

SUMMARY
Genetic studies have revealed significant overlaps of risk genes among psychiatric disorders. However, it is not clear how different mutations of the same gene contribute to different disorders. We characterized two lines of mutant mice with Shank3 mutations linked to ASD and schizophrenia. We found both shared and distinct synaptic and behavioral phenotypes. Mice with the ASD-linked InsG3680 mutation manifest striatal synaptic transmission defects before weaning age and impaired juvenile social interaction, coinciding with the early onset of ASD symptoms. On the other hand, adult mice carrying the schizophrenia-linked R1117X mutation show profound synaptic defects in prefrontal cortex and social dominance behavior. Furthermore, we found differential Shank3 mRNA stability and SHANK1/2 upregulation in these two lines. These data demonstrate that different alleles of the same gene may have distinct phenotypes at molecular, synaptic, and circuit levels in mice, which may inform exploration of these relationships in human patients.
INTRODUCTION
Although schizophrenia and autism are two different disorders (DSM-5), it has long been proposed that they share some common pathologies and symptoms (de Lacy and King, 2013). Currently, the etiologies of schizophrenia and autism are largely unknown, but recent human studies highlight the contribution of genetic risk factors to both disorders (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; De Rubeis et al., 2014). In particular, mutations in a group of genes linked to synaptic development, function, and plasticity were frequently identified in patients diagnosed with either schizophrenia or autism (Kenny et al., 2014), suggesting that genetic mutations leading to dysregulation of synaptic transmission play critical roles in the pathophysiology of both disorders (De Rubeis et al., 2014; Fromer et al., 2014). Interestingly, recent genetic studies further revealed significant overlaps of risk genes across major psychiatric disorders including schizophrenia, bipolar disorder, major depressive disorder, and autism (Lee et al., 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). Furthermore, large-scale exome sequencing of autism spectrum disorder (ASD) and schizophrenia patient DNA samples has identified many of the same genes in both disorders, suggesting that different mutations in the same gene can cause/contribute to different disorders (Guilmatre et al., 2014; McCarthy et al., 2014).
One such example is the SHANK3 gene (Boeckers et al., 1999; Naisbitt et al., 1999). SHANK family members share five main domain regions: N-terminal ankyrin repeats, SH3 domain, PDZ domain, proline-rich region, and a C-terminal SAM domain. Through these functional domains, SHANK interacts with many postsynaptic density (PSD) proteins. Most notably, SHANK binds to SAPAP, which in turn binds to PSD95 to form the PSD95/SAPAP/SHANK postsynaptic complex (Kim and Sheng, 2004). Together, these three groups of multi-domain proteins are proposed to form a key scaffold, orchestrating the assembly of the macromolecular postsynaptic signaling complex at glutamatergic synapses. This complex has been shown to play an important role in targeting, anchoring, and dynamically regulating synaptic localization of neurotransmitter receptors and signaling molecules (McAllister, 2007). SHANK is also connected to the mGluR pathway through its binding to Homer (Tu et al., 1999). In addition, given its link to actin-binding proteins, SHANK has been shown to regulate spine development (Roussignol et al., 2005; Sala et al., 2001).
Deletion of SHANK3 has been shown to be the cause of core neurodevelopmental and neurobehavioral deficits in Phelan-McDermid syndrome (PMS), an autism spectrum disorder with symptoms that include intellectual disability, autistic behaviors, hypotonia, and impaired development of speech and language (Wilson et al., 2003). Subsequent genetic screens also identified a variety of mutations in the SHANK3 gene in ASD patients not diagnosed with PMS including a guanine nucleotide insertion in exon 21 of SHANK3 gene (position 3680) from two brothers diagnosed with ASD accompanied by severe mental retardation (Durand et al., 2007; Gauthier et al., 2009; Moessner et al., 2007). These data implicate SHANK3 gene disruption/mutation as a monogenic cause of ASD. In support of these genetic findings, studies of Shank3 mutant mice from our laboratory and others have revealed various degrees of synaptic dysfunction and autistic-like behaviors (Bozdagi et al., 2010; Kouser et al., 2013; Peça et al., 2011; Wang et al., 2011b). In addition, duplication of the SHANK3 gene was found in patients diagnosed with bipolar disorders and mice with Shank3 overexpression exhibit synaptic dysfunction and manic-like phenotypes (Han et al., 2013).
Interestingly, a non-sense mutation of SHANK3 changing an arginine to stop codon (R1117X) was identified from three brothers diagnosed with schizophrenia/schizoaffective disorder between ages 16 and 21 without showing obvious autistic features during their childhood (Gauthier et al., 2010). The three brothers also had mild-to-moderate mental retardation, which is often seen, and generally more severe, in ASD patients with SHANK3 mutations. Understanding the mechanisms by which different mutations in the same gene lead to different disorders will likely shed light on both shared and unique neural mechanisms of these disorders. We therefore created two mutant mouse lines. The first line harbors the ASD patient-linked single guanine nucleotide (G) insertion at cDNA position 3680 and leads to a frameshift and downstream stop codon (InsG3680 mutation). The second line contains the schizophrenia patient-linked point mutation and changes arginine 1117 to a stop codon (R1117X mutation). We performed systematic comparisons between the two mutant lines at molecular, cellular, synaptic, and behavioral levels and found both distinct and shared defects in these two mutant models. In particular, we found that mutant mice with the ASD-linked InsG3680 mutation, but not with the schizophrenia-linked R1117X mutation, manifest defective synaptic transmission in the striatum before weaning age, as well as impaired juvenile social play behavior, coinciding with the early onset of ASD symptoms in human patients. On the other hand, adult mice with the R1117X mutation, but not with the InsG3680 mutation, show synaptic defects in prefrontal cortex, consistent with clinical findings implicating prefrontal cortex defects in schizophrenia patients. Biochemical studies revealed both common and differential defects in postsynaptic signaling complexes and differential compensatory mechanisms in these two mutant lines. Behaviorally, both lines of mutant mice exhibit anxiety-like behavior and social interaction deficits. However, InsG3680 mutant mice show stronger repetitive/compulsive grooming behavior, whereas R1117X mutant mice show stronger allogrooming and social dominance-like behavior. Together, our study potentially provides a mechanistic explanation on how distinct mutations of the Shank3 gene may lead to distinct molecular, synaptic, and circuit defects and relevant behavior abnormalities.
RESULTS
Distinct Effects of InsG3680 and R1117X Mutations on SHANK3 Protein and mRNA
Shank3 is a very complex gene at the transcript level because of multiple intragenic promotors and alternative splicing (Wang et al., 2011b; Wang et al., 2014b). Both InsG3680 and R1117X mutations are in exon 21, which is common to most if not all isoforms, and the two mutations were separated by only 325 nucleotides (Figure 1A). For InsG3680 mutation, we placed the G insertion at position 3680 in Shank3, which causes a frameshift and a stop codon immediately after the G insertion (Figures 1A and 1B). For the R1117X mutation, we changed arginine (R) codon “CGG” to stop codon “TGA” to introduce the “R” to “X” mutation at amino acid position 1117 as described in the finding from schizophrenia patients (Figures 1A and 1B). We generated a C57 B6/S129 Sv mixed background mice population for all experiments performed in this study unless otherwise specified. Both homozygous InsG3680 and R1117X Shank3 mutant mice are viable and fertile.
Figure 1. Genetically Engineered Mice with InsG3680 or R1117X Shank3 Mutation Differentially Express SHANK3 Protein and mRNA.
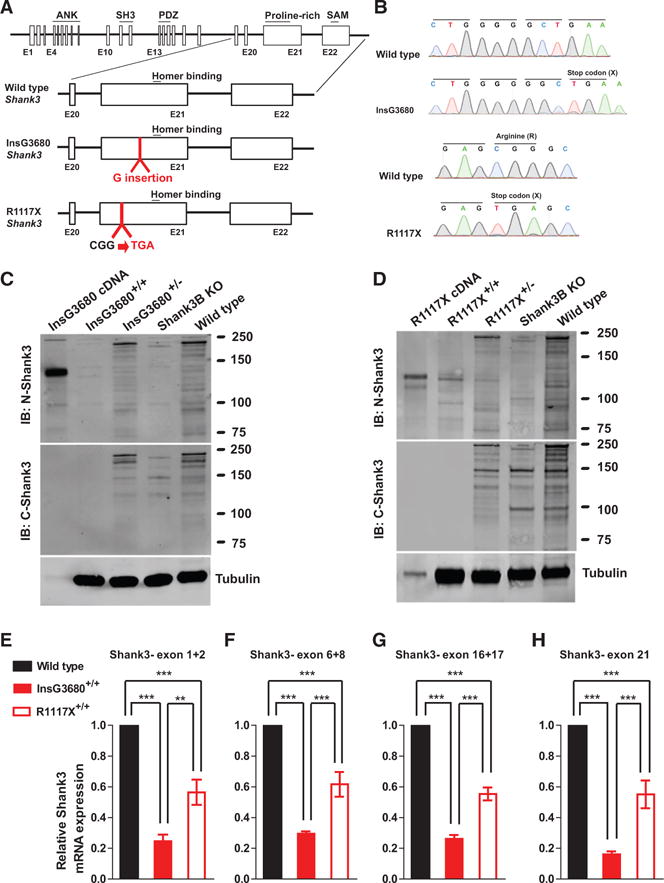
(A) Schematic diagram for wild-type Shank3-, InsG3680-, and R1117X-targeted Shank3 alleles. Gene structure of wild-type mouse Shank3 gene and magnified panels on the structure between exon 20 and exon 22 are shown below. Top: wild-type Shank3; middle: autism-associated InsG3680 Shank3 mutation with an insertion of “guanine” nucleotide at position 3680; bottom: schizophrenia-associated R1117X Shank3 mutation changing the “CGG” codon for arginine to a “TGA” stop codon.
