

Abstract
Huntington's disease (HD) is a progressive neurological disorder whose non-motor symptoms include sleep disturbances. Whether sleep and activity abnormalities are primary molecular disruptions of mutant Huntingtin (mutHtt) expression or result from neurodegeneration is unclear. Here, we report Drosophila models of HD exhibit sleep and activity disruptions very early in adulthood, as soon as sleep patterns have developed. Pan-neuronal expression of full-length or N-terminally truncated mutHtt recapitulates sleep phenotypes of HD patients: impaired sleep initiation, fragmented and diminished sleep, and nighttime hyperactivity. Sleep deprivation of HD model flies results in exacerbated sleep deficits, indicating that homeostatic regulation of sleep is impaired. Elevated PKA/CREB activity in healthy flies produces patterns of sleep and activity similar to those in our HD models. We were curious whether aberrations in PKA/CREB signaling were responsible for our early-onset sleep/activity phenotypes. Decreasing signaling through the cAMP/PKA pathway suppresses mutHtt-induced developmental lethality. Genetically reducing PKA abolishes sleep/activity deficits in HD model flies, restores the homeostatic response and extends median lifespan. In vivo reporters, however, show dCREB2 activity is unchanged, or decreased when sleep/activity patterns are abnormal, suggesting dissociation of PKA and dCREB2 occurs early in pathogenesis. Collectively, our data suggest that sleep defects may reflect a primary pathological process in HD, and that measurements of sleep and cAMP/PKA could be prodromal indicators of disease, and serve as therapeutic targets for intervention.
Introduction
One goal of neurodegenerative disease research is to identify early, prodromal phenotypes. Early diagnosis could lead to earlier intervention, increasing the odds of success. A related advantage is that early pathogenesis may also be less complex, with fewer compensatory cellular and molecular changes that may complicate later intervention (1). Sleep disruptions are evident in pre-symptomatic HD carriers and early HD patients, and include altered sleep architecture (2), fragmented sleep–wake behavior or insomnia with decreased random eye movement (3), increased sleep latency (4) and increased nocturnal activity (5). Sleep disturbances correlate with early cognitive alterations (6,7) and are progressive (8,9). Whether these sleep disruptions contribute to disease advancement is unknown, but longitudinal MRI studies among HD carriers revealed significant loss of gray matter in the hypothalamus, an area critical in sleep regulation, that precedes clinical diagnosis by a decade or more (10). Transgenic animal models of HD also exhibit sleep disruptions, from Drosophila melanogaster (11), mice (12,13), to sheep (14), indicating that they reflect a conserved disturbance of cellular or molecular function.
As a model organism, Drosophila has been critical to the molecular and genetic dissection of the circadian clock, and has emerged as a similarly powerful system for understanding the regulation of sleep (15,16). Sleep has two major, partially independent regulatory mechanisms: circadian regulation, which limits sleep behavior to ecologically appropriate times, and homeostatic regulation, which controls how sleep pressure accumulates during wake and dissipates during sleep. When homeostatic mechanisms are intact, extended wakefulness or sleep deprivation (SD) results in compensatory ‘rebound’ sleep that is longer and more consolidated than at baseline (17,18). Mutants have been identified that selectively decrease basal sleep without changing the homeostatic response (19), demonstrating that altered sleep patterns do not necessarily reflect impaired homeostatic regulation. Since sleep was first described in Drosophila (20,21), a number of molecules involved in sleep regulation have been identified in flies (16,22,23). Among these, the cAMP/PKA/CREB pathway occupies a key role in sleep/wake and homeostatic regulation in flies (24–27) and sleep/wake in mammals (28–31).
Canonical signaling through the highly conserved cAMP/PKA/CREB pathway initiates with ligand binding to G-protein-coupled receptors (GPCRs). This binding leads to dissociation of the heterotrimeric G protein complex, and the subsequent activation or inhibition of transmembrane adenylyl cyclase molecules. When the cyclase is activated, it increases the synthesis of cyclic adenosine monophosphate (cAMP), which binds to the regulatory subunit of protein kinase A (PKA-R). Binding causes the dissociation of the catalytic subunits (PKA-C) from tetramers consisting of regulatory and catalytic subunits. Free PKA-C then phosphorylates substrate proteins, including the transcription factor CREB. Phosphorylation of CREB is required for interaction with the CBP co-activator, and activation of transcription from genes that contain CRE sites in their promoters (32). In general, elevation of cAMP, PKA and CREB activity decreases sleep in the fly (24,25,27).
The gene mutated in Huntington's disease (HD), HTT, encodes the ∼348 kDa Huntingtin protein (Htt) whose wild-type functions are still being elucidated (33). In addition to the exon 1 polyglutamine tract that is expanded in HD, Htt contains a number of well-characterized proteolytic cleavage sites whose contributions to HD pathogenesis are under active investigation (34,35). The first transgenic murine HD models, the R6/1 and R6/2 mice, express exon 1 of the human HTT gene with ∼115 and ∼150 CAG repeats, respectively, and toxicity of the transgenes corresponds to repeat length (36,37). Nuclear aggregates of transgenic N-terminal HTT protein (mutHtt) corresponding to symptomatology were first observed in R6/2 and R6/1mice (38), leading to the discovery of polyglutamine-expanded Htt proteolytic fragments in the cortices and striata of HD patients (39). To this day, most widely used transgenic models of HD express polyglutamine expanded N-terminal truncations of HTT; there are fewer full-length mutHtt transgenic animal models. Patterns of pathology vary within, and across, N-terminal and full-length mutHtt transgenic models (34,40,41). For these reasons, we elected to use Drosophila models expressing both full length and N-terminally truncated human mutHtt transgenes with distinct subcellular distribution and aggregation patterns (42,43).
Molecular conservation, a short lifespan and genetic malleability of the fly also provide experimental advantages for modeling HD pathogenesis. We have previously demonstrated that sleep disruptions are present in Drosophila models of HD. Here, we demonstrate that sleep and activity abnormalities are an early adult behavioral phenotype in full-length and N-terminal mutHtt fly models. We also describe disrupted sleep homeostasis in HD model animals for the first time. These sleep defects implicate pathological up-regulation of the cAMP/PKA signaling pathway early in HD pathogenesis. We tested PKA pathway components for suppression of a mutHtt-associated developmental lethality phenotype. Genetic reduction in PKA signaling suppresses larval lethality and reverses sleep and activity phenotypes accompanying neuronal mutHtt expression. We also find that PKA reduction extends the median lifespan of HD model flies. Using in vivo reporters of dCREB2-responsive transcription, we find that dCREB2 activity is not elevated in HD model flies during periods when PKA-associated sleep disturbances are evident. These data are consistent with earlier results indicating depressed CREB transcription in HD pathogenesis, and provide further evidence that CREB pathway dysregulation in HD may include early dissociation of PKA signaling activity from CREB activation. These studies suggest that dysregulated sleep could serve as a preliminary screen for modifiers of mutHtt pathology, or serve as a systemic outcome measures in the process of drug development. In addition, this provides entry points to analyze early pathology in the disease progression pathway.
Results
Sleep and activity defects present in early adulthood in HD model flies
Flies do not exhibit adult sleep patterns until 3–4 days post-eclosion (DPE) (21). We have previously shown that fly models of HD exhibit sleep abnormalities, but these measurements began with animals 5–7 DPE. Since the climbing deficits and increased mortality that have been reported for these HD model lines occur considerably later (42,44,45), we wanted to determine precisely when sleep abnormalities are first detectable in HD model flies. Light entrained, virgin females were collected and sleep recording initiated within 12 ho of eclosion. Sleep over the first 6 DPE is shown for UAS-Htt128QFL M36 (‘M36’) (Fig. 1A), and UAS-Htt128QN-terminus F27B (‘F27B’) (Fig. 1A′) lines in the presence (elav-Gal4>) or absence (UAS-Htt128Q/+) of a pan-neuronal elav-Gal4 driver. There is a clear, persistent decrease in nighttime sleep in both lines within 4 days, and this effect is dependent upon HD transgene expression. Sleep latency, a measure of sleep initiation, is consistently increased by 4 DPE in elav-Gal4>UAS-Htt128QFL M36 (Fig. 1B) and elav-Gal4>UAS-Htt128QNT F27B flies (Fig. 1B′) relative to controls.
Figure 1.

Sleep defects are evident early in adulthood in HD model flies. (A) Sleep for the first 6 DPE among UAS-Htt128QFL M36 flies in the presence (blue) or absence (gray) of the elav-Gal4G28 driver. (A′) Sleep for the first six DPE in UAS-Htt128QNT F27B flies in the presence (red) or absence (white) of the elav-Gal4G28 driver. Gray box from ZT 0–9 represents the period of fly collection and experiment set-up. Recording began at ZT 10 on the day of eclosion. X-axes are labeled with ZT values relative to the beginning of the experiment. Matings and flies were maintained on a 12:12 light:dark cycle, with dark periods indicated by dark rectangles along the x-axes. Sleep latency is significantly increased in elav-Gal4>UAS-Htt128QFL M36 (B) and elav-Gal4>UAS-Htt128QNT F27B flies (B′) relative to controls by 4 DPE (corresponding to hours 72–96 of recording). (C) Average nighttime bout length is selectively reduced in elav-Gal4>UAS-Htt128QFL M36 flies by 5 DPE. (C′) Average day sleep bout is reduced in elav-Gal4>UAS-Htt128QNT F27B flies compared with sibling controls and night bout length trends lower at 5 DPE. (D) Day and night activity in elav-Gal4>UAS-Htt128QFL M36 flies and controls from 3 to 5 DPE. Elav-Gal4>UAS-Htt128QFL M36 flies show persistent nighttime hyperactivity by 3DPE. (D′) Nighttime hyperactivity is evident in elav-Gal4>UAS-Htt128QNT F27B flies by 4 DPE, while daytime activity is comparable to that of controls. UAS-Htt128QFL M36/+, n = 31. Elav-Gal4>UAS-Htt128QFL M36, n = 30. UAS-Htt128QNT F27B/+, n = 19. Elav-Gal4>UAS-Htt128QNT F27B, n = 27. Data are presented as averages ± SEM. P-values using Student's t-test, *≤0.05; **≤0.01; ***≤0.005.
