Liu CB, Cao HL, et al. Prevalence of hepatitis B and C virus infections in the general Chinese population: Results from a nationwide cross-sectional seroepidemiologic study of hepatitis A, B, C, D, and E virus infections in China, 1992. Intern Hepatol Comm 1996; 5:62–73. [Google Scholar]
- 5.Shan H, Wang JX, Ren FR, et al. Blood banking in China. Lancet 2002; 360:1770–1775. [DOI] [PubMed] [Google Scholar]
- 6.Chen YS, Li L, Cui FQ, et al. A sero-epidemiological study on hepatitis C in China. Chin J Epidemiol 2011; 32:888–891. [PubMed] [Google Scholar]
- 7.Xia X, Luo J, Bai J, et al. Epidemiology of hepatitis C virus infection among injection drug users in China: systematic review and meta-analysis. J Public Health 2008; 122:990–1003. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Assanangkornchai S, Duo L, et al. Risk behaviors, prevalence of HIV and hepatitis C virus infection and population size of current injection drug users in a China-Myanmar border city: results from a Respondent-Driven Sampling Survey in 2012. PLoS One 2014; 9:e106899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Chen Z, Cao Y, et al. Molecular characterization of human immunodeficiency virus type 1 and hepatitis C virus in paid blood donors and injection drug users in china. J Virol 2004; 78:13591–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou X, Chow EP, Zhao P, et al. Systematic review of HIV and HCV infection among drug users in China. BMC Infect Dis 2014; 14:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao YP, Liu ZM, Lu L. Review of HIV and HCV infection among drug users in China. Curr Opin Psychiatry 2010; 23:187–194. [DOI] [PubMed] [Google Scholar]
- 12.Danta M, Brown D, Bhagani S, et al. Recent epidemic of acute hepatitis C virus in HIV-positive men who have sex with men linked to high-risk sexual behaviours. AIDS 2007; 21:983–991. [DOI] [PubMed] [Google Scholar]
- 13.Gambotti L, Batisse D, Colin-de-Verdiere N, et al. Acute hepatitis C infection in HIV positive men who have sex with men in Paris, France. Euro Surveill 2005; 10:115–117. [PubMed] [Google Scholar]
- 14.Giraudon I, Ruf M, Maguire H, et al. Increase in diagnosed newly acquired hepatitis C in HIV-positive men who have sex with men across London and Brighton, 2002–2006: is this an outbreak? Sex Transm Infect 2008; 84:111–115. [DOI] [PubMed] [Google Scholar]
- 15.Serpaggi J, Chaix ML, Batisse D, et al. Sexually transmitted acute infection with a clustered genotype 4 hepatitis C virus in HIV-1-infected men and inefficacy of early antiviral therapy. AIDS 2006; 20:233–240. [DOI] [PubMed] [Google Scholar]
- 16.van de Laar TJ, van der Bij AK, Prins M, et al. Increase in HCV incidence among men who have sex with men in Amsterdam most likely caused by sexual transmission. J Infect Dis 2007; 196:230–238. [DOI] [PubMed] [Google Scholar]
- 17.Montoya-Ferrer A, Fierer DS, Alvarez-Alvarez B, et al. Acute hepatitis C outbreak among HIV-infected men, Madrid, Spain. Emerg Infect Dis 2011; 17:1560–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthews GV, Pham ST, Hellard M, et al. Patterns and characteristics of hepatitis C transmission clusters among HIV-positive and HIV-negative individuals in the Australian trial in acute hepatitis C. Clin Infect Dis 2011; 52:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention. Sexual transmission of hepatitis C virus among HIV-infected men who have sex with men: New York City, 2005–2010. MMWR Morb Mortal Wkly Rep 2011; 60:945–950. [PubMed] [Google Scholar]
- 20.Ruan Y, Jia Y, Zhang X, et al. Incidence of HIV-1, syphilis, hepatitis B, and hepatitis C virus infections and predictors associated with retention in a 12-month follow-up study among men who have sex with men in Beijing, China. J Acquir Immune Defic Syndr 2009; 52:604–610. [DOI] [PubMed] [Google Scholar]