(B) Representative sequencing chromatograms of wild-type and InsG3680 mutated alleles; wild-type and R1117X mutated alleles showing the point mutations.
(C) Representative western blots using striatal PSD fractions prepared from wild-type, Shank3B KO mice, InsG3680+/− mice, InsG3680+/+ mice; lysate from HEK239 cells expressing cDNA plasmid encoding the InsG3680 mutated Shank3. Note that neither the antibody against the N nor C terminus detected SHANK3 protein in InsG3680+/+ mice.
(D) Representative western blots using striatal PSD fractions prepared from wild-type, Shank3B KO mice, R1117X+/− mice, R1117X+/+ mice; lysate from HEK239 cells expressing cDNA plasmid encoding the R1117X mutated Shank3. Note that SHANK3 protein expression is almost abolished except for a prominent truncated isoform in the R1117X+/+ line that is detected with the antibody against the N terminus.
(E–H) The relative level of Shank3 mRNA in striatum is different between the two lines as quantified by primers amplifying exon 1 to 2, exon 6 to 8, 16 to 17, and partial exon 21 before both Shank3 mutation sites. Data are normalized to Gapdh mRNA and presented as mean ± SEM. **p < 0.01, ***p < 0.001; one-way ANOVA with Bonferroni post hoc test, WT mice (n = 5), R1117X+/+ mice (n = 5) and InsG3680+/+ mice (n = 5).
The predicted sizes of the resulting truncated proteins are 135 kDa for InsG3680 mutation and 122 kDa for R1117X mutation, respectively. To examine whether such truncated SHANK3 proteins exist in the brain, we prepared postsynaptic density (PSD) fractions from striatal tissue and probed with antibodies recognizing epitopes located at either the N terminus (located upstream of both mutations) or C terminus of the SHANK3 protein (located downstream of both mutations; Table S1). When probed with the C terminus antibody, no signals above 75 kDa were detected in striatal PSD preparations from either homozygous InsG3680 mutant mice (InsG3680+/+) or R1117X mutant mice (R1117X+/+) (Figures 1C and 1D), consistent with the fact that the C terminus antibody recognizes epitopes that are beyond the premature stop codons caused by the InsG3680 and R1117X mutations. In contrast, when probed with an antibody raised against the N terminus of SHANK3 (Neuromab 367/62), we detected truncated SHANK3 bands in striatal PSD preparations from the R1117X mutant mice with the major band matching the predicted 122 kDa expressed in HEK293 cells (Figure 1D). However, no clear signals were detected in striatal PSD preparations from InsG3680 mutant mice (Figure 1C). Similar results were obtained in PSD preparations from the cortex of R1117X and InsG3680 mutant mice using N terminus and C terminus antibodies. Together, our results reveal that R1117X and InsG3680 mutations have distinct effects on SHANK3 protein expression.
Since non-sense mutations could lead to reduced mRNA levels through non-sense-mediated decay (NMD) of abnormal mRNAs (Frischmeyer and Dietz, 1999), we examined Shank3 mRNA levels in the striatum of InsG3680 and R1117X mutant mice by quantitative real-time PCR. Due to the extensive alternative splicing of multiple coding exons in the Shank3 gene (Wang et al., 2011b), we designed four pairs of primers probing different coding regions to minimize the potential detection bias caused by alternative splicing. We consistently observed dramatically reduced levels of Shank3 mRNA from striatal tissue of InsG3680+/+ mice with all four pairs of probes (Figures 1E–1H). Interestingly, striatal tissues from R1117X+/+ mice showed significantly higher levels of Shank3 mRNA than from InsG3680+/+ mice (Figures 1E–1H). These results suggest that Shank3 mRNAs with the R1117X mutation are more stable than mRNAs with the InsG3680 mutation, consistent with our result that truncated SHANK3 proteins are present in R1117X+/+ mice. Together, our data suggest that the ASD-linked InsG3680 mutation results in an almost complete loss of SHANK3 protein, which is consistent with the full deletion of the SHANK3 gene identified in most Phelan-McDermid Syndrome patients (Bonaglia et al., 2011; Wilson et al., 2003). In contrast, the schizophrenia-linked R1117X mutation results in the generation of truncated SHANK3 protein, which could potentially either be partially functional or act in a dominant-negative form.
InsG3680 Mutants Exhibit Early Striatal Synaptic Transmission Defects and Impaired Social Interaction
Since ASD patients with SHANK3 mutations are usually diagnosed before the age of 3, whereas the schizophrenia patients carrying the R1117X mutation were diagnosed between ages 16 and 21, we wondered whether these two mutated mice exhibit any differential defects at early developmental stages. Since Shank3 is the only Shank family gene highly expressed in the striatum, we first examined the strength of evoked population spike responses in the striatum by performing field recordings in dorsolateral striatum at postnatal day 14 (P14). We found that InsG3680G+/+ mice have reduced field population spikes at postnatal day 14 (Figures 2A and 2B). In contrast, no difference was found between R1117X+/+ mice and their wild-type littermate controls (Figures 2D and 2E). Presynaptic function seems unaffected, as indicated by the relationship of stimulation intensity to amplitude of negative peak 1 (Figures 2C and 2F). We further measured other synaptic parameters including evoked AMPA to NMDA current ratio and miniature EPSCs by whole-cell patch-clamping recordings of dorsolateral striatal medium spiny neurons (MSNs). Through pharmacologically isolating evoked AMPA and NMDA current as described previously (Saal et al., 2003), we detected no change of AMPA to NMDA current ratio in both mutants as compared to their wild-type (Figure S1). No change of miniature EPSC (mEPSC) frequency was detected from both mutants as compared to wild-type controls (Figures 2G–2J). Interestingly, we observed significant increase of mEPSC amplitude in InsG3680+/+ mutants (Figures 2G and 2I), which may suggest a compensatory mechanism of MSNs in the presence of evoked synaptic transmission defects.
Figure 2. InsG3680+/+ but Not R1117X+/+ Mice Display Altered Striatal Synaptic Transmission at P14 and Reduced Social Interaction at P23.
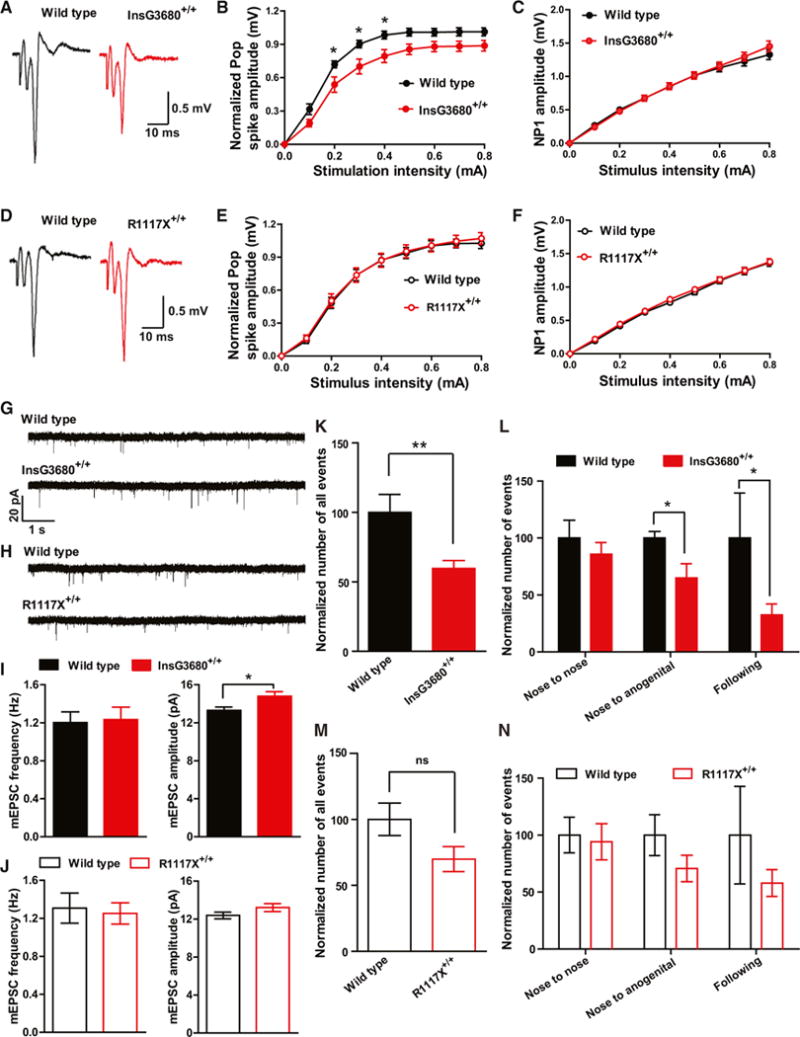
(A and D) Representative cortico-striatal pop spike traces recorded from P14 mice with indicated genotypes.