Sleep consolidation is also altered in HD models, with decreases in the average duration of day and night sleep bouts (Fig. 1C and C′), and an increase in the number of night bouts (data not shown). Night sleep is selectively fragmented in elav-Gal4>UAS-Htt128QFL M36 flies (Fig. 1C), while elav-Gal4>UAS-Htt128QNT F27B flies exhibit decreases in day and night bout duration (Fig. 1C′), indicating a more general fragmentation of sleep patterns. Therefore, sleep disturbances appear in the HD flies as soon as the animal shows normal adult behavioral rhythms.
The number of infrared beam breaks per waking minute is a measure of the fly's basal activity state. This metric allows us to distinguish between animals that are awake, but largely inactive, and animals that are very active when awake but sleep much of the hour (46). Figure 1D and D′ shows that elav-Gal4>UAS-Htt128QFL M36 and elav-Gal4>UAS-Htt128QNT F27B flies exhibit persistent nighttime hyperactivity relative to controls by 5DPE. Basal sleep and activity parameters in elav-Gal4/+ flies, an additional genetic control, overlapped with UAS-Htt128Q/+ controls, demonstrating that sleep disturbances in HD model flies resulted from neuronal mutHtt expression (Supplementary Material, Fig. S1).
HD model flies exhibit disturbed sleep homeostasis
Mutations in genes regulating sleep do not necessarily alter sleep homeostasis (16,19), and it is not currently known whether sleep homeostasis is disrupted in humans with HD (47). To determine whether sleep homeostasis is intact in HD model flies, we assessed their response to SD using the Sleep Nullifying Apparatus (SNAP). The SNAP produces a mild repetitive physical stimulus that does not induce non-specific stress pathways in healthy flies (48). Flies were deprived of sleep for 12 h during a dark period, when M36 and F27B animals exhibit reduced and fragmented sleep, followed by 72 h of recovery recording. Elav-Gal4/+ controls exhibit robust sleep recovery following deprivation as expected (Supplementary Material, Fig. S2). Figure 2A shows the percentage change in sleep over baseline during the SD period, and during the recovery days and nights for M36 flies in the presence (violet) and absence (gray) of the elav-Gal4 driver. While both elav-Gal4>UAS-Htt128QFL M36 flies and controls show increased daytime sleep in the 12 h following deprivation, daytime recovery trends lower in elav-Gal4>UAS-Htt128QFL M36, and nighttime sleep remains depressed. Elav-Gal4>UAS-Htt128QNT F27B flies (Fig. 2B, red) have significantly decreased daytime sleep recovery relative to sibling controls (Fig. 2B, white), and show further depression of nighttime sleep following SD. Interestingly, night sleep consolidation is not increased in HD models after deprivation: average night bout duration remains depressed in both M36 (Fig. 2C) and F27B (Fig. 2D) relative to controls, and the number of night sleep bouts increases post-SD in elav-Gal4>UAS-Htt128QFL M36 flies (Fig. 2E). There is no change in the number of bouts in elav-Gal4>UAS-Htt128QNT F27B flies after deprivation (Fig. 2F). These data show that SD does not increase night sleep in HD model flies.
Figure 2.

Sleep homeostasis is disrupted in HD flies. Percentage sleep lost during 12 h SD and regained in the first 24 h of recovery in UAS-Htt128QFL M36 (A) and UAS-Htt128QNT F27B flies (B). (A) Elav-Gal4>UAS-Htt128QFL M36 flies (violet) exhibit a trend toward decreased SD recovery from sibling controls (gray). (B) Elav-Gal4>UAS-Htt128QNT F27B flies (red) exhibit significant decreases in day and night sleep recovered after deprivation compared with controls (white). Depressed average night bout duration is not increased after SD in elav-Gal4>UAS-Htt128QFL M36 (C) or in elav-Gal4>UAS-Htt128QNT F27B (D) relative to controls. (E) Night bout number further increases following SD in elav-Gal4>UAS-Htt128QFL M36 flies and remains high but stable in elav-Gal4>UAS-Htt128QNT F27B (F) relative to controls. (G) Daytime activity (left) is depressed in controls after SD but remains elevated in elav-Gal4>UAS-Htt128QFL M36 flies. Nighttime hyperactivity (right) relative to controls persists in elav-Gal4>UAS-Htt128QFL M36 following SD. (H) Daytime activity (left) decreases in controls after SD but is unchanged in elav-Gal4>UAS-Htt128Q NT F27B flies. Nighttime hyperactivity (right) remains pronounced in F27B flies relative to controls before and after SD. UAS-Htt128QFL M36/+, n = 31. Elav-Gal4>UAS-Htt128QFL M36, n = 31. UAS-Htt128QNT F27B/+, n = 32. Elav-Gal4>UAS-Htt128QNT F27B, n = 48. Data are presented as averages ± SEM. P-values using Student's t-test, *≤0.05; **≤0.01; ***≤0.005.
Heightened sensitivity to physical stimulation might account for further disruption of sleep following SD in HD model flies and hyperarousal has been associated with mutations affecting sleep maintenance (49). If HD model flies were hypersensitive to mechanical stimulation, we would expect to see increased waking activity after the end of SNAP-induced SD. Hyperactivity is present in HD flies prior to SD, and persists after it ends, but does not increase (Fig. 2G and H, colored bars). In addition to increasing sleep in the day period following SD, control flies depress waking activity, but SD does not change day or night activity in elav-Gal4>UAS-Htt128QFL M36 flies or elav-Gal4>UAS-Htt128QNT F27B (Fig. 2G and H). Hypersensitivity may be assessed by the use of mild physical stimuli to induce geotactic behavior (50). We found no change in geotactic response in young M36 and F27B flies (Supplementary Material, Fig. S3). These data suggest that heightened sleep disruptions after SD in HD model flies do not result from sensitivity to physical stimulation. Together, our data demonstrate that HD model flies have disrupted sleep homeostasis, including exacerbated nighttime fragmentation and sleep loss following SD.
Neuronal mutHtt expression results in sleep and activity defects across HD models
In total, we assayed sleep and activity phenotypes of one full-length mutHtt model and three different genomic insertions of an N-terminally truncated mutHtt transgene (42,43) and the elav-Gal4 driver alone. Basal sleep and SD response for elav-Gal4/+ flies are shown in Supplementary Material, Figures S1 and S2. Data for the elav-driven HD models are summarized in Table 1. Neuronal expression of mutHtt is sufficient to produce these phenotypes; glial mutHtt expression does not result in sleep and activity changes in young flies (data not shown). Variations in severity of the sleep and activity phenotypes among the three N-terminal transgene insertions predominantly affected daytime activity and consolidation. The F27B line exhibited behavior similar to that of the full-length line, M36, where disruptions were primarily limited to nighttime sleep and activity. We have previously shown that M64, a second insertion on the same chromosome, exhibits broad disruptions of day and night sleep and activity (11). It also shows high mortality during SD (Table 1). A third insertion on the X chromosome exhibited intermediate phenotypes between F27B and M64 (data not shown). The variability among these lines provides the opportunity to screen potential modifiers of weak (F27B) and strong (M64) sleep and activity phenotypes.
Table 1.
Comparison of sleep phenotypes among HD full-length and N-terminal insertion lines
| UAS-Htt128Q | Full length (M36) | N-term (F27B) | N-term (NtX) | N-term (M64) |
|---|---|---|---|---|
| Chromosome insertion | II | III | X | III |
| Total day sleep change | NS | NS | ↑ | ↑ |
| Average day bout | NS | ↑ | ↑ | ↑ |
| Day bout # | NS | NS | NS | ↑ |
| Average day activity | NS | ↑ | ↓ | ↓ |
| Total night sleep change | ↓ | ↓ | ↓ | ↓ |
| Average night bout | ↓ | ↓ | ↓ | ↓ |
| Night bout # | ↑ | ↑ | ↑ | ↑ |
| Average night activity | ↑ | ↑ | ↑ | ↑ |
| Rebound sleep after SD | ↓ | ↓ | Not tested | Not determined*** |
| Day bout # after SD | ↑ | ↑ | Not tested | Not determined*** |
| Average day bout after SD | ↓ | ↓ | Not tested | Not determined*** |
| Night bout # after SD | ↑ | ↑ | Not tested | Not determined*** |
| Average night bout after SD | ↓ | ↓ | Not tested | Not determined*** |
| Lethality of SD | 5–10% | 10–20% | Not tested | >60% |
SD, sleep deprivation (12 h).
NS, not significantly different from -elav controls.
Not determined***, insufficient n due to lethality of sleep deprivation.
Comparisons were made between flies expressing the specified HD transgene under the control of the elavG28-Gal4 driver and siblings lacking the Gal4 driver.
Genetically decreasing PKA suppresses mutHtt phenotypes
The sleep phenotypes of these HD model flies are striking because few genetic or pharmacological interventions selectively alter nighttime sleep in Drosophila. Many involve dopaminergic signaling or its effectors, cAMP/PKA signaling (25,27,51,52). Consistent with elevated dopaminergic signaling through GαS, feeding a tyrosine hydroxylase inhibitor (3-iodo-tyrosine or 3IY) to adult HD model flies restores their sleep to control levels (Supplementary Material, Fig. S4). Interestingly, feeding 3IY to HD model flies beginning at an advanced physiological age also reverses sleep and activity deficits (Supplementary Material, Fig. S5). Chronic administration of 3IY, however, resulted in significantly decreased lifespan relative to control food for HD model flies as well as genetic controls (data not shown). In addition, HD models exhibit reduced response to caffeine (Supplementary Material, Fig. S6), which affects sleep in flies by acting as a phosphodiesterase (PDE) inhibitor (27). Furthermore, LC-MS/MS analysis of head extracts suggests that basal cAMP is increased in HD models compared with controls (Supplementary Material, Fig. S7). Collectively, these data implicate pathological elevation of the cAMP/PKA signaling pathway as a factor in early-onset sleep abnormalities in HD model flies.
If aberrant cAMP/PKA/CREB pathway signaling occurs early in HD pathogenesis, manipulation of that pathway should suppress sleep/activity defects and other disease-related phenotypes, such as shortened lifespan. Strong neuronal mutHtt expression causes developmental lethality (44), and we used this phenotype to test PKA pathway components for suppression. Elav-Gal4C155 (C155) is a stronger X-chromosome driver than the second chromosome elav-Gal4 insertion used in our sleep experiments. C155 expression of F27B results in ∼85% lethality during metamorphosis. When M64 is expressed using C155, there is 100% pupal lethality. Death occurs at multiple developmental transitions and fewer larvae pupate, indicating that lethality occurs earlier in development in this insertion line than in F27B (C.S. Wesley and E.D.G., unpublished data). The severity of the developmental lethality parallels that of sleep and activity phenotypes in the UAS-Htt128QNT insertion lines (Table 1) and permits us to test transgenes that affect the cAMP/PKA/CREB pathway relatively rapidly.