- 21.An MH, Han XX, Liu J, et al. Study on the rates of infection and spontaneous clearance on HCV among HIV-infected men who have sex with men in China. Chin J Epidemiol 2013; 34:15–18. [PubMed] [Google Scholar]
- 22.Chan DP, Lin AW, Wong KH, et al. Diverse origins of hepatitis C virus in HIV co-infected men who have sex with men in Hong Kong. Virol J 2015; 12:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment Web resource. Hepatol 2014; 59:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatol 2005; 42:962–973. [DOI] [PubMed] [Google Scholar]
- 25.Mahaney K, Tedeschi V, Maertens G, et al. Genotypic analysis of hepatitis C virus in American patients. Hepatol 1994; 20:1405–1411. [DOI] [PubMed] [Google Scholar]
- 26.Grassi E, Aghemo A. How to optimize HCV therapy in genotype 2 patients. Liver Int 2013; 33 Suppl 1:35–40. [DOI] [PubMed] [Google Scholar]
- 27.Lagging M, Rembeck K, Rauning Buhl M, et al. Retreatment with peg-interferon and ribavirin in patients with chronic hepatitis C virus genotype 2 or 3 infection with prior relapse. Scand J Gastroenterol 2013; 48:839–847. [DOI] [PubMed] [Google Scholar]
- 28.Abe H, Aida Y, Ishiguro H, et al. New proposal for response-guided peg-interferon- plus-ribavirin combination therapy for chronic hepatitis C virus genotype 2 infection. J Med Virol 2013; 85:1523–1533. [DOI] [PubMed] [Google Scholar]
- 29.InamullahF IM, Ahmed H, Sajidulg AM, et al. Hepatitis C virus genotypes circulating in district Swat of Khyber Pakhtoonkhaw, Pakistan. Virol 2011; J8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakravarti A, Ashraf A, Malik S. A study of changing trends of prevalence and genotypic distribution of hepatitis C virus among high risk groups in North India. Indian J Med Microbiol 2013; 31:354–359. [DOI] [PubMed] [Google Scholar]
- 31.Verbeeck J, Maes P, Lemey P, et al. Investigating the origin and spread of hepatitis C virus genotype 5a. J Virol 2006; 80:4220–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akkarathamrongsin S, Praianantathavorn K, Hacharoen N, et al. Geographic distribution of hepatitis C virus genotype 6 subtypes in Thailand. J Med Virol 2010; 82:257–262. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen NH, Vutien P, Trinh HN, et al. Risk factors, genotype 6 prevalence, and clinical characteristics of chronic hepatitis C in Southeast Asian Americans. Hepatol Int 2010; 4:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu L, Nakano T, He Y, et al. Hepatitis C virus genotype distribution in China: predominance of closely related subtype 1b isolates and existence of new genotype 6 variants. J Med Virol 2005; 75:538–549. [DOI] [PubMed] [Google Scholar]
- 35.Fu Y, Qin W, Cao H, et al. HCV 6a prevalence in Guangdong Province had the origin from Vietnam and recent dissemination to other regions of China: phylogeographic analyses. PLoS One 2012; 7:e28006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y, Wang Y, Xia W, et al. New trends of HCV infection in China revealed by genetic analysis of viral sequences determined from first-time volunteer blood donors. J Viral Hepat 2011; 18:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu L, Tong W, Yuan M, et al. An increased diversity of HCV isolates were characterized among 393 patients with liver disease in China representing six genotypes, 12 subtypes, and two novel genotype 6 variants. J ClinVirol 2013; 57:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao J, Liang J, Zhang H, et al. The molecular epidemiological study of HCV subtypes among intravenous drug users and non-injection drug users in China. PLoS One 2015; 10:e0140263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Xia X, Li C, et al. A new HCV genotype 6 subtype designated 6 v was confirmed with three complete genome sequences. J Clin Virol 2009; 44:195–199. [DOI] [PubMed] [Google Scholar]