(B and C) Cortical-striatal input-output curve shows reduced pop spike responses in InsG3680+/+ mice compared to wild-type littermates. NP1 amplitude is similar between the genotypes, suggesting that presynaptic input is not different.
(E and F) Cortical-striatal input-output curve shows similar pop spike responses and NP1 amplitude between R1117X+/+ mice and their wild-type littermates. In (B), (C), (E), and (F), data are presented as mean ± SEM. *p < 0.05; two-way ANOVA repeated-measures with Bonferroni post hoc test. n = 12 slices from 4 pairs of littermates for each cohort.
(G and H) Typical AMPA receptor-mediated mEPSC traces recorded from P14 MSNs with indicated genotypes.
(I and J) Amplitude but not frequency of mEPSC is increased in InsG3680+/+ MSNs compared to wild-type. No change of amplitude or frequency of mEPSC in R1117X+/+ MSNs. In InsG3680 cohort, n = 29 neurons for wild-type; n = 29 neurons for InsG3680+/+ from 3 pairs of littermates; in R1117X cohort, n = 30 neurons for wild-type, n = 31 neurons for R1117X+/+ from 3 pairs of littermates. Data are presented as mean ± SEM; two-tailed t test.
(K and M) Total number of interaction events from mice pairs with indicated genotypes as normalized to wild-type control.
(L and N) Normalized number of categorized interaction events from mice pairs with indicated genotypes as normalized to wild-type control.
In (K), (L), (M), and (N), in R1117X cohort, n = 9 pairs of mice for wild-type; n = 9 pairs of mice for R1117X+/+; in InsG3680 cohort, n = 8 pairs of mice for wild-type; n = 10 pairs of mice for InsG3680+/+. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01; two-tailed t test.
We next examined how the two mutations may affect the expression of synaptic proteins in the striatum at P14. We found significantly reduced Homer protein in both R1117X+/+ and InsG3680+/+ mice (Figure S2). Interestingly, we found that in the striatum of P14 InsG3680+/+ mice, GluR1 is significantly upregulated, consistent with the increased AMPA-mediated mEPSC amplitude observed. Several other synaptic proteins including SynGAP, Shank2, and NMDA receptor subunits also show a trend of upregulation (Figure S2). Together, our biochemistry data and electrophysiological measurements from evoked and basal synaptic transmission suggest a complex scenario in young InsG3680+/+ mice in which striatal-evoked population spike responses are reduced but basal AMPA receptor-mediated mEPSC amplitude and GluR1 are increased. This may suggest that at this developmental stage, MSNs are trying to compensate for the defective evoked synaptic transmission by upregulating AMPR receptors in remaining functional synapses.
Although no basic synaptic transmission defects were detected in the striatum of P14 R1117X+/+ mice, we found that PSD93, SynGAP, and NMDA receptor subunits are reduced in a striatal synaptosomal plasma membrane (SPM) preparation from these mice (Figure S2). Furthermore, unlike in the P14 InsG3680+/+ mice, we did not see a trend of upregulation of any of the synaptic proteins tested. Together, these data suggest that the truncated R1117X Shank3 protein might be partially functional and its presence is sufficient to maintain basic normal synaptic transmission. To test this hypothesis, we evaluated the ability of the R1117X Shank3 mutant to rescue previously characterized cortico-striatal synaptic dysfunction in our Shank3B knockout mice (Peça et al., 2011) by using a cortico-striatal co-culture system. GFP plasmid alone or GFP plasmid together with R1117X Shank3 mutant plasmid were introduced into primary MSNs derived from Shank3B knockout mice through Nucleofection before plating. Transfected MSNs were then co-plated with cortical neurons. Whole-cell patch-clamp recordings of transfected neurons showed that expression of R1117X mutants significantly increased the frequency of mEPSCs in Shank3 knockout MSNs when compared to GFP control neurons (Figure S3). These in vitro data support the hypothesis that R1117X Shank3 mutant is partially functional in developing MSNs.
To evaluate the behavioral consequence caused by R1117X and InsG3680 Shank3 mutations at an early developmental stage, we first measured the maternal separation-induced ultrasonic vocalization behavior in pups between postnatal days 2 and 12. We found no significant differences in total number, total duration, mean duration, and peak amplitude of calls among genotypes. We next examined juvenile social play behavior at P23. We found significantly reduced numbers of all interactive events between mouse pairs carrying InsG3680+/+ mutation as compared to their wild-type littermate controls (Figure 2K). By categorizing the reciprocal play behavior into nose-to-nose, anogenital sniffing, and following behavior, we found both the nose-to-anogenital sniffing and following behavior are significantly reduced in InsG3680+/+ mice (Figure 2L). We found a similar trend in R1117X mice but it did not reach statistical significance (Figures 2M and 2N). Together, these data indicate an early-onset social interaction deficit in InsG3680+/+ Shank3 mutant mice.
Reduced Striatal Synaptic Transmission in Both Adult Mutant Lines
Although Shank3 is expressed in many brain regions, it is the only Shank family member enriched in the striatum. Our previous studies of young adult homozygous Shank3B knockout mice revealed significant synaptic defects in MSNs of the striatum including reduced pop spike responses by field recordings and reduced frequency and amplitude of miniature excitatory postsynaptic current (mEPSC) by whole-cell recordings (Peça et al., 2011). Here we compared the effects of InsG3680 and R1117X mutations on striatal synaptic function using electrophysiological recording on acutely isolated brain slices. We found both InsG3680 and R1117X homozygous but not heterozygous mice showed reduced pop spike responses (Figures 3A, 3B, 3D, and 3E). No differences of presynaptic function were observed among all genotypes as indicated by similar NP1 response among genotypes (Figures 3C and 3F).
Figure 3. Striatal Synaptic Transmission Is Reduced in Adult R1117X and InsG3680 Mutant Mice.
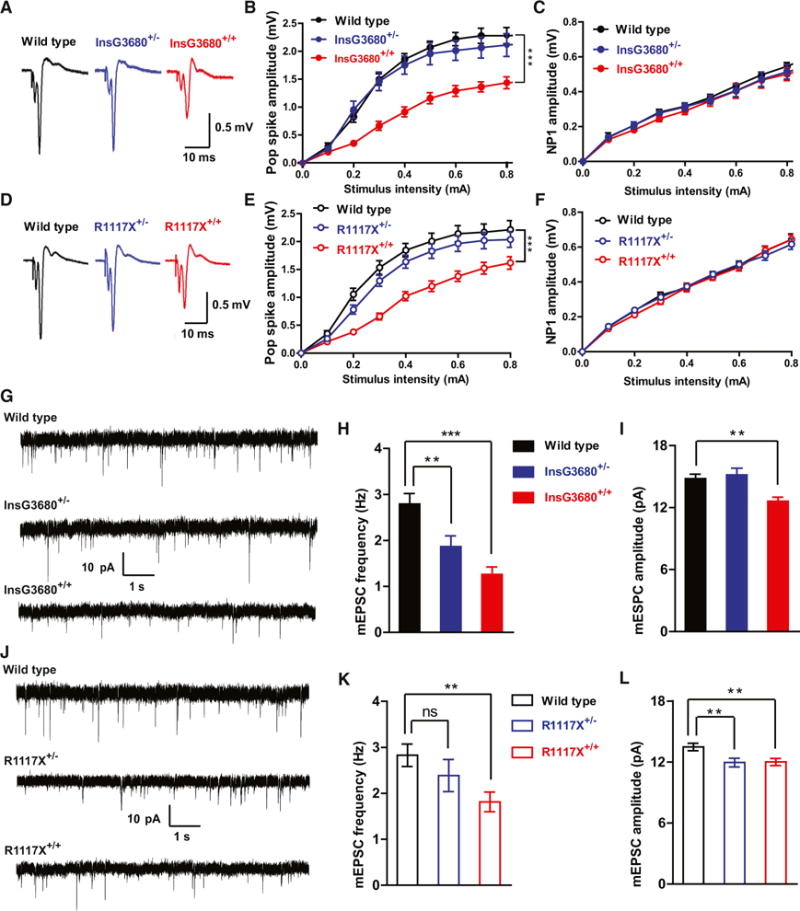
(A and D) Representative cortical-striatal pop spike traces from mice with indicated genotypes.
(B and E) Reduced striatal pop spike amplitude in both InsG3680+/+ and R1117X+/+ mice.
(C and F) No difference of NP1 amplitude among genotypes.
In (B), (C), (E), and (F) data are presented as mean ± SEM. ***p < 0.001; two-way ANOVA repeated-measures with Bonferroni post hoc test. n = 12 slices per genotype from 4 pairs of mice.
(G and J) Typical AMPA receptor-mediated mEPSC traces recorded from MSNs with indicated genotypes.