Two different methods were used to genetically alter PKA levels: UAS-driven transgenes that express RNAi directed against the catalytic and regulatory subunits of PKA (53) (Vienna Drosophila Resource Center) or overexpression of a regulatory subunit (54); RNAi lines targeting PKA catalytic subunits were selected for their absence of predicted OFF-target effects. Table 2 shows that decreasing PKA in HD model flies, either by knocking down PKA catalytic subunits or overexpressing regulatory subunit, suppresses lethality. As controls, we use UAS-FLP (whose product is neutral) or UAS-PKA-RIIRNAi to increase PKA activity. Co-expression of UAS-FLP or UAS PKA-RIIRNAi with UAS-Htt128QNT F27B or M64 does not suppress developmental lethality. These results demonstrate that the beneficial effects of knocking down PKA are due to the directionality of altered PKA activity, and not off-target effects of RNAi or dilution of available Gal4 across multiple UAS-driven targets. Transgenes that rescue lethality, i.e. permitted eclosion of adult flies, were subsequently tested for their effects on sleep and longevity.
Table 2.
Suppression of c155 developmental lethality by PKA manipulations
| Gal4 driver | UAS-Htt128QN-term line | Potential suppressor | Suppresses lethality |
|---|---|---|---|
| Elavc155 | F27B (weak) | N | |
| Elavc155 | F27B (weak) | UAS-FLPa | N |
| Elavc155 | F27B (weak) | UAS-PKA-RII RNAi | N |
| Elavc155 | F27B (weak) | UAS-PKA-C2 RNAi | Y |
| Elavc155 | F27B (weak) | UAS-PKA-C1 RNAi | Y |
| Elavc155 | F27B (weak) | UAS-PKAraBDK35b | Y |
| Elavc155 | M64 (strong) | N | |
| Elavc155 | M64 (strong) | UAS-FLPa | N |
| Elavc155 | M64 (strong) | UAS-PKA-RII RNAi | N |
| Elavc155 | M64 (strong) | UAS-PKA-C2 RNAi | Y |
| Elavc155 | M64 (strong) | UAS-PKA-C1 RNAi | Delays lethality |
| Elavc155 | M64 (strong) | UAS-PKAraBDK35b | Not tested |
aGal4 dilution control: co-expresssion of an unrelated UAS-driven transgene is not sufficient to suppress lethality.
bPKA-RI overexpression transgene.
Genetically decreasing PKA suppresses sleep defects of HD N-term models
PKA-C1 mRNA is present and enriched relative to other mRNAs in adult brains, whereas PKA-C2 is expressed at very low levels (55). Since RNAi directed against both PKA-C1 and -C2 suppressed C155-associated mutHtt lethality (Table 2), we used the milder second chromosome elav-Gal4G28 driver to assess their effects on sleep. Disruptions of sleep and activity in elav-Gal4>UAS-Htt128QNT F27B animals at 7–8 DPE are summarized in Figure 3A–D. To control for potential location effects of the suppressor and mutHttN-term transgenes, comparisons are made between animals with the elav-Gal4 driver and their driver-less siblings. Co-expression of the PKA-C1RNAi transgene in F27B flies (Fig. 3E) prevents the nighttime sleep loss seen in animals expressing F27B alone (Fig. 3A). Likewise, PKA-C1RNAi suppresses (Fig. 3F–H) diminished nighttime sleep consolidation (Fig. 3B and C) and nighttime hyperactivity (Fig. 3D). Pan-neuronal co-expression of a PKA-C2RNAi transgene produces similar effects (Fig. 4B, and data not shown). RNAi-mediated suppression of PKA is sufficient to prevent F27B-mediated sleep consolidation and activity defects, in addition to suppressing pupal lethality.
Figure 3.

Decreasing PKA activity in neurons suppresses sleep and activity defects in HD model flies. (A–D) Sleep phenotypes of UAS-Htt128QNT F27B flies at 7–8 DPE. (A) Nighttime sleep is decreased in elav-Gal4>UAS-Htt128QNT F27B flies (red) relative to sibling controls (white). (B) Duration of average night sleep bout is reduced in elav-Gal4>UAS-Htt128QNT F27B flies. (C) Nighttime sleep bout number is elevated in elav-Gal4>UAS-Htt128QNT F27B flies. (D) Day and night activity patterns are disrupted in elav-Gal4>UAS-Htt128QNT F27B flies. (E) Co-expression of RNAi against PKA-C1 with mutHtt restores nighttime sleep in elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B flies to control levels. (F) Average night sleep bout duration is increased in elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B flies so that it is comparable to that of controls. (G) Number of night sleep bouts is reduced in elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B flies. (H) Day and night activity patterns in elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B flies are indistinguishable from controls. (I) Co-expression of PKA regulatory subunit PKAr*BDK35 with mutHtt restores nighttime sleep in elav-Gal4>UAS-Htt128QNT F27B/UAS-PKAr*BDK35 flies. (J) Average night sleep bout duration is comparable in elav-Gal4>UAS-Htt128QNTF27B/UAS-PKAr*BDK35 flies to sibling controls. (G) Number of night sleep bouts is reduced in elav-Gal4>UAS-Htt128QNT F27B/UAS-PKAr*BDK35 flies. (H) Day and night activity patterns in elav-Gal4>UAS-Htt128QNT F27B/UAS-PKAr*BDK35 flies are indistinguishable from controls. UAS-Htt128QNT F27B/+, n = 28. Elav-Gal4>UAS-Htt128QNT F27B, n = 31. UAS-PKA-C1RNAi/+; UAS-Htt128QNT F27B/+, n = 21. Elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B/+, n = 25. UAS-PKAr* BDK35/UAS-Htt128QNT F27B, n = 22. Elav-Gal4>UAS-PKAr* BDK35/UAS-Htt128QNT F27B, n = 31. Data are presented as averages ± SEM. P-values using Student's t-test, *≤0.05; **≤0.01; ***≤0.005.
Figure 4.

Genetically decreasing PKA in neurons partially suppresses sleep homeostasis defects. (A) Co-expression of PKA regulatory subunit UAS-PKAr*BDK35 with UAS-Htt128QNT F27B makes total day and night sleep in elav-Gal4>UAS-PKAr*BDK35/UAS-Htt128QNT F27B flies indistinguishable from that of controls. (B) The percentage of sleep recovery after SD in elav-Gal4>UAS-PKAr*BDK35/UAS-Htt128QNT F27B flies (gray) is not significantly different from sibling controls (red). (C) Number of sleep bouts is not significantly different between elav-Gal4>UAS-PKAr* BDK35/UAS-Htt128QNT F27B flies and controls before SD (white and light red, ‘pre’) or after SD (gray and dark red, ‘post’). (D) UAS-PKA-C2 RNAi expression with UAS-Htt128QNT F27B (white) restores day and night sleep to control levels (red). (E) Sleep recovery following SD in elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT F27B flies is not significantly different from driverless siblings. (F) Number of night sleep bouts is not significantly different between elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT F27B flies and controls before SD (white and light red, ‘pre’) or after SD (gray and dark red, ‘post’). (G) Co-expression of UAS-PKA-C2RNAi with the strong mutHtt insertion UAS-Htt128QNT M64 restores day and night sleep to control levels. (H) Percentage recovery sleep following SD in flies co-expressing PKA-C2RNAi with M64 (green) is comparable to controls (gray). (I) Night bout number is not significantly different between elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT M64 flies and control siblings before SD (light gray and green, ‘pre’) or after SD (dark gray and green, ‘post’). UAS-PKAr* BDK35/UAS-Htt128QNT F27B, n = 28. Elav-Gal4>UAS-PKAr* BDK35/UAS-Htt128QNT F27B, n = 30. UAS-PKA-C2RNAi/+; UAS-Htt128QNT F27B/+, n = 16. Elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT F27B/+, n = 15. UAS-PKA-C2RNAi/+; UAS-Htt128QNT M64/+, n = 14. Elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT M64/+, n = 12. Data are presented as averages ± SEM. P-values using Student's t-test, *≤0.05; **≤0.01; ***≤0.005.
As a complementary means of decreasing PKA activity, we can increase PKA-RI levels, whose expression is enriched in the adult Drosophila brain (55). UAS-PKAr*BDK35 expresses an RI subunit whose cAMP binding site has been mutated to prevent PKA-RI dissociation from PKA catalytic subunits regardless of cAMP levels (54). Co-expression of UAS-PKAr*BDK35 with F27B in neurons suppresses nighttime sleep loss (Fig. 3I) present in animals expressing F27B alone (Fig. 3A). PKAr*BDK35 rescues sleep consolidation defects accompanying mutHtt expression, restoring night bout length (Fig. 3J) and number (Fig. 3K) to control levels, and restores normal activity (Fig. 3L).
Can decreasing PKA restore sleep homeostasis in HD model flies? PKAr*BDK35 co-expression with F27B not only restores baseline day and night sleep to that of controls (Fig. 4A), but there is no further loss or fragmentation of nighttime sleep following SD. Recovery following SD is comparable between elav-Gal4>UAS-Htt128QNT F27B/UAS-PKAr*BDK35 flies and UAS-Htt128QNT F27B/UAS-PKAr*BDK35 siblings (Fig. 4B), as is night bout number (Fig. 4C), night bout length and activity (data not shown). UAS-PKA-C2RNAi, the sole transgene that suppresses developmental lethality in both the weak F27B and strong M64 mutHtt insertion lines, also suppresses the effects of F27B alone on baseline day and night sleep (Fig. 4D) and restores normal amounts of recovery sleep (Fig. 4E). In addition, PKA-C2RNAi co-expression with F27B returns nighttime sleep consolidation before and after SD to control levels (Fig. 4F). Therefore, in addition to restoring basal sleep and activity levels, genetically reducing PKA is sufficient to suppress sleep homeostasis defects.
While elav-Gal4G28-driven M64 flies are viable and survive to adulthood, they have dramatically altered sleep and activity patterns (Table 1). SD experiments in elav-Gal4>UAS-Htt128QNT M64 flies were confounded by high mortality rates (60–80%) when subjected to our standard 12 h deprivation protocol, which results in 5–10% mortality in elav-Gal4>UAS-Htt128QNT F27B flies (Table 1). When UAS-PKA-C2RNAi is co-expressed with M64, SD-associated mortality is reduced to 20% and day and night sleep return to control levels (Fig. 4G). Similar to its effects on the weaker F27B insertion, PKA-C2RNAi co-expression with M64 results in recovery sleep that is not significantly different from sibling controls (Fig. 4H) with no significant difference in sleep consolidation or activity after SD (Fig. 4I, and data not shown). Hence, reducing PKA in neurons expressing mutHtt limits the enhanced sensitivity to SD exhibited by HD models (Table 1 and Fig. 2).