- 40.Xia X, Lu L, Tee KK, et al. The unique HCV genotype distribution and the discovery of a novel subtype 6u among IDUs co-infected with HIV-1 in Yunnan, China. J Med Virol 2008; 80:1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia X, Zhao W, Tee KK, et al. Complete genome sequencing and phylogenetic analysis of HCV isolates from China reveals a new subtype, designated 6u. J Med Virol 2008; 80:1740–1746. [DOI] [PubMed] [Google Scholar]
- 42.Su YY, Liu HX, Wang N. Hepatitis C virus genotypes in China: a systematic review. Chin J Epidemiol 2013; 34:80–84. [PubMed] [Google Scholar]
- 43.Lu L, Wang M, Xia W, et al. Migration patterns of hepatitis C virus in China characterized for five major subtypes based on samples from 411 volunteer blood donors from 17 provinces and municipalities. J Virol 2014; 88:7120–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang CH, Zhang Y. Investigating HCV-Ab and genotypes of HCV distribution among residents in a “blood donation” village in Hebei province. Chin J Exp Clin Virol 2009; 23:8–10. [PubMed] [Google Scholar]
- 45.De Rosa FG, Bargiacchi O, Audagnotto S, et al. The early HCV RNA dynamics in patients with acute hepatitis C treated with pegylated interferon-alpha2b. Antivir Ther 2006; 11:165–171. [PubMed] [Google Scholar]
- 46.Zhang YH, Bao YG, Sun JP, et al. Analysis of HIV/Syphilis/HCV infection among drug users in 15 cities, China. Chin J Prev Med 2010; 44:969–974. [PubMed] [Google Scholar]
- 47.Liu YH, Yan L, Hu Y, et al. Analysis on the status of HIV, HCV and Syphilis infection among drug addicts in Beijing City. Dis Surveill 2007; 22:10–12. [Google Scholar]
- 48.Solgi G, Sabouri Ghannad M, Khalilian A, et al. Molecular epidemiology of hepatitis C virus and its relation with persistence or clearance of infection in Hamadan, West-Iran. Iran J Microbiol 2015; 7:109–117. [PMC free article] [PubMed] [Google Scholar]
- 49.Cote P, Baril JG, Hebert MN, et al. Management and treatment of hepatitis C virus in patients with HIV and hepatitis C virus coinfection: A practical guide for health care professionals. Can J Infect Dis Med Microbiol 2007; 18:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busch MP, Glynn SA, Stramer SL, et al. Correlates of hepatitis C virus (HCV) RNA negativity among HCV-seropositive blood donors. Transfusion 2006; 46:469–475. [DOI] [PubMed] [Google Scholar]
- 51.Schott E, Witt H, Hinrichsen H, et al. Gender-dependent association of CTLA4 polymorphisms with resolution of hepatitis C virus infection. J Hepatol 2007; 46:372–380. [DOI] [PubMed] [Google Scholar]
- 52.Strasak AM, Kim AY, Lauer GM, et al. Antibody dynamics and spontaneous viral clearance in patients with acute hepatitis C infection in Rio de Janeiro, Brazil. BMC Infect Dis 2011; 11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan Y, Li Z, Guo XH, et al. A study on HCV-RNA quantitative detection and genotype in Peking. J Med Theory Prac 2007; 20:756–757. [Google Scholar]
- 54.Zhang C, Wu N, Liu J, et al. HCV subtype characterization among injection drug users: implication for a crucial role of Zhenjiang in HCV transmission in China. PLoS One 2011; 6:e16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jansen K, Thamm M, Bock CT, et al. High prevalence and high incidence of coinfection with hepatitis B, hepatitis C, and syphilis and low rate of effective vaccination against hepatitis B in HIV-positive men who have sex with men with known date of HIV seroconversion in Germany. PLoS One 2015; 10:e0142515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanhommerig JW, Thomas XV, van der Meer JT, et al. Hepatitis C virus (HCV) antibody dynamics following acute HCV infection and reinfection among HIV-infected men who have sex with men. Clin Infect Dis 2014; 59:1678–1685. [DOI] [PubMed] [Google Scholar]