(H and I) mEPSC frequency of MSNs is reduced in both homozygous and heterozygous InsG3680 mutant mice compared to wild-type littermates. mEPSC amplitude is also reduced in homozygous InsG3680 mutant mice. n = 26 neurons for WT; n = 24 neurons for InsG3680+/−; n = 21 neurons for InsG3680+/+ from three pairs of mice.
(K and L) mEPSC frequency of MSNs is reduced in homozygous R1117X mutant mice compared to wild-type littermates. mEPSC amplitude is reduced in both homozygous and heterozygous R1117X mutant mice. n = 23 neurons for WT; n = 24 neurons for R1117X+/−; n = 26 neurons for R1117X+/+ from three pairs of mice. In (H), (I), (K), and (L), data are presented as mean ± SEM. **p < 0.01, ***p < 0.001; one-way ANOVA with Bonferroni post hoc test.
We further measured the AMPA receptor-mediated mEPSCs by performing whole-cell patch-clamping recordings on dorsolateral striatal MSNs. We found that MSNs of both InsG3680 homozygous and heterozygous mice showed significant reduction of mEPSC frequency (Figures 3G and 3H). This reduction of mEPSC frequency was also observed in R1117X homozygous mice (Figures 3J and 3K). There is also a small but statistically significant reduction of mEPSC amplitude in MSNs from InsG3680+/+ and R1117X+/+ homozygous as well as R1117X+/− mice (Figures 3I and 3L). In addition, we found significantly reduced NMDA receptor-mediated currents in both R1117X+/+ and InsG3680+/+ as compared to their wild-type controls (Figure 4). Together, these results suggest that both InsG3680 and R1117X mutations cause significant synaptic dysfunction in the adult striatum.
Figure 4. Reduced NMDA Receptors Mediated Synaptic Transmission in Both R1117X+/+and InsG3680+/+ Shank3 Mutant Mice.
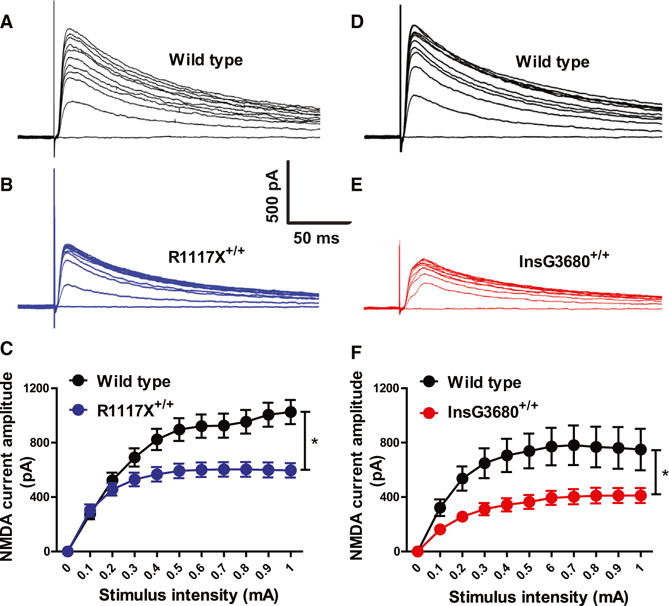
(A–C) Typical NMDA receptors mediated currents from striatal MSNs by stimulating corpus callosum and quantification. n = 16 neurons for wild-type; n = 18 neurons for R1117X+/+ from three pairs of littermates at 7 week’s age.
(D–F) Typical NMDA receptors mediated currents from striatal MSNs by stimulating corpus callosum and quantification. n = 14 neurons for wild-type; n = 12 neurons for InsG3680+/+ from three pairs of littermates at 8 week’s age. In both (C) and (F), data are presented as mean ± SEM. *p < 0.05; two-way ANOVA test.
Distinct Alteration of Synaptic Transmission in Prefrontal Cortex of R1117X Mutant Mice
Although pathological mechanisms of schizophrenia are still not known, numerous patient studies have implicated the dysfunction of prefrontal cortex (PFC) as an important pathogenic source in patients (Anderson et al., 1999). We found a significant reduction of mEPSC frequency in pyramidal neurons of mPFC in both R1117X+/− and R1117X+/+ mutant mice (Figures 5A and 5B). In addition, we observed a significant reduction of mEPSC amplitude in R1117X+/+ mice (Figure 5C). In contrast, no significant differences of either frequency or amplitude of mEPSC were found in InsG3680+/− or InsG3680+/+ mutant mice when compared to WT controls (Figures 5D–5F), although there is a trend of reduction in mEPSC frequency in InsG3680+/+ mutant mice. These results indicate that the R1117X mutation, but not the InsG3680 mutation, causes a profound deficit of synaptic transmission in mPFC.
Figure 5. Profound Cortical Synaptic Defects Manifest in Mice Carrying the Schizophrenia-Associated R1117X Mutation.
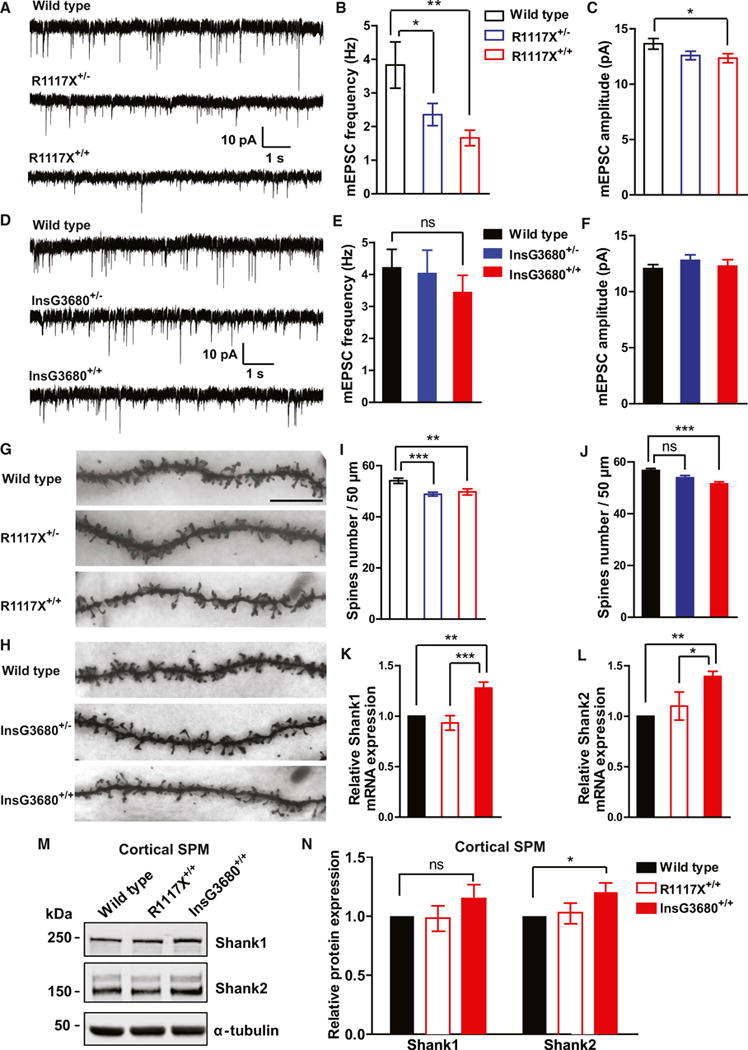
(A–C) Typical AMPA receptor-mediated mEPSC recordings in the prefrontal cortex and statistical results for R1117X mice. n = 13 neurons for WT; n = 20 neurons for R1117X+/−; n = 18 neurons for R1117X+/+ from three pairs of mice. Note the highly significant reduction of mEPSC frequency in both heterozygous and homozygous mice and a modest reduction of amplitude in homozygotes.
(D–F) Typical AMPA receptor-mediated mEPSC recordings in the prefrontal cortex and statistical results for InsG3680 cohort. n = 18 neurons for WT; n = 16 neurons for InsG3680+/−; n = 17 neurons for InsG3680+/+ from three pairs of mice. Both heterozygous and homozygous mice display comparable miniature events to wild-type mice.
In (B), (C), (E), and (F), data are presented as mean ± SEM. *p < 0.05, **p < 0.01; one-way ANOVA with Bonferroni post hoc test.
(G and H) Representative confocal images of secondary dendrites of pyramidal neurons from frontal association area of mice with indicated genotypes (scale bar, 10 μm).
(I and J) Quantification of spine number from neurons with indicated genotypes from three littermate pairs indicates reduced spine numbers in R1117X+/−, R1117X+/+, and InsG3680+/+ mice.
(K and L) mRNAs of Shank3 homologs Shank1 and Shank2 are upregulated in cortical tissue from InsG3680+/+ compared to wild-type mice. Data are normalized to Gapdh mRNA and presented as mean ± SEM. WT mice (n = 5), R1117X+/+ mice (n = 5), and InsG3680+/+ mice (n = 5). In all the panels, data were collected from 2-month-old mice. In (I), (J), (K), and (L), *p < 0.05, **p < 0.01, **p < 0.001; one-way ANOVA with Bonferroni post hoc test.
(M) Representative blots for proteins detected by specific antibodies in the cortical SPM fraction from adult wild-type, R1117X+/+, and InsG3680+/+ mice.