Decreasing PKA restores immediate memory impairments in HD model flies
Early cognitive deficits in R6/1 model mice have been linked to pathological increases in hippocampal PKA activity (56,57). We used a classical conditioning olfactory avoidance paradigm (58,59) to determine whether HD model flies have cognitive deficits when sleep and activity problems are readily apparent. We assessed learning and memory in the F27B or M64 lines at 8–10 DPE under the control of the elav-Gal4G28 driver used in sleep experiments. Learning (or immediate memory after a single training trial) was intact in both lines compared with controls (data not shown). This led us to test immediate memory in response to 3× massed training (60,61). The M64 line exhibits an immediate memory deficit after three cycles of massed training (Supplementary Material, Fig. S8, black bar), but the weaker F27B line does not (data not shown). PKA-C2 is a minor transcript in the adult brain (55) when compared with DC0/PKA-C1, which has a well-established role in memory (62,63). Therefore, decreasing PKA-C2 is less likely to exert a dominant negative effect on memory. Indeed, elav-Gal4G28-driven expression of PKA-C2RNAi does not have an effect on immediate memory (Supplementary Material, Fig. S8, white and dark gray bars). Co-expression of PKA-C2RNAi with M64 partially reverses the immediate memory deficit of M64 alone (Supplementary Material, Fig. S8, light gray bar). These data suggest that the immediate memory disruption may be delayed relative to sleep and activity abnormalities in HD model flies.
Knockdown of PKA catalytic subunits extends longevity of HD model flies
Diminishing PKA suppresses developmental lethality and sleep defects in HD fly models. Are these genetic manipulations able to suppress later-onset, disease-related phenotypes? Genetic reduction in PKA activity to ∼60% of WT levels has no effect on lifespan in aged flies (64), and knockdown of PKA in-and-of itself would not be expected to extend survival unless PKA activity plays a role in HD pathogenesis. We compared lifespan of flies expressing mutHtt alone or with UAS-PKA-C1 or -C2RNAi transgenes under elav-Gal4G28 control. Co-expression of either PKA-C1RNAi or PKA-C2RNAi with F27B (Fig. 5A, red triangles and red circles, respectively) increases survival over-expression of F27B alone (Fig. 5A, red squares). The median lifespan in flies co-expressing PKA-C1RNAi or PKA-C2RNAi with F27B is restored to that of sibling controls lacking driver (Fig. 5A, white squares, gray triangles and gray circles). Likewise, co-expression of PKA-C2RNAi with M64 extends the median survival (Fig. 5B, green triangles) over that of M64 neuronal expression alone (Fig. 5B, green squares). This effect is comparable to the life expectancy of sibling controls that lack a driver (Fig. 5B, gray triangles). Thus, decreasing PKA in HD models not only suppresses sleep and activity defects, but also extends survival.
Figure 5.
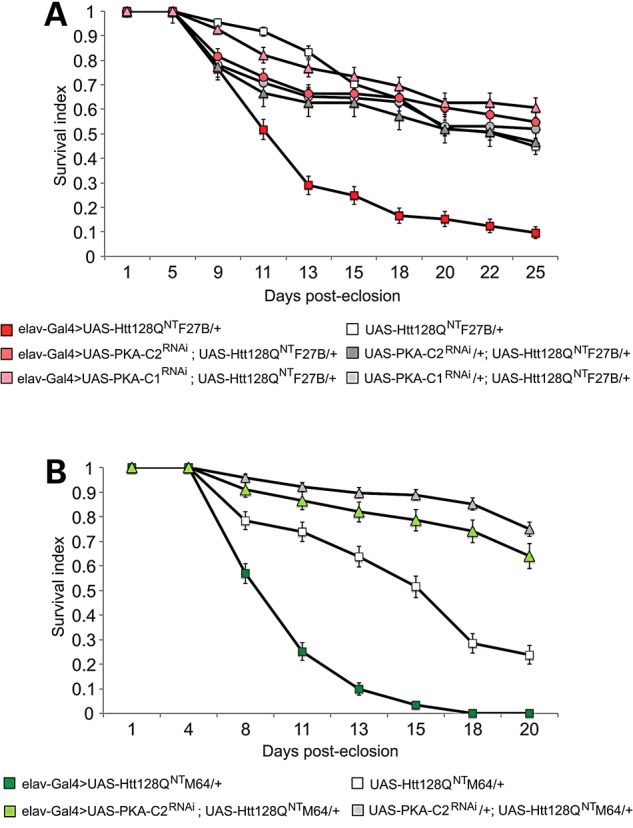
RNAi knockdown of PKA-C1 and -C2 extends lifespan in HD model flies. (A) Survival of UAS-Htt128QNT F27B flies in the presence (dark red) of the elav-Gal4G28 driver (red squares) is extended by co-expression of UAS-PKA-C2RNAi (red circles) or UAS-PKA-C1RNAi (red triangles) to lifespan of Gal4 driverless controls (UAS-Htt128QNT F27B, white squares; UAS-PKA-C2RNAi; UAS-Htt128QNT F27B, gray circles; UAS-PKA-C1RNAi; UAS-Htt128QNT F27B, gray triangles). (B) Survival of UAS-Htt128QNT M64 flies in the presence (green squares) of the elav-Gal4G28 driver is extended by co-expression of UAS-PKA-C2RNAi (green triangles). Elav-Gal4 driverless controls are in white squares (UAS-Htt128QNT M64) and gray triangles (UAS-PKA-C2RNAi; UAS-Htt128QNT M64). Longevity assays were conducted at 25°C. UAS-Htt128QNT F27B/+, n = 149. Elav-Gal4>UAS-Htt128QNT F27B, n = 237. UAS-PKA-C1RNAi/+; UAS-Htt128QNT F27B/+, n = 74. Elav-Gal4>UAS-PKA-C1RNAi; UAS-Htt128QNT F27B/+, n = 74. UAS-PKA-C2RNAi/+; UAS-Htt128QNT F27B/+, n = 174. Elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT F27B/+, n = 174. UAS-Htt128QNT M64/+, n = 143. Elav-Gal4>UAS-Htt128QNT M64, n = 132. UAS-PKA-C2RNAi/+; UAS-Htt128QNT M64/+, n = 216. Elav-Gal4>UAS-PKA-C2RNAi; UAS-Htt128QNT M64/+, n = 96. Data are presented as averages ± SEM.
Patterns of dCREB2 activity are altered in HD models
Our data indicate that when mutHtt is expressed at high levels in neurons, early and persistent elevations in PKA underlie pupal lethality, sleep defects and early mortality in adults. The canonical nuclear target of PKA phosphorylation is CREB, a transcription factor implicated in neuronal function and neuroprotection (65–67). Based on our PKA suppression data, a simple prediction is that CREB activity may be increased in the HD models. Drosophila contains a single CREB/CREM gene, dCREB2, which encodes multiple protein isoforms generated through alternative splicing and the use of alternative translation start sites (68,69). How does mutHtt expression affect dCREB2 activity when sleep defects are present? To measure dCREB2 activity in vivo in HD model flies, we employ a genetically encoded CRE-luciferase reporter (70,71). dCREB2 activity undergoes circadian oscillations in vivo, with a large transcriptional peak during the day and a smaller peak in the nighttime. We do not detect changes in basal dCREB2 protein levels or phosphorylation state by immunoblot at 7–10 DPE when sleep defects are present (data not shown).
Growth temperature was used to control HS-Gal4-responsive M36 expression. Larvae were maintained at 19°C throughout development to repress M36 expression or shifted to 30°C as 3rd instar to induce expression. Upon eclosion, both groups of flies were switched to and maintained at 19°C during the measurements of reporter activity. CRE-luciferase activity is almost indistinguishable between M36 flies and controls maintained at 19°C throughout development (Fig. 6A). In contrast, there are decreases in dCREB2 activity that persist days after the end of mutHtt induction in flies raised at 30°C (Fig. 6B). Interestingly, these decreases in dCREB2 activity are more pronounced during the day. These data show that organism-wide dCREB2 activity is altered even after the end of ubiquitous mutHtt induction, suggesting that changes to transcriptional patterns in HD may be established early in disease.
Figure 6.

CREB activity patterns are altered in HD model flies. (A) Whole fly CRE-luciferase activity over 4 days (∼7–11 DPE) in flies maintained at 19°C throughout development. (B) Whole fly CRE-luciferase activity over 7–11 DPE in flies induced at 30°C from 3rd instar to 3 DPE. Altered CREB activity is specific to mutHtt induction. A and B were recorded simultaneously; data are shown starting ∼40 h after transfer to luciferin food and the beginning of recording, i.e. 7–8 DPE. (C) Schematic of the CRE-luciferase reporter used to measure tissue-specific CREB activity in vivo in HD model flies. (1) Three CRE binding sites are shown in green upstream of the TATA box in blue. A start codon is 5′ of an FRT-flanked mCherry ORF ending in three tandem stop codons (red) 5′ of the second FRT to end translation prior to the luciferase ORF (yellow) lacking a start codon. In the absence of FLP recombinase, only mCherry is produced. (2) In the presence of the UAS-FLP recombinase, the mCherry sequence is excised and the recombined transgene produces only luciferase in response to CRE-binding. (3) Introduction of an enhancer-Gal4 driver provides tissue specificity to UAS-recombinase expression. All three transgenes are required for CRE-luciferase expression. (D) Genotypes of flies used in neuron-specific CRE-luciferase recording; values ≤200 RLU were excluded as background activity and those genotypes are not shown on graphs. (E) Neuronal CRE-luciferase activity in the presence or absence of UAS-Htt128Q NT F27B. (F) Neuronal CRE-luciferase activity in the presence or absence of UAS-Htt128Q NT M64. X-axis shows ZT and shaded areas indicate dark periods. Y-axis gives relative light units (RLU) produced by luciferin hydrolysis. Flies were entrained to a 12:12 L:D cycle as larvae and were collected as adults within 48 h. Recording for E and F began at 5–6 DPE, when sleep phenotypes are evident. Data shown in E and F were recorded simultaneously and represent the period 24–108 h after transfer to luciferin food and beginning of recording. Flies that did not survive the duration of the experiments were excluded from analysis. CRE-luci (785-3)/Y; HS-Gal4/III flies maintained at 19°C throughout experiment, n = 48. CRE-luci (785-3)/Y; UAS-Htt128QFL M36/II; HS-Gal4/III flies maintained at 19°C throughout experiment, n = 48. CRE-luci (785-3)/Y; HS-Gal4/III flies induced at 30°C from 3rd instar to 3 DPE, n = 46. CRE-luci (785-3)/Y; UAS-Htt128QFL M36/II; HS-Gal4/III flies induced at 30°C from 3rd instar to 3 DPE, n = 48. Elav-Gal4>UAS-FLP; FRT-CRE-luci/III, n = 43. Elav-Gal4>UAS-FLP; FRT-CRE-luci/UAS-Htt128QNT F27B, n = 46. Elav-Gal4>UAS-FLP; FRT-CRE-luci/III, n = 25. Elav-Gal4>UAS-FLP; FRT-CRE-luci/UAS-Htt128QNT M64, n = 22. Data are presented as averages ± SEM. Lines with # indicate periods of significantly different CRE luciferase activity with P ≤ 0.05.