(N) Adult InsG3680+/+ but not R1117X+/+ mice show increased Shank2 expression in cortical tissue. Quantification of relative levels of proteins as normalized to tubulin from cortical SPM. Data are presented as mean ± SEM *p < 0.05; one sample t test. (n = 8 samples per protein per genotype, each n being pooled tissue from two mice).
One of the pathological findings in postmortem brains of schizophrenia patients is the reduction of spine density of layer 3 pyramidal neurons in the prefrontal cortex (Glantz and Lewis, 2000). To examine whether a similar defect exists in the two lines of Shank3 mutant mice, we used Golgi staining to measure the spine density of layer 2/3 pyramidal neurons in the frontal association area. We found a significant reduction of spine density in both R1117X heterozygous and homozygous mutant mice (Figures 5G and 5I). We also found a significant reduction of spine density in InsG3680 homozygous but not heterozygous mutant mice (Figures 5H and 5J).
All three SHANK family members are highly homologous and expressed in the cortex (Böckers et al., 2004; Lim et al., 1999; Peça et al., 2011). It has been reported that SHANK3 was upregulated in Shank2 knockout mice (Schmeisser et al., 2012). Thus, it is possible that SHANK1-2 may be upregulated in Shank3 mutants to compensate for the loss of SHANK3. To explore this possibility, we quantified the expression level of Shank1 and Shank2 mRNA in the cortex of InsG3680+/+ and R1117X+/+ mutant mice. We found both Shank1 and Shank2 mRNAs were significantly upregulated in InsG3680+/+ but not R1117X+/+ mutant mice (Figures 5K and 5L), suggesting that the two mutations have differential effects on Shank1 and Shank2 mRNA upregulation. Furthermore, we observed significantly upregulated SHANK2 protein and a trend of increased SHANK1 protein in the cortex of InsG3680+/+ but not R1117X+/+ mutant mice (Figures 5M and 5N). However, we did not observe upregulation of either mRNA or protein of Shank1 or Shank2 in the striatum of either mutant line. Together, these results suggest both mutation-specific and circuit-specific upregulation of other Shank family members, and the upregulation of SHANK1 and SHANK2 mRNAs and proteins in the cortex of InsG3680+/+ mutant mice may partially compensate for the loss of SHANK3 protein and thus alleviate synaptic defects in mPFC of InsG3680+/+ mutant mice.
Given the fact that patients carrying the schizophrenia-associated R1117X Shank3 mutation were diagnosed at their late adolescence stage (Gauthier et al., 2010), we examined whether there are synaptic defects in the mPFC of young mice (P14) from either mutant line. Similar to our findings in the mEPSC of adult InsG3680+/+ mice, we did not detect significant differences of either mEPSC frequency or amplitude in the mPFC of P14 InsG3680+/+ mice (Figures S4D–S4F). Interestingly, we detected a significant increase in mEPSC frequency in R1117X+/+ mice at P14 (Figures S4A–S4C), suggesting there is already some degree of aberrant synaptic connection/function in the mPFC region at this age, albeit different from adult defects.
We next examined synaptic protein expression in cortical SPM preparations at P14. As shown in Figure S4, we did not detect significant changes in synaptic protein levels except a small reduction of Homer protein in R1117X+/+ mice (Figure S4H). Interestingly, we observed a significant upregulation of Shank2 protein in InsG3680+/+ mice (Figure S4H), which we also observed in adult cortical SPM preparations of InsG3680+/+ mice (Figures 5M and 5N). Together, these data indicate much minor molecular defects at cortical synapses at P14 in both lines.
Alteration of PSD Composition in Adult R1117X and InsG3680 Mutant Mice
The PSD95-SAPAP-SHANK scaffolding complex has been proposed to play important roles in the trafficking, assembly, and anchoring of signaling proteins to the PSD as well as in regulating the dynamic plasticity of the PSD (Sheng and Kim, 2011). Our previous studies showed that several scaffolding proteins and glutamate receptor subunits were reduced in the striatal PSD of Shank3B knockout mice (Peça et al., 2011). Here we examined levels of several scaffolding and signaling proteins as well as glutamate receptor subunits in the SPM of InsG3680+/+ and R1117X+/+ mutant mice. In an SPM preparation from striatal tissue, we found that Homer1b/c is dramatically reduced in both InsG3680+/+ and R1117X+/+ mutant mice (~14% of WT; Figures 6A and 6B). This is consistent with the fact that the Homer binding domain of Shank3 is located downstream of the premature stop codons caused by InsG3680 and R1117X mutation (Tu et al., 1999). Interestingly, we found that in InsG3680 and R1117X heterozygous mice, which has about 50% of full-length Shank3 protein, Homer protein is also reduced to 50% of the wild-type level (Figure S5), indicating the dependence of Homer synaptic localization on Shank3 and their one-to-one stoichiometry.
Figure 6. InsG3680 and R1117X Mutant Mice Display Common and Differential Disruptions of Post-synaptic Signaling Complexes.
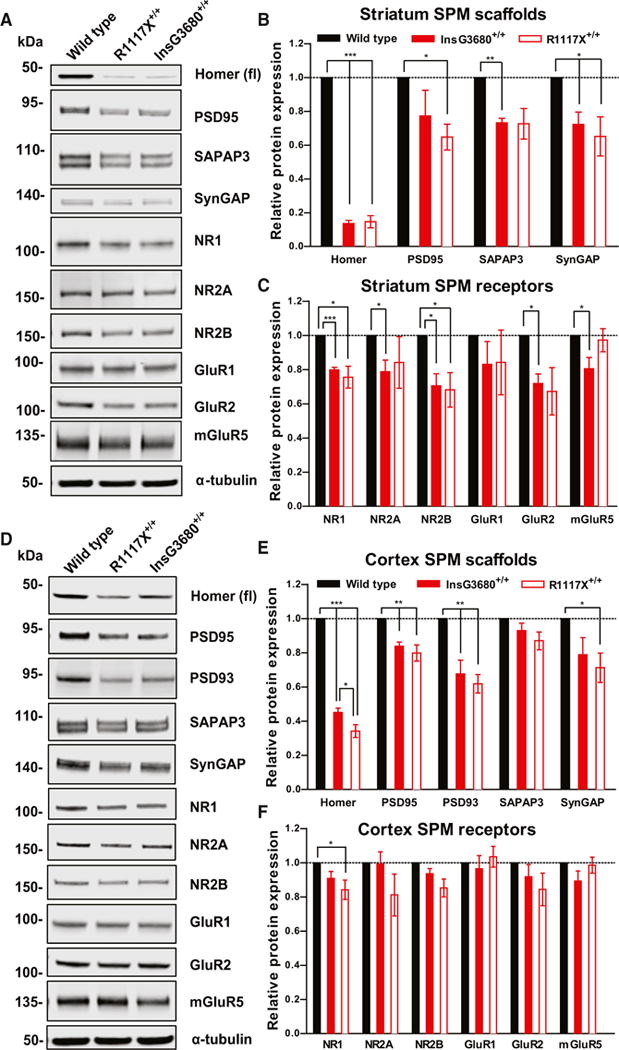
(A) Representative blots for proteins detected by specific antibodies in the striatal SPM fraction from wild-type, InsG3680+/+, and R1117X+/+ mice.
(B and C) Quantification of relative levels of proteins as normalized to tubulin protein expression from striatal SPM. (n = 4–6 samples per protein per genotype, each n being pooled tissue from three mice).
(D) Representative blots for proteins detected by specific antibodies in the cortical SPM fraction from wild-type, InsG3680+/+, and R1117X+/+ mice.
(E and F) Quantification of relative levels of proteins as normalized to tubulin protein expression from cortex SPM. (n = 4–11 samples per protein per genotype, each n being pooled tissue from two mice).
In (B), (C), (E), and (F), data are presented as mean ± SEM. *p < 0.05, **p < 0.01, **p < 0.001; one sample t test.
In general, we observed a very similar pattern of reduced synaptic protein in the striatal SPM of the two mutant lines. Syn-GAP1, PSD95, SAPAP3, NR1, NR2A, NR2B, and GluR2 are all either significantly reduced or have the similar trend of reduction in both mutant lines (Figures 6A–6C). However, a small reduction of mGluR5 protein was only observed in the InsG3680+/+ line (Figures 6A and 6C). This correlates with the trend for impaired LTD maintenance 15~25 min after DHPG application compared to wild-type (Figure S6D) and mildly impaired PPR increase after DHPG-LTD in InsG3680+/+ (Figures S6G and S6H) but not R1117X+/+ mice (Figures S6C, S6E, and S6F). These data are consistent with our electrophysiological results showing severe striatal synaptic defects in adults of both mutant lines.
In the cortex, we found that Homer1b/c, PSD 95, and PSD 93 were significantly reduced in both InsG3680+/+ and R1117X+/+ mutant mice as compared to WT controls (Figures 6D and 6E). Interestingly, the level of Homer1b/c reduction is slightly more significant in R1117X+/+ mutant mice than in InsG3680+/+ mice (Figure 6E). In addition, we observed significant reduction of NR1 and its close interacting partner SynGAP1 only in the R1117X+/+ mutant mice (Figures 6D–6F), in line with previous findings using postmortem cortex tissue from schizophrenia patients (Funk et al., 2009; Weickert et al., 2013). Together, these data indicate that although cortical synaptic transmission defects are very different in the adults of the two mutant lines, differences in molecular defects are not as dramatic, raising the possibility that developmental defects in connectivity may contribute significantly to observed synaptic transmission and behavioral defects.
InsG3680 and R1117X Mutant Mice Show Both Common and Differential Behavioral Phenotypes
As described above, our molecular and electrophysiological studies of InsG3680 and R1117X mutant mice showed that they have both shared and distinct synaptic defects. We next tested whether these synaptic defects are accompanied by behavioral changes. We found that both R1117X+/+ and InsG3680+/+ mice show significantly reduced explorative activity in an open field arena (total distance) when compared to wild-type littermates (Figures 7A and 7B). In addition, InsG3680+/− mice also showed significantly reduced activity (Figure 7A). Both lines of mutant mice show a very similar habituation time course compared to their wild-type littermates (Figures S7A and S7B), suggesting that the reduced explorative activity is caused by reduced locomotion rather than faster habituation of mutant mice. These reduced locomotion and hypoactive features in mutant mice were further supported by rotarod test findings showing that both R1117X+/+ and InsG3680+/+ Shank3 mice have impaired motor learning and coordination capability (Figures S8A and S8B).
Figure 7. InsG3680 and R1117X Mutant Mice Display Anxiety Behavior and Social Interaction Deficits.
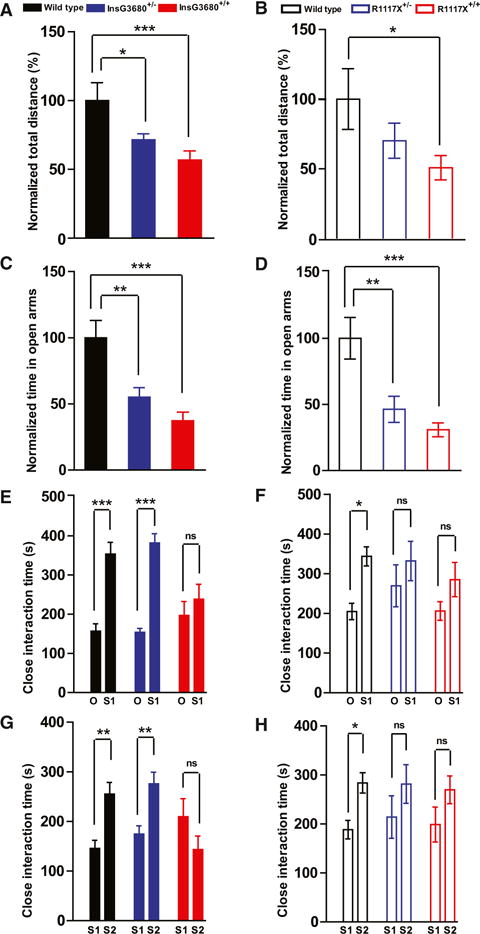
(A and B) Total distance traveled in the open field test as normalized to wild-type littermates.
(C and D) Time spent in the open arms in the elevated zero maze test as normalized to wild-type littermates.
In the InsG3680 cohort, n = 17 mice for wild-type; n = 19 mice for InsG3680+/−; n = 18 mice for InsG3680+/+ group. In the R1117X cohort, n = 15 mice for wild-type, n = 15 mice for R1117X+/−; n = 15 mice for R1117X+/+.
(E and F) Time spent on close interaction with an object (O) versus stranger mice (S1) in the phase II social interaction test.
(G and H) Time spent on close interaction with a familiar mouse (S1) versus stranger mouse (S2) in the phase III social interaction test.
In the InsG3680 cohort, n = 17 mice for wild-type; n = 19 mice for InsG3680+/−; n = 18 mice for InsG3680+/+ group; in the R1117X cohort, n = 23 for wild-type, n = 25 for R1117X+/−; n = 24 for R1117X+/+ group.
In all the panels, data are presented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Bonferroni post hoc test.
In addition to the hypoactive phenotype, both R1117X and InsG3680 Shank3 mutant mice spent much less time exploring the center in an open area test compared to wild-type littermates (Figures S7C and S7D), suggesting an anxiety-like behavior. In the elevated zero maze test, we found that both R1117X+/+ and InsG3680+/+ mice spent much less time in the open arms as compared to their wild-type littermates (Figures 7C and 7D). In addition, heterozygous mutants from both lines also spent less time in the open arms as compared to their wild-type littermates (Figures 7C and 7D). These results indicate that both R1117X and InsG3680G mutant mice exhibit increased levels of anxiety.
Pre-pulse inhibition (PPI) is commonly used to test sensorymotor gating function in animal models of schizophrenia, although defects in PPI are not specific to schizophrenia and are present in many other psychiatric disorders, both in patients and animal models (Swerdlow et al., 1993; Joober et al., 2002). We found that both R1117X+/+ and InsG3680+/+ mice show profound defects in acoustic startle response (Figures S8C and S8D). Thus, although we found significantly reduced PPI in InsG3680+/+ and slightly impaired PPI in R1117X+/+ mice (Figures S8E and S8F), the interpretation of these data is complicated by the significant defects in startle response, because any PPI results are primarily confounded by a decrease in absolute startle to a 120 dB auditory stimulus. In addition, we found no difference in performance in the T maze spontaneous alternation test between wild-type and either line of mutant mice, suggesting normal basic working memory in both lines of mutant mice.
Since impaired social interaction is one of the core features of ASD patients and social withdraw is a characteristic negative symptom in schizophrenia patients, we tested social interactions of R1117X and InsG3680 mutant mice with a slightly modified standard three chamber social interaction paradigm (Chao et al., 2010; Silverman et al., 2010). As expected, wild-type mice showed significant preference for strange mouse (S1) to a novel object (O) (Figures 7E and 7F). However, both InsG3680 and R1117X homozygous mutant mice showed no significant preference for other mice compared to the novel object side (Figures 7E and 7F). Interesting, heterozygous R1117X mutant mice, but not heterozygous InG3680 mutant mice, also displayed social interaction deficits (Figure 7F). Similarly, in the social novelty test, both homozygous and heterozygous R1117X mutant mice exhibited deficits, while only homozygous InsG3680 mutant mice showed the defect (Figures 7G and 7H). Together, these results demonstrated that both the InsG3680 mutation and R1117X mutation lead to social interaction deficits, similar to Shank3 mutant mice with deletion of either the ankyrin repeats or PDZ domain (Peça et al., 2011; Wang et al., 2011b).
Repetitive behavior/restricted interest is another key feature of ASD. Several mouse models of psychiatric disorders including ASD, OCD, and Tourette syndrome show repetitive grooming phenotypes and some of them develop skin lesions due to overgrooming (Karayannis et al., 2014; Peça et al., 2011; Rothwell et al., 2014; Welch et al., 2007). These repetitive/compulsive-like behaviors have been strongly linked to cortico-striatal-thalamo-cortical circuitry dysfunction (Ahmari et al., 2013; Burguière et al., 2013; Peça et al., 2011; Rothwell et al., 2014; Shmelkov et al., 2010; Welch et al., 2007). In our breeding colony, we found that 28.1% (18/64) of InsG3680+/+ mice develop lesions between 4 and 6 months of age (Figure 8A), a similar rate to our previous finding in Shank3B knockout mice (Peça et al., 2011). In contrast, only 8.7% (6/69) of R1117X+/+ mice developed skin lesions at the same age. Statistical analysis of lesion penetrance using chi-square test revealed significant difference between R1117X+/+ and InsG3680+/+ mice (p = 0.0036). No skin lesions were found in R1117X+/−, InsG3680+/− and wild-type groups. We next quantified the percentage of time R1117X and InsG3680 mice spent on grooming during a 2-hr session. We found that InsG3680+/+ mice spend twice as much time on grooming when compared to their WT littermates (Figure 8B), and a similar degree of increase in grooming time was also observed during the social interaction test. In contrast, R1117X+/+ mice did not show a statistically significant increase in grooming time compared to their WT littermates (Figure 8C), although a trend of increase was observed. These results suggest that only InsG3680+/+ mice show robust repetitive/compulsive grooming behavior.
Figure 8. InsG3680 Mice Show More Profound Repetitive Self-Grooming, whereas R1117X Mice Display Allogrooming and Dominance-like Behavior.