In HD, transcriptional irregularities can vary in different tissue types (34,72). To restrict our analysis to tissues where we have demonstrated the effects of mutHtt expression, we used a novel CRE-luciferase reporter system, FRT-CRE-luci (73). This Flippase-dependent reporter system (Fig. 6C) allows us to generate CRE-luciferase mosaics in a temporally or tissue-specific fashion. Figure 6D shows the genotypes of the animals used for in vivo FRT-CRE-luciferase monitoring beginning 5–6 DPE when sleep defects are evident. In these experiments, neuronal CRE-luciferase activity is compared between animals with or without the mutHtt transgene. Animals lacking either the UAS-FLP or FRT-CRE-luci transgenes exhibit minimal activity as expected.
Interestingly, there is negligible change in CRE activity when F27B is expressed (Fig. 6E, red) relative to controls (Fig. 6E, gray). In contrast, neuronal M64 expression depresses and disrupts CRE-luciferase activity patterns (Fig. 6F, green). The daytime peaks are split or absent, and there is a reduction in amplitude relative to controls (Fig. 6F, gray). The nighttime peaks of activity are reduced in amplitude but appear otherwise normal in M64 flies. These data demonstrate that, despite genetic evidence implicating PKA elevation accompanying mutHtt expression, dCREB2 activity is not increased, and is likely depressed in neurons when sleep defects are present. Together, our data suggest that PKA activity may be elevated in HD concurrent with a decrease in dCREB2 activity.
Discussion
The identification of prodromal disease states and their biomarkers is a shared concern across neurodegenerative conditions because early therapeutic intervention is presumably most effective in limiting later order pathology. Sleep disruptions present in HD carriers in the earliest stages of disease and have been detected in animal HD models, but the molecular pathways underlying these sleep defects have not been identified. Here, we use multiple fly models of HD to connect early adult sleep and activity disruptions with the cAMP/PKA signaling pathway. We show that early-onset sleep fragmentation, nighttime hyperactivity and altered sleep architecture accompany diminished sleep homeostasis in fly models. Neuronal expression of mutHtt is sufficient to induce these phenotypes. These sleep and activity phenotypes manifest early in adulthood and precede climbing and memory defects, suggesting that sleep is particularly sensitive to disturbances of cell and molecular dysfunction induced by mutHtt. Abnormal sleep characteristics are evident in humans with HD (5,8,74), even in early stages of disease (2,3). Decreasing PKA signaling partially or fully suppresses all of these phenotypes, suggesting a relationship between aberrant signaling, sleep and early disease progression. The early onset of these sleep defects suggest that they may be a systemic readout of primary molecular dysfunction in HD.
The pattern of sleep and activity disturbances in HD model flies is consistent with elevations in cAMP/PKA signaling (24–27). The inhibition of dopaminergic signaling in adults suppresses the sleep and activity phenotypes, demonstrating that they are reversible. HD model flies also exhibit reduced responses to caffeine, a PDE inhibitor in flies, and they show elevated cAMP levels in the head. Strong neuronal mutHtt expression results in developmental lethality, which is suppressed by genetic reduction in PKA activity. Genetic reductions in PKA also suppress sleep and activity phenotypes accompanying weaker pan-neuronal mutHtt expression, and extend the median lifespan of HD model flies. Notably, suppression of developmental lethality is a reliable predictor of whether sleep phenotypes will be suppressed. Both developmental lethality and early-onset sleep disruptions present opportunities to screen for potential pharmacological or genetic modifiers.
Sleep has two major, partially independent regulatory mechanisms: circadian regulation, which limits sleep behavior to ecologically appropriate times, and homeostatic regulation, which controls how sleep pressure accumulates during wake and dissipates during sleep. When homeostatic mechanisms are intact, extended wakefulness or SD results in compensatory ‘rebound’ sleep that is longer and more consolidated than at baseline (17,18). Our data show that HD flies either experience further sleep loss in the night period following deprivation, in the case of the M36 model, or that fragmentation of nighttime sleep becomes more pronounced, in the case of the F27B model. We are the first to report disrupted sleep homeostasis and increased fragmentation following SD in HD model flies. These disruptions of sleep homeostasis may mean that neuronal mutHtt expression confers a heightened sensitivity to external disturbances of sleep or that recovery from SD is compromised in HD.
Genetic down-regulation of PKA signaling in HD flies restores the amount and consolidation of recovery sleep, suppressing fragmentation before and after SD. Co-expression of the regulatory subunit PKAr*BDK35 with F27B returns recovery sleep to control levels and prevents increase in night bout number or duration before and after deprivation. Likewise, PKA-C2 RNAi suppresses fragmentation of baseline and post-deprivation sleep in mutHtt F27B flies, as well as restoring the amount of recovery sleep. PKA-C2 RNAi suppresses sleep-deprivation-induced mortality in our strong mutHttN-term model, M64, and renders baseline and recovery sleep indistinguishable from controls. Earlier work in Drosophila has shown that inducibly overexpressing PKA-CI results in accumulated sleep debt that is discharged when PKA induction ends, indicating that high PKA activity in otherwise healthy animals can occlude homeostatic output (25). Taken all together, our data and previously reported work are consistent with the possibility that PKA is an important player in sleep homeostasis, perhaps through a role in GPCR signaling and cAMP signal transduction.
There are parallels between the sleep abnormalities in HD patients and those seen in elderly populations (75). This raises the question whether accelerated aging is part of HD pathogenesis (47). Shared sleep and activity phenotypes between our young HD models and aged wild-type flies include increases in bout number and daytime sleep (76), decreases in bout duration and disruptions in sleep homeostasis (77). Intriguingly, reducing PKA is effective in ameliorating aging as well as mutHtt-associated phenotypes. In aged flies, genetic reductions in PKA activity reduce circadian instability and arrhythmia, consolidate sleep (78) and suppress age-related memory impairment without altering lifespan (64). Decreasing PKA also improves aspects of health in aged mammals (64,79,80). PKA activity is not elevated in the heads of aged flies, however (64), suggesting that age-related increases in PKA signaling may occur downstream of PKA, perhaps by reduced phosphatase activity or diminished substrate degradation. Alternatively, reducing PKA may counteract dysregulation of a PKA-independent signaling pathway that shares substrates with PKA. Decreasing PKA in HD model flies may work by a similar, as-yet unidentified mechanism as it does in aged flies. The efficacy of PKA reductions in ameliorating mutHtt- and age-related phenotypes suggest PKA dysregulation may be a shared molecular disruption in aging and HD pathogenesis.
We show that decreasing PKA signaling in HD model flies suppresses several disease-related phenotypes. At the same time, in vivo CRE reporters suggest that dCREB2 activity is unchanged or mildly reduced in HD model flies. Our reporter data are consistent with reports of mutHtt interfering with CREB co-activators (81–91), but appear to be at odds with the beneficial effects of reducing PKA in the fly HD models. One possibility is that in HD, CREB activity becomes dissociated from PKA activity. Several groups have reported that elevations in cAMP and PKA are not correlated with increased CREB activity in HD model systems. Ariano et al. (92) showed that cAMP is elevated in the striata of five different HD models at the onset of motor symptoms, while expression of known CREB targets D1R and D2R is reduced. Alberch and colleagues (56) have demonstrated that PKA activity is elevated in hippocampi from Stage IV HD patients and in R6/1 and R6/2 mice prior to the onset of motor dysfunction. Intriguingly, this increase in PKA activity was not associated with increased S133-P CREB, although phosphorylation of other, membrane-associated PKA targets was elevated. Subsequent work by this group shows that nuclear PKA activity is unaffected in R6/1 mice at the same time point when membrane-associated PKA targets exhibit increased phosphorylation (57). Our findings are consistent with a model in which PKA activity increases phosphorylation of non-CREB targets but is accompanied by a depression in CREB activity. This dissociation of PKA and dCREB2 may represent a parallel mechanism of CREB dysfunction in HD pathogenesis.
There is a report of decreased cAMP and CREB phosphorylation in the striatum and cerebral cortex of HdhQ111 knock-in model mice well before mutHtt aggregates and neurodegeneration are evident (93). One likely reason for the apparent discrepancy between their results and ours is that our genetic manipulations are pan-neuronal, and our cAMP measurements are made from whole heads. The basis of the differential susceptibility of subsets of neurons to HD pathology is unclear, and it is possible that cAMP/PKA/CREB signaling is selectively altered in the tissues most susceptible to HD pathology. Indeed, cerebellar cAMP is unchanged in HdhQ111 knock-in mice even at 16 months (93). This suggests that the effects of mutHtt expression on alterations in cAMP/PKA signaling may be region- or tissue-specific. Because our current work relies primarily upon pan-neuronal genetic manipulations using the UAS-Gal4 system, mutHtt and PKA signaling are altered during the same developmental periods. It is possible that the co-expression of PKA with mutHtt prevents a developmental defect that results in sleep and activity deficits in adults. Reversal of sleep deficits in adult HD models by acute administration of a dopaminergic signaling inhibitor 3IY (Supplementary Material, Figs S4 and S5) suggests that this is not the case, and that sleep alterations are an early-adult onset phenotype.