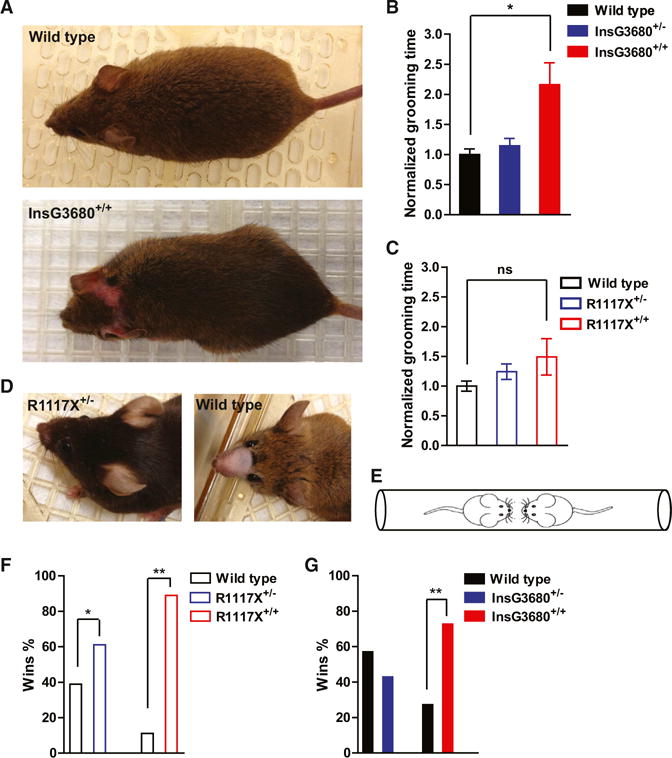
(A) Representative pictures from an adult wild-type and an InsG3680+/+ mouse that developed a lesion in the head/neck area.
(B and C) Time spent on grooming during 2 hr videotaping of indicated genotypes as normalized to their wild-type littermates. In the InsG3680 cohort, n = 9 mice for wild-type; n = 9 mice for InsG3680+/−; n = 10 mice from InsG3680+/+ group. In the R1117X cohort, n = 9 mice for wild-type, n = 9 mice for R1117X+/−; n = 9 mice for R1117X+/+. Data are presented as mean ± SEM. *p < 0.05; Kruskal-Wallis test with Dunn’s post hoc comparison.
(D) Representative pictures of an R1117X+/− mouse with intact facial hair and a wild-type mouse shaved by its cage mate as an indication for allogrooming behavior.
(E) Diagram of tube test task between two unfamiliar mice with different genotypes.
(F and G) Percentage of wins in test pairs between indicated genotypes, 11/18 (61%) of R1117X+/− versus WT; 16/18 (89%) of R1117X+/+ versus WT; 6/14 (43%) of InsG3680+/− versus WT; 8/11 (73%) of InsG3680+/+ versus WT. Note that both R1117X+/− and R1117X+/+ mice perform significantly above chance level. One-sample chi-square test was used to determine the significant difference. “*” indicates significantly different from an expected chance (50:50 win-lose outcome).
In our R1117X+/− × WT mating cages, we frequently observed partial or complete facial hair loss of WT mice without any lesions, suggesting allogrooming/barbering by R1117X+/− mice (Figure 8D). We confirmed that loss of whisker and facial hair was caused by allogrooming by a cage mate instead of selfgrooming by separating the mating pairs with the phenotype. We observed nearly full regrowth of facial hairs after weeks of single housing. Furthermore, pairing the recovered animals with their original cagemates induced robust loss of whisker and facial hair again within 2 weeks (data not shown). This allogrooming/barbering phenotype also occurred in R1117X+/− × R1117X+/− mating cages, in which one of the R1117X+/− mice and/or offspring would lose their facial hair. We observed allogrooming/barbering in 48% (14 of 29) mating cages with R1117X+/− mice. In contrast, we only observed allogrooming/barbering phenotype in 14% (5 of 37) mating cages with InsG3680+/− mice. Analysis of allogrooming phenotype penetrance using chi-square test revealed a significant difference between R1117X+/− and InsG3680+/− mice (p = 0.002). These results suggest that R1117X and InsG3680 mutations have differential effects on the expression of allogrooming/barbering phenotype.
Previous studies in rodents have implicated the impairment of PFC function in allogrooming/social dominance (Jiang-Xie et al., 2014; Wang et al., 2011a). Our electrophysiological findings from mPFC indicated that both R1117X+/− and R1117X+/+ have profound deficits of synaptic transmission (Figures 5A–5C). To further evaluate the social dominance phenotype, we performed standard tube test (Figure 8E) between wild-type versus heterozygous and wild-type versus homozygous mice in the two mutant lines. When the two stranger mice meet in the middle of the tube, the dominant mouse will advance and drive the other mouse out of the tube (Wang et al., 2011a). We found that both R1117X+/+ and R1117X+/− mice show much higher probability of winning the matches when tested with their wild-type opponents (Figure 8F). We also observed a significantly increased winning percentage in InsG3680+/+ but not in InsG3680+/− mice against their wild-type opponents (Figure 8G). These results are consistent with our electrophysiological and morphological findings in the prefrontal cortex showing synaptic deficits in both heterozygous and homozygous mice with R1117X mutation but only in homozygous mice with InsG3680 mutation.
DISCUSSION
Although schizophrenia and autism are two differential disorders based on phenomenological diagnosis (DSM-5), it has long been recognized that they share some common features and comorbidity (de Lacy and King, 2013; King and Lord, 2011). This is particularly true between child onset schizophrenia and autism (Meyer et al., 2011; Rapoport et al., 2009). For example, in a recent study, out of 46 schizophrenia patients with normal intelligence, about 50% met the diagnostic criteria of ASD (Unenge Hallerbäck et al., 2012). Recent human genetic studies have provided strong biological support for these clinical findings. GWAS, CNV, and exome sequencing have all identified many of the same genes in both disorders (Lee et al., 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Krystal and State, 2014). Despite strong clinical and genetic evidence, however, it is not clear how different mutations of the same gene may contribute to different disorders. In this study, we generated new mouse models that harbor the highly penetrant SHANK3 mutations found in ASD and schizophrenia patients. Our study revealed both distinct and shared defects in the two lines of mutant mice at molecular, synaptic, circuit, and behavioral levels (summarized in Table S2). Our results may provide, for the first time, some neurobiological insights on how different mutations in the SHANK3 gene may lead to mutation-specific defects and relevant behavior abnormalities.
Early Synaptic Transmission Defects in the Striatum of Mice with ASD-Associated Mutation
One of our interesting findings is that mice with the ASD-associated InsG3680 mutation exhibit earlier synaptic transmission defects when compared with mice with the schizophrenia-associated R1117X mutation. We found a reduction of population spike responses in P14 InsG3680+/+ mutant mice, indicating defects in evoked striatal synaptic transmission. The lack of obvious presynaptic defects suggests that there are fewer functional synapses in these mutant mice. Interestingly, we found a surprising increase of GluR1 protein and mEPSC amplitude in striatal MSNs from P14 InsG3680+/+ mutant mice. Along with the trend of upregulation of many other synaptic proteins (Shank2, SynGAP, NR1, and NR2) in the striatum of P14 InsG3680+/+ mutant mice, this may reflect a potential compensatory mechanism by the remaining functional synapses to counteract defects in evoked synaptic transmission.
A puzzling finding in P14 striatum of R1117X mutant mice is the reduction of PSD93, SynGAP, NR1, NR2A, and NR2B proteins but normal basic synaptic transmission including population spike responses, AMPA/NMDA ratio, mEPSC frequency, and amplitude. We do not have a clear answer regarding this discrepancy between the electrophysiological and biochemical findings. One possible explanation is that the remaining truncated R1117X Shank3 protein is partially functional in MSNs at this developmental stage, as suggested by our study in cultured neurons, and its presence is sufficient to maintain a basic level of synaptic transmission. However, it is also highly possible that striatal synaptic transmission in R1117X+/+ mutant mice at P14 is already mildly dysregulated and more extensive studies may reveal defects of other synaptic parameters.
Distinct Prefrontal Cortex Defects in R1117X Mutant Mice
Currently, the pathological mechanisms underlying schizophrenia are not well understood. However, clinical studies from human patients have implicated the dysfunction of prefrontal cortex (PFC), in particular dorsolateral prefrontal cortex (dlPFC), as an important cause of deficits in working memory, executive function, and social impairment (Anderson et al., 1999; Euston et al., 2012). In addition, postmortem studies have revealed reduced numbers of dendritic spines in pyramidal neurons of dlPFC region from schizophrenia patients (Glantz and Lewis, 2000). Rodents do not have an anatomically distinct dlPFC (Uylings et al., 2003). However, electrophysiological and behavioral studies suggest that medial prefrontal cortex (mPFC) in rodents may perform some equivalent functions to primate dlPFC at a rudimentary level (Seamans et al., 2008), such as working memory, decision making, and social interaction (Adhikari et al., 2010; Amodio and Frith, 2006; Wang et al., 2011a). Consistent with these findings, our study revealed profound synaptic transmission defects in mPFC of mice with the schizophrenia-associated R1117X mutation, but not with the ASD-associated InsG3680 mutation, indicating that these two mutations affect distinct circuits.