Several open questions remain for future experimentation. Can reducing PKA pathway signaling only in adults reverse sleep/activity deficits and ameliorate later onset phenotypes? Is cAMP/PKA/dCREB2 signaling differentially affected in sleep/wake circuits versus the whole brain? When does the dissociation of cAMP/PKA and dCREB2 take place? The ever-expanding genetic toolkit in Drosophila makes these tractable questions. To determine whether altering cAMP/PKA signaling in adults can reverse sleep/activity deficits as well as later onset phenotypes, we could inducibly alter adenylyl cyclase, PKA components or PDE expression in adult HD model flies. Alternatively, we could modulate the activity of this pathway selectively in sleep/wake circuits. Additionally, by manipulating excitability of defined subsets of the neuronal sleep circuitry in HD model flies, for example, we could genetically induce sleep in adults at defined time points and look for delay of climbing deficits, neurodegeneration or extension of lifespan. These intersectional genetic manipulations will require generating novel lines that either constitutively express mutHtt or express mutHtt under the control of an alternative to the Gal4-UAS system.
Circadian disturbances are a conserved feature across HD patients (5) and HD model systems, including mice (5,94,95) and sheep (14). In the R6/2 mice, there is decreased expression of central clock genes mPer2 and Bmal1 in several brain regions (5). Importantly, Morton and colleagues (96) demonstrated that electrophysiological function and mPer1 transcription patterns in the suprachiasmatic nucleus (SCN) of R6/2 mice are normal in vitro, indicating that circadian anomalies arise from circuit dysfunction, not cell autonomous molecular problems. Pharmacological imposition of sleep with a short-acting benzodiazepine improved motor and cognitive performance, extended lifespan and partially restored SCN circadian gene expression. The authors attribute their results to the imposition of regular sleep–wake cycles, concluding that sleep abnormalities outside the central clock may feed back onto circadian circuitry, inducing aberrations of circadian rhythms (96). These results suggest that a pathologic mechanism extrinsic to the SCN underlies circadian and sleep abnormalities in HD, both of which may contribute to cognitive dysfunction, and that increasing sleep may provide a significant therapeutic opportunity in HD.
In this report, we present data that link early-onset sleep and activity abnormalities in HD models to disruptions of PKA/dCREB2 signaling. Suppression of developmental lethality by PKA manipulations, along with suppression of sleep and activity deficits, suggests that disturbed PKA/CREB signaling are early events in HD pathogenesis. PKA is also a molecular connection between sleep homeostasis and circadian regulation, the key regulatory elements of sleep behavior. Sleep deficits impair cognitive function in neurologically normal people (97) and have been linked to metabolic and hormonal disruptions (98) and may have broad consequences for maintaining health in people affected by HD. We propose that modulation of PKA activity might provide a pharmacological target to ameliorate sleep and circadian dysfunction in HD. Increasing sleep in canonical memory mutants can improve cognitive performance without specifically rescuing the molecular lesion underlying the memory defects (99), demonstrating that sleep plays a central role in modulating neural plasticity. Therefore, improving even subclinical sleep deficits could have an array of benefits for HD patients and their caregivers.
Materials and Methods
Stocks
Homozygous yw; pUAS-Htt128QFL full-length [M36] and pUAS-Htt128Q1-208 N-terminal insertion lines [F27B], [M64] and hemizyogus [NtX] were generous gifts of Dr Juan Botas (Baylor College of Medicine) and are described in (43). UAS-driven transgenic flies (PKA-C1RNAi line 101524, PKA-C2RNAi line 108424 and PKA-RIIRNAi line 39437) all on chromosome II were obtained from the Vienna Drosophila Resource Center (VDRC) (53). The UAS-PKAr*BDK35 (III) (stock #35550) (100), UAS-Epac1-camps (stock #25407) and the UAS-FLP (stock #4539) (101) lines were obtained from the Bloomington Drosophila Stock Center (Bloomington, IA, USA). The elavc155-Gal4, a gift of Dr Kanae Iijima-Ando (Thomas Jefferson University), resides on the X chromosome. Additional lab stocks include the weaker second chromosome insertion elavG28-GAL4/CyO line, HSP70-Gal4 line, CRE-luciferase (pJY785-3) line and FRT-CRE-luciferase (pJY2036-4) lines.
All stocks were backcrossed five times into a Cantonized w iso(CJ1) lab stock known colloquially as 2202U and abbreviated as 2U. A double balancer stock [w(2U); Sp/CyO; Sb/Ser] was used to produce the following homozygous stocks:
w(2U); Sp/CyO; pUAS-Htt128QN-term [M64]
w(2U); Sp/CyO; pUAS-Htt128QN-term F27B [F27B]
w(2U); pUAS-PKA-C1RNAi; Sb/Ser
w(2U); pUAS-PKA-C2RNAi; Sb/Ser
w(2U); Sp/CyO; pUAS-PKAr*
w(2U); pUAS-PKA-RIIRNAi; Sb/Ser
w(2U); UAS-FLP; Sb/Ser
w(2U); Epac1-camps; Sb/Ser
Balanced homozygous stocks were crossed together to produce doubly homozygous lines with potential rescue transgenes on II and HD transgenes on III.
Developmental lethality assessment and rescue
Fifty elavc155-Gal4 (C155) virgin females were mated to 25 homozygous males of either mutHttN-term [M64] or [F27B] genotypes, and progeny were assayed for viability. Parents were moved to new vials every 24 h and vials were maintained at 22°C. The numbers of pupae were counted 12 days after the beginning of the mating. Assessment of progress of metamorphosis was done visually and qualitatively. The C155/+; II; F27B/+ pupae were in a 4:1 ratio with C155/+; II; M64/+ pupae and metamorphosis advanced further in C155/+; II; F27B/+ pupae. To determine whether C155/+; II; mutHttN-term/+ were viable in low frequencies, the direction of the matings was reversed and the scale increased. Females but not males produced by this mating carry both C155 and the mutHttN-term transgene, permitting determination of viability ratios. Approximately 1:100 flies was a C155/w; II; F27B/III female (13 of >1100 scored), but no females eclosed from the M64 mating. For rescue experiments, 50 virgin elavc155-Gal4 virgin females were mated to 25 males of the doubly homozygous w(2U); (Rescue Transgene); UAS-Htt128QNT [M64] or [F27B] genotypes to assay viability. Pupae were counted and scored as before. If adults eclosed and survived 3 DPE, the rescue transgene was scored as providing complete rescue.
Sleep experiments
Virgin females were isolated within hours post-eclosion using CO2 anesthesia and placed in vials containing standard cornmeal-molasses-agar food in cohorts of 6–20 flies. Flies were socialized for 24–48 h following selection before being placed in TriKinetics (Waltham, MA, USA) monitors in incubators at 22°C. Data were excluded from analysis for 36–48 h after the end of CO2 anesthesia to eliminate the effects of anesthesia on sleep. All monitoring was performed under 12:12 light:dark conditions at 22°C with 60% humidity unless another temperature is specified. SD was performed using a SNAP (48) for 12 h at 22°C during the dark period after two complete days of baseline monitoring. Recovery from SD was monitored for 72 h following end of deprivation. All experiments were performed with a minimum n = 16 for each genotype; data show representative results of experiments that have been replicated at least once.
Sleep analysis
Sleep and activity data were collected by the TriKinetics Drosophila Activity Monitoring System (DAMS) and analyzed using an Excel-based Macro (30). Percent changes in sleep were calculated as described in (25). Briefly, sleep lost following deprivation was determined by subtracting baseline nighttime sleep from nighttime sleep during deprivation. Daytime sleep regained was determined by subtracting baseline daytime sleep from daytime sleep in the first 12 h following deprivation. Data are expressed as averages ± SEM. Statistical significance was determined by two-tailed Student's t-test; *P < 0.5; **P < 0.1, ***P < 0.005.
Longevity assays
Flies were collected within 24 h of eclosion and kept together in mixed sex groups for 2 days to ensure that females had mated, as virgin females have been reported to have extended lifespan relative to mated females (102). Males and females were then separated under CO2 anesthesia and counted into three groups of 15–20 flies/vial. Vials were flipped three times weekly and flies that escaped were removed from analysis. Longevity assays were maintained at 22°C under 12:12 L:D conditions. Experiments were repeated at least twice and the results combined to generate final figures.
In vivo CRE-luciferase assays
In vivo luciferase assays were performed as previously described (70,73). Briefly, circadian-entrained flies were loaded under light CO2 anesthesia into a 96-well microtiter plate containing luciferin media: 1% agar 5% sucrose food containing d-Luciferin (Gold BioTechnology). For CRE-luci reporter assays, 5 mm d-Luciferin was used, and 25 mm d-Luciferin was used for FRT-CRE-luci reporter assays. Plates were maintained at 22°C under a 12:12 LD cycle, and automated luminescence measurements were taken hourly using a Packard TopCount luminescence counter (PerkinElmer). For all light conditions, plates were in the dark during detection periods (∼20 min/h). The first 24 h of data are excluded from analysis to allow flies to recover from anesthesia unless otherwise specified. A smoothing function is applied such that each data point represents the average of three measurements. For comparisons of overall activity between groups, the mean hourly reading is calculated for each fly and compared using Student's t-test.
Supplementary Material
Funding
This work was supported by the National Institutes of Health (MH067774 and NS09541) and funding from the CHDI/HighQ Foundation. A.K.T. was partially supported through the Neurosciences Training Program NIH Training Grant T32GM007507.
Supplementary Material
Acknowledgements
The authors wish to thank members of the Yin Lab, particularly Kaitlin Dye, for their comments on this work. E.D.G. is grateful to Dr Richard Daniels and Dr Carin Loewen of Dr Barry Ganetzky's laboratory, Dr Jamie Elliott and Dr Grace Boekhoff-Falk for discussions of this manuscript. We thank Dr Juan Botas (Baylor College of Medicine) for HD model lines, Dr Kanae Iijima-Ando (Thomas Jefferson University) for the c155 driver line and the Vienna Drosophila Resource Center for RNAi lines. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study.
Conflict of Interest statement. None declared.
References
- 1.Ross C.A., Tabrizi S.J. (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol., 10, 83–98. [DOI] [PubMed] [Google Scholar]
- 2.Goodman A.O.G., Rogers L., Pilsworth S., McAllister C.J., Shneerson J.M., Morton A.J., Barker R.A. (2011) Asymptomatic sleep abnormalities are a common early feature in patients with Huntington's disease. Curr. Neurol. Neurosci. Rep., 11, 211–217. [DOI] [PubMed] [Google Scholar]
- 3.Arnulf I., Nielsen J., Lohmann E., Schiefer J., Schieffer J., Wild E., Jennum P., Konofal E., Walker M., Oudiette D. et al. (2008) Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol., 65, 482–488. [DOI] [PubMed] [Google Scholar]
- 4.Cuturic M., Abramson R.K., Vallini D., Frank E.M., Shamsnia M. (2009) Sleep patterns in patients with Huntington's disease and their unaffected first-degree relatives: a brief report. Behav. Sleep Med., 7, 245–254. [DOI] [PubMed] [Google Scholar]
- 5.Morton A.J., Wood N.I., Hastings M.H., Hurelbrink C., Barker R.A., Maywood E.S. (2005) Disintegration of the sleep–wake cycle and circadian timing in Huntington's disease. J. Neurosci. Off. J. Soc. Neurosci., 25, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aziz N.A., Anguelova G.V., Marinus J., Lammers G.J., Roos R.A.C. (2010) Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington's disease. Parkinsonism Relat. Disord., 16, 345–350. [DOI] [PubMed] [Google Scholar]
- 7.Videnovic A., Leurgans S., Fan W., Jaglin J., Shannon K.M. (2009) Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat. Disord., 15, 471–474. [DOI] [PubMed] [Google Scholar]
- 8.Hansotia P., Wall R., Berendes J. (1985) Sleep disturbances and severity of Huntington's disease. Neurology, 35, 1672–1674. [DOI] [PubMed] [Google Scholar]
- 9.Wiegand M., Möller A.A., Lauer C.J., Stolz S., Schreiber W., Dose M., Krieg J.C. (1991) Nocturnal sleep in Huntington's disease. J. Neurol., 238, 203–208. [DOI] [PubMed] [Google Scholar]
- 10.Soneson C., Fontes M., Zhou Y., Denisov V., Paulsen J.S., Kirik D., Petersén A. and Huntington Study Group PREDICT-HD Investigators. (2010) Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol. Dis., 40, 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzales E.D., Yin J.C. (2010) Drosophila models of Huntington's disease exhibit sleep abnormalities. PLoS Curr., 2, pii RR1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh D.H., Kudo T., Truong D., Wu Y., Colwell C.S. (2013) The Q175 mouse model of Huntington's disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PloS One, 8, e69993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantor S., Szabo L., Varga J., Cuesta M., Morton A.J. (2013) Progressive sleep and electroencephalogram changes in mice carrying the Huntington's disease mutation. Brain J. Neurol., 136, 2147–2158. [DOI] [PubMed] [Google Scholar]
- 14.Morton A.J., Rudiger S.R., Wood N.I., Sawiak S.J., Brown G.C., McLaughlan C.J., Kuchel T.R., Snell R.G., Faull R.L.M., Bawden C.S. (2014) Early and progressive circadian abnormalities in Huntington's disease sheep are unmasked by social environment. Hum. Mol., 23, 3375–3383. [DOI] [PubMed] [Google Scholar]
- 15.Hendricks J.C., Sehgal A. (2004) Why a fly? Using Drosophila to understand the genetics of circadian rhythms and sleep. Sleep, 27, 334–342. [DOI] [PubMed] [Google Scholar]
- 16.Cirelli C. (2009) The genetic and molecular regulation of sleep: from fruit flies to humans. Nat. Rev. Neurosci., 10, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber R., Hill S.L., Holladay C., Biesiadecki M., Tononi G., Cirelli C. (2004) Sleep homeostasis in Drosophila melanogaster. Sleep, 27, 628–639. [DOI] [PubMed] [Google Scholar]
- 18.Saper C.B., Cano G., Scammell T.E. (2005) Homeostatic, circadian, and emotional regulation of sleep. J. Comp. Neurol., 493, 92–98. [DOI] [PubMed] [Google Scholar]
- 19.Cirelli C., Bushey D., Hill S., Huber R., Kreber R., Ganetzky B., Tononi G. (2005) Reduced sleep in Drosophila Shaker mutants. Nature, 434, 1087–1092. [DOI] [PubMed] [Google Scholar]
- 20.Hendricks J.C., Finn S.M., Panckeri K.A., Chavkin J., Williams J.A., Sehgal A., Pack A.I. (2000) Rest in Drosophila is a sleep-like state. Neuron, 25, 129–138. [DOI] [PubMed] [Google Scholar]
- 21.Shaw P.J., Cirelli C., Greenspan R.J., Tononi G. (2000) Correlates of sleep and waking in Drosophila melanogaster. Science, 287, 1834–1837. [DOI] [PubMed] [Google Scholar]
- 22.Harbison S.T., Mackay T.F.C., Anholt R.R.H. (2009) Understanding the neurogenetics of sleep: progress from Drosophila. Trends Genet. TIG, 25, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sehgal A., Mignot E. (2011) Genetics of sleep and sleep disorders. Cell, 146, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendricks J.C., Williams J.A., Panckeri K., Kirk D., Tello M., Yin J.C., Sehgal A. (2001) A non-circadian role for cAMP signaling and CREB activity in Drosophila rest homeostasis. Nat. Neurosci., 4, 1108–1115. [DOI] [PubMed] [Google Scholar]
- 25.Joiner W.J., Crocker A., White B.H., Sehgal A. (2006) Sleep in Drosophila is regulated by adult mushroom bodies. Nature, 441, 757–760. [DOI] [PubMed] [Google Scholar]
- 26.Crocker A., Sehgal A. (2008) Octopamine regulates sleep in Drosophila through protein kinase A-dependent mechanisms. J. Neurosci., 28, 9377–9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu M.N., Ho K., Crocker A., Yue Z., Koh K., Sehgal A. (2009) The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J. Neurosci. Off. J. Soc. Neurosci., 29, 11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capece M.L., Lydic R. (1997) cAMP and protein kinase A modulate cholinergic rapid eye movement sleep generation. Am. J. Physiol., 273, R1430–R1440. [DOI] [PubMed] [Google Scholar]
- 29.Graves L.A., Hellman K., Veasey S., Blendy J.A., Pack A.I., Abel T. (2003) Genetic evidence for a role of CREB in sustained cortical arousal. J. Neurophysiol., 90, 1152–1159. [DOI] [PubMed] [Google Scholar]
- 30.Hellman K., Hernandez P., Park A., Abel T. (2010) Genetic evidence for a role for protein kinase A in the maintenance of sleep and thalamocortical oscillations. Sleep, 33, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo J., Phan T.X., Yang Y., Garelick M.G., Storm D.R. (2013) Increases in cAMP, MAPK activity, and CREB phosphorylation during REM sleep: implications for REM sleep and memory consolidation. J. Neurosci. Off. J. Soc. Neurosci., 33, 6460–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sands W.A., Palmer T.M. (2008) Regulating gene transcription in response to cyclic AMP elevation. Cell. Signal., 20, 460–466. [DOI] [PubMed] [Google Scholar]
- 33.Schulte J., Littleton J.T. (2011) The biological function of the Huntingtin protein and its relevance to Huntington's disease pathology. Curr. Trends Neurol., 5, 65–78. [PMC free article] [PubMed] [Google Scholar]
- 34.Zuccato C., Valenza M., Cattaneo E. (2010) Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol. Rev., 90, 905–981. [DOI] [PubMed] [Google Scholar]
- 35.Lee C.Y.D., Cantle J.P., Yang X.W. (2013) Genetic manipulations of mutant huntingtin in mice: new insights into Huntington's disease pathogenesis. FEBS J., 280, 4382–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W. et al. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell, 87, 493–506. [DOI] [PubMed] [Google Scholar]
- 37.Li J.Y., Popovic N., Brundin P. (2005) The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. NeuroRx J. Am. Soc. Exp. Neurother., 2, 447–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell, 90, 537–548. [DOI] [PubMed] [Google Scholar]
- 39.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science, 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- 40.Vonsattel J.P.G. (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. (Berl.), 115, 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pouladi M.A., Morton A.J., Hayden M.R. (2013) Choosing an animal model for the study of Huntington's disease. Nat. Rev. Neurosci., 14, 708–721. [DOI] [PubMed] [Google Scholar]
- 42.Kaltenbach L.S., Romero E., Becklin R.R., Chettier R., Bell R., Phansalkar A., Strand A., Torcassi C., Savage J., Hurlburt A. et al. (2007) Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet., 3, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romero E., Cha G.-H., Verstreken P., Ly C.V., Hughes R.E., Bellen H.J., Botas J. (2008) Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron, 57, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee W.-C.M., Yoshihara M., Littleton J.T. (2004) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc. Natl Acad. Sci. USA, 101, 3224–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu X.-H., Mattis V.B., Wang N., Al-Ramahi I., van den Berg N., Fratantoni S.A., Waldvogel H., Greiner E., Osmand A., Elzein K. et al. (2014) Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington's disease. Sci. Transl. Med., 6, 268ra178. [DOI] [PubMed] [Google Scholar]
- 46.Andretic R., Shaw P.J. (2005) Essentials of sleep recordings in Drosophila: moving beyond sleep time. Methods Enzymol., 393, 759–772. [DOI] [PubMed] [Google Scholar]
- 47.Morton A.J. (2013) Circadian and sleep disorder in Huntington's disease. Exp. Neurol., 243, 34–44. [DOI] [PubMed] [Google Scholar]
- 48.Shaw P.J., Tononi G., Greenspan R.J., Robinson D.F. (2002) Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature, 417, 287–291. [DOI] [PubMed] [Google Scholar]
- 49.Wu M.N., Koh K., Yue Z., Joiner W.J., Sehgal A. (2008) A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep, 31, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seugnet L., Suzuki Y., Thimgan M., Donlea J., Gimbel S.I., Gottschalk L., Duntley S.P., Shaw P.J. (2009) Identifying sleep regulatory genes using a Drosophila model of insomnia. J. Neurosci. Off. J. Soc. Neurosci., 29, 7148–7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheeba V., Fogle K.J., Kaneko M., Rashid S., Chou Y.-T., Sharma V.K., Holmes T.C. (2008) Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr. Biol., 18, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lebestky T., Chang J.-S.C., Dankert H., Zelnik L., Kim Y.-C., Han K.-A., Wolf F.W., Perona P., Anderson D.J. (2009) Two different forms of arousal in Drosophila are oppositely regulated by the dopamine D1 receptor ortholog DopR via distinct neural circuits. Neuron, 64, 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dietzl G., Chen D., Schnorrer F., Su K.-C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature, 448, 151–156. [DOI] [PubMed] [Google Scholar]
- 54.Li W., Ohlmeyer J.T., Lane M.E., Kalderon D. (1995) Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell, 80, 553–562. [DOI] [PubMed] [Google Scholar]
- 55.Chintapalli V.R., Wang J., Dow J.A.T. (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet., 39, 715–720. [DOI] [PubMed] [Google Scholar]
- 56.Giralt A., Saavedra A., Carretón O., Xifró X., Alberch J., Pérez-Navarro E. (2011) Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington's disease. Hum. Mol. Genet., 20, 4232–4247. [DOI] [PubMed] [Google Scholar]
- 57.Giralt A., Saavedra A., Carretón O., Arumí H., Tyebji S., Alberch J., Pérez-Navarro E. (2013) PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington's disease mouse model. Hippocampus, 23, 684–695. [DOI] [PubMed] [Google Scholar]
- 58.Tully T., Quinn W.G. (1985) Classical conditioning and retention in normal and mutant Drosophila melanogaster. J. Comp. Physiol. [A], 157, 263–277. [DOI] [PubMed] [Google Scholar]
- 59.Drier E.A., Tello M.K., Cowan M., Wu P., Blace N., Sacktor T.C., Yin J.C.P. (2002) Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat. Neurosci., 5, 316–324. [DOI] [PubMed] [Google Scholar]
- 60.Skoulakis E.M., Davis R.L. (1996) Olfactory learning deficits in mutants for leonardo, a Drosophila gene encoding a 14-3-3 protein. Neuron, 17, 931–944. [DOI] [PubMed] [Google Scholar]
- 61.Beck C.D., Schroeder B., Davis R.L. (2000) Learning performance of normal and mutant Drosophila after repeated conditioning trials with discrete stimuli. J. Neurosci. Off. J. Soc. Neurosci., 20, 2944–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis R.L., Cherry J., Dauwalder B., Han P.L., Skoulakis E. (1995) The cyclic AMP system and Drosophila learning. Mol. Cell. Biochem., 149–150, 271–278. [DOI] [PubMed] [Google Scholar]
- 63.Li W., Tully T., Kalderon D. (1996) Effects of a conditional Drosophila PKA mutant on olfactory learning and memory. Learn. Mem., 2, 320–333. [DOI] [PubMed] [Google Scholar]
- 64.Yamazaki D., Horiuchi J., Nakagami Y., Nagano S., Tamura T., Saitoe M. (2007) The Drosophila DCO mutation suppresses age-related memory impairment without affecting lifespan. Nat. Neurosci., 10, 478–484. [DOI] [PubMed] [Google Scholar]
- 65.Lonze B.E., Ginty D.D. (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron, 35, 605–623. [DOI] [PubMed] [Google Scholar]
- 66.Shaywitz A.J., Greenberg M.E. (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem., 68, 821–861. [DOI] [PubMed] [Google Scholar]
- 67.Sakamoto K., Karelina K., Obrietan K. (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem., 116, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin J.C., Wallach J.S., Wilder E.L., Klingensmith J., Dang D., Perrimon N., Zhou H., Tully T., Quinn W.G. (1995) A Drosophila CREB/CREM homolog encodes multiple isoforms, including a cyclic AMP-dependent protein kinase-responsive transcriptional activator and antagonist. Mol. Cell. Biol., 15, 5123–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tubon T.C. Jr, Zhang J., Friedman E.L., Jin H., Gonzales E.D., Zhou H., Drier D., Gerstner J.R., Paulson E.A., Fropf R. et al. (2013) dCREB2-mediated enhancement of memory formation. J. Neurosci. Off. J. Soc. Neurosci., 33, 7475–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belvin M.P., Zhou H., Yin J.C. (1999) The Drosophila dCREB2 gene affects the circadian clock. Neuron, 22, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iijima-Ando K., Yin J.C.P. (2005) Transgenic cAMP response element reporter flies for monitoring circadian rhythms. Methods Enzymol., 393, 302–315. [DOI] [PubMed] [Google Scholar]
- 72.Cha J.-H.J. (2007) Transcriptional signatures in Huntington's disease. Prog. Neurobiol., 83, 228–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanenhaus A.K., Zhang J., Yin J.C.P. (2012) In vivo circadian oscillation of dCREB2 and NF-κB activity in the Drosophila nervous system. PloS One, 7, e45130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Silvestri R., Raffaele M., De Domenico P., Tisano A., Mento G., Casella C., Tripoli M.C., Serra S., Di Perri R. (1995) Sleep features in Tourette's syndrome, neuroacanthocytosis and Huntington's chorea. Neurophysiol. Clin., 25, 66–77. [DOI] [PubMed] [Google Scholar]
- 75.Ohayon M.M. (2004) Sleep and the elderly. J. Psychosom. Res., 56, 463–464. [DOI] [PubMed] [Google Scholar]
- 76.Koh K., Evans J.M., Hendricks J.C., Sehgal A. (2006) A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl Acad. Sci. USA, 103, 13843–13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brown M.K., Chan M.T., Zimmerman J.E., Pack A.I., Jackson N.E., Naidoo N. (2014) Aging induced endoplasmic reticulum stress alters sleep and sleep homeostasis. Neurobiol. Aging, 35, 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo W., Chen W.-F., Yue Z., Chen D., Sowcik M., Sehgal A., Zheng X. (2012) Old flies have a robust central oscillator but weaker behavioral rhythms that can be improved by genetic and environmental manipulations. Aging Cell, 11, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramos B.P., Birnbaum S.G., Lindenmayer I., Newton S.S., Duman R.S., Arnsten A.F.T. (2003) Dysregulation of protein kinase a signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron, 40, 835–845. [DOI] [PubMed] [Google Scholar]
- 80.Yamazaki D., Horiuchi J., Miyashita T., Saitoe M. (2010) Acute inhibition of PKA activity at old ages ameliorates age-related memory impairment in Drosophila. J. Neurosci. Off. J. Soc. Neurosci., 30, 15573–15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kazantsev A., Preisinger E., Dranovsky A., Goldgaber D., Housman D. (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl Acad. Sci. USA, 96, 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCampbell A., Taylor J.P., Taye A.A., Robitschek J., Li M., Walcott J., Merry D., Chai Y., Paulson H., Sobue G. et al. (2000) CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet., 9, 2197–2202. [DOI] [PubMed] [Google Scholar]
- 83.Nucifora F.C., Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L. et al. (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science, 291, 2423–2428. [DOI] [PubMed] [Google Scholar]
- 84.Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M. et al. (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature, 413, 739–743. [DOI] [PubMed] [Google Scholar]
- 85.Jiang H., Nucifora F.C., Ross C.A., DeFranco D.B. (2003) Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet., 12, 1–12. [DOI] [PubMed] [Google Scholar]
- 86.Sugars K.L., Brown R., Cook L.J., Swartz J., Rubinsztein D.C. (2004) Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J. Biol. Chem., 279, 4988–4999. [DOI] [PubMed] [Google Scholar]
- 87.Cong S.-Y., Pepers B.A., Evert B.O., Rubinsztein D.C., Roos R.A.C., van Ommen G.-J.B., Dorsman J.C. (2005) Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol. Cell. Neurosci., 30, 560–571. [PubMed] [Google Scholar]
- 88.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell, 127, 59–69. [DOI] [PubMed] [Google Scholar]
- 89.Jiang H., Poirier M.A., Liang Y., Pei Z., Weiskittel C.E., Smith W.W., DeFranco D.B., Ross C.A. (2006) Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis., 23, 543–551. [DOI] [PubMed] [Google Scholar]
- 90.Chaturvedi R.K., Hennessey T., Johri A., Tiwari S.K., Mishra D., Agarwal S., Kim Y.S., Beal M.F. (2012) Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington's disease. Hum. Mol. Genet., 21, 3474–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeong H., Cohen D.E., Cui L., Supinski A., Savas J.N., Mazzulli J.R., Yates J.R., Bordone L., Guarente L., Krainc D. (2012) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med., 18, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ariano M.A., Aronin N., Difiglia M., Tagle D.A., Sibley D.R., Leavitt B.R., Hayden M.R., Levine M.S. (2002) Striatal neurochemical changes in transgenic models of Huntington's disease. J. Neurosci. Res., 68, 716–729. [DOI] [PubMed] [Google Scholar]
- 93.Gines S., Seong I.S., Fossale E., Ivanova E., Trettel F., Gusella J.F., Wheeler V.C., Persichetti F., MacDonald M.E. (2003) Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum. Mol. Genet., 12, 497–508. [DOI] [PubMed] [Google Scholar]
- 94.Rudenko O., Tkach V., Berezin V., Bock E. (2009) Detection of early behavioral markers of Huntington's disease in R6/2 mice employing an automated social home cage. Behav. Brain Res., 203, 188–199. [DOI] [PubMed] [Google Scholar]
- 95.Kudo T., Schroeder A., Loh D.H., Kuljis D., Jordan M.C., Roos K.P., Colwell C.S. (2011) Dysfunctions in circadian behavior and physiology in mouse models of Huntington's disease. Exp. Neurol., 228, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pallier P.N., Maywood E.S., Zheng Z., Chesham J.E., Inyushkin A.N., Dyball R., Hastings M.H., Morton A.J. (2007) Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington's disease. J. Neurosci. Off. J. Soc. Neurosci., 27, 7869–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang G., Grone B., Colas D., Appelbaum L., Mourrain P. (2011) Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci., 34, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim T.W., Jeong J.-H., Hong S.-C. (2015) The impact of sleep and circadian disturbance on hormones and metabolism. Int. J. Endocrinol., 2015, 591729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dissel S., Angadi V., Kirszenblat L., Suzuki Y., Donlea J., Klose M., Koch Z., English D., Winsky-Sommerer R., van Swinderen B. et al. (2015) Sleep restores behavioral plasticity to Drosophila mutants. Curr. Biol., 25, 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kiger J.A., Eklund J.L., Younger S.H., O'Kane C.J. (1999) Transgenic inhibitors identify two roles for protein kinase A in Drosophila development. Genetics, 152, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duffy J.B., Harrison D.A., Perrimon N. (1998) Identifying loci required for follicular patterning using directed mosaics. Dev. Camb. Engl., 125, 2263–2271. [DOI] [PubMed] [Google Scholar]
- 102.Chapman T., Liddle L.F., Kalb J.M., Wolfner M.F., Partridge L. (1995) Cost of mating in Drosophila melanogaster females is mediated by male accessory gland products. Nature, 373, 241–244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.