Since all three members of the Shank family genes are expressed in the cortex (Böckers et al., 2004; Lim et al., 1999; Peça et al., 2011), it is possible that they may have functional redundancy to compensate for the loss of other members. Very interestingly, we found that upregulation of Shank1 and Shank2 happens only in the cortex of InsG3680 mutants but not in R1117X mutants. The upregulation of Shank1-2 in the cortex of InsG3680 mutants may compensate for the loss of SHANK3 and thus alleviate synaptic defects. In addition, the truncated SHANK3 protein resulting from the R1117X mutation may have a dominant-negative effect, preventing SHANK1 and/or SHANK2, expressed at normal level, from assembling into the postsynaptic signaling complex. Such a dominant-negative effect would prevent compensation by Shank1 and Shank2 in R1117X mutants. Interestingly, Shank1 and Shank2 are normally not highly expressed in the striatum (Peça et al., 2011) and we did not observe upregulation of Shank1 and Shank2 in the striatum of either mutant line. Thus, upregulation and compensation by Shank1 and Shank2 is not only mutation specific, but also cell type/brain region specific, adding another layer of complexity to the study of neurobiological mechanisms underlying genetically overlapping psychiatric disorders.
Commonality and Differences in Behavioral Phenotypes
One of the most common phenotypes in mouse models with mutations of ASD risk genes is repetitive behavior, and previous studies have strongly linked repetitive/compulsive behaviors to defects in cortico-striatal-thalamo-cortical (CSTC) circuits (Burguière et al., 2013; Peça et al., 2011; Rothwell et al., 2014; Shmelkov et al., 2010; Welch et al., 2007). Similar to our reported Shank3B knockout mice, the repetitive/compulsive grooming behaviors in InsG3680 mutant mice are very robust. R1117X mutant mice do not show significant repetitive grooming behavior, although both mutant lines show striatal molecular and synaptic defects. There are several possible explanations for this phenotypic difference. First of all, the striatum is only one part of the CSTC circuit, and differential defects in other parts of the brain, such as the cortex, may significantly affect the behavioral output. Second, recent studies have begun to reveal specific changes in subcircuits/microcircuits of the CSTC that may underlie repetitive behaviors, and the two mutations may differentially affect these sub-circuits/micro-circuits. Finally and probably most importantly, striatal synaptic transmission defects at early developmental stages in InsG3680 mutants may lead to long-lasting alterations in striatal connectivity. These connectivity defects may lead to InsG3680-specific circuit dysfunction and overgrooming even though synaptic transmission defects are similar between the two mutant lines in adults.
A behavioral phenotype predominantly found in R1117X mutants is the allogrooming and social dominance behavior. Although the exact mechanisms/circuits involved in dominance behaviors in mice are still not well understood, recent studies have provided evidence implicating mPFC in social dominance (Lin et al., 2011; Wang et al., 2014a). Our studies demonstrated significant synaptic transmission defects in the mPFC of R1117X, but not InsG3680 mutant mice. These correlative data may provide a partial explanation for the difference in social dominance behavior between these two mutant lines.
Previous studies have demonstrated that two different mutations of the MECP2 gene in Rett Syndrome patients differentially affect the function of a key domain in MeCP2 and result in different severity of phenotypes in mice similar to the disease progression and symptoms observed in patients with these two mutations (Baker et al., 2013). Although it is difficult to directly correlate mouse behaviors with patient symptoms and diagnosis, our study at least provides neurobiological evidences and mechanisms that the two SHANK3 mutations associated with ASD and schizophrenia cause both common and differential defects at molecular, synaptic, and behavioral levels. More broadly, we demonstrated that different mutations of the same gene may elicit neurobiological changes at different developmental stages, brain regions, and cell types through a variety of potential mechanisms including differential mRNA stability, differential regulation of compensatory gene expression, and different degrees of signaling complex disruption. Thus, future detailed analysis of such mutations will help to gain a more precise understanding of synaptic development and function.
EXPERIMENTAL PROCEDURES
Animal Work Statement
All animal-related work was performed under the guidelines of Division of Comparative Medicine (DCM), with protocol (# 0513-044-16 of Feng laboratory and #1012-102-15 of Lewis laboratory) approved by Committee for Animal Care (CAC) of Massachusetts Institute of Technology and was consistent with the Guide for Care and Use of Laboratory Animals, National Research Council 1996 (institutional animal welfare assurance no. A-3125-01). Only aged-matched male mice were used for all behavioral experiments; all other tests included age-matched males and females in proportional contribution across groups.
Behavioral Studies
All behavioral studies were carried out and analyzed with experimenter blinded to genotype. For all assays, mice were habituated in the test facility for 1 hr prior to starting the task. Each cohort of mice was used for maximally three behavioral tests with at least 5 days’ break between tasks.
Electrophysiological Recordings
All electrophysiological measurements were performed and analyzed with experimenter blinded to genotype.
Data Analysis
All comparisons between groups were collected from littermate animals with experiments performed at the same time. All results were presented as mean ± SEM and were analyzed statistically using Student’s t test, one-way or two-way analysis of variance with proper post hoc test as specified in legend of each figure (GraphPad Prism, RRID: rid_000081, SCR_002798). *p < 0.05; **p < 0.01; ***p < 0.001.
Supplementary Material
Highlights.
Major psychiatric disorders share many risk genes
Two mouse lines with ASD-linked and schizophrenia-linked mutations were studied
Two mutant lines show both shared and distinct synaptic and behavioral phenotypes
Both developmental and molecular differences were detected in the two mutant lines
Acknowledgments
We thank Ms. Peimin Qi of MIT transgenic facility for support with ES cell injection, Ms. Elaine Kun, Heather Zaniewski and Ms. Triana Dalia for help with animal care and experiments, Alexandra Krol for critical reading of the manuscript and the Feng lab members for helpful discussion. We are also grateful to Dr. Paul Worley (Johns Hopkins University) for generous sharing their SHANK3 antibodies and Dr. Hailan Hu (Shanghai Institute of Neuroscience, Chinese Academy of Sciences) for advice on tube test assay. This work is supported by the Simons Center for the Social Brain at MIT and the Stanley Center for Psychiatric Research at Broad Institute. G.F. is supported by the National Institute of Mental Health (5R01MH097104), the Poitras Center for Affective Disorders Research at MIT, Stanley Center for Psychiatric Research at Broad Institute of MIT and Harvard, Nancy Lurie Marks Family Foundation, Simons Foundation Autism Research Initiative (SFARI), and Simons Center for the Social Brain at MIT. Additional support is provided by Robert Buxton, Marshall Tulin, and John and Valerie Stelling. Yang Zhou is supported by postdoc fellowships from the Simons Center for the Social Brain at MIT, Nancy Lurie Marks Family Foundation and Shenzhen Overseas Innovation Team Project (No. KQTD20140630180249366). P.M. was supported by the Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard and a doctoral fellowship from the Portuguese Foundation for Science and Technology (SFRH/BD/33894/2009). B.B. is supported by postdoc fellowships from the Simons Center for the Social Brain at MIT and the Autism Science Foundation. Mingjie Zhang is supported by grants from Research Grant Council of Hong Kong (AoE/M09/12). Z.F. is supported by Stanley Center for Psychiatric Research at Broad Institute of MIT and Harvard and NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation. T.K. is supported by the Leonardo Kolleg, the Dr. Ernst und Anita Bauer-Stiftung and the Henry E. Singleton Fellowship. F.Z. is supported by the NIMH through a NIH Director’s Pioneer Award (DP1-MH100706), the NINDS through a NIH Transformative R01 grant (R01-NS 07312401), NSF Waterman Award, the Keck, Damon Runyon, Searle Scholars, Klingenstein, Vallee, Merkin, and Simons Foundations, and Bob Metcalfe.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, eight figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2015.11.023.
AUTHOR CONTRIBUTIONS
Yang Z. and G.F. conceived the study. Yang Z., Yongdi Z., Mingjie Z., and G.F. designed the experiments and oversaw the project. Yang Z. generated the mutant mice. F.Z. and N.S. provided resource at the early stage of this project. T.K. and Yang Z. performed the biochemical studies of SPM/PSD and data analysis. Z.F., P.M., Yang Z., M.S.V.d.G., and Chenchen L. performed electrophysiological recording. D.W. imaged Golgi slices and analyzed the spine density. Congyi L. performed cultured neuron experiments. X.Z. and Yang Z. conducted the mRNA Q-PCR assay. Yang Z., X.Z., Menglong Z., D.W., B.B., A.A., S.N., M.L., and M.W. designed, performed and analyzed the behavioral experiments. Yang Z. and G.F. wrote the manuscript with inputs from all authors. Tobias Kaiser, Patrícia Monteiro and Xiangyu Zhang contributed equally to this work.
References
- Adhikari A, Topiwala MA, Gordon JA. Synchronized activity between the ventral hippocampus and the medial prefrontal cortex during anxiety. Neuron. 2010;65:257–269. doi: 10.1016/j.neuron.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, Gordon JA, Hen R. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013;340:1234–1239. doi: 10.1126/science.1234733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006;7:268–277. doi: 10.1038/nrn1884. [DOI] [PubMed] [Google Scholar]
- Anderson SW, Bechara A, Damasio H, Tranel D, Damasio AR. Impairment of social and moral behavior related to early damage in human prefrontal cortex. Nat Neurosci. 1999;2:1032–1037. doi: 10.1038/14833. [DOI] [PubMed] [Google Scholar]
- Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckers TM, Segger-Junius M, Iglauer P, Bockmann J, Gundelfinger ED, Kreutz MR, Richter D, Kindler S, Kreienkamp HJ. Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol Cell Neurosci. 2004;26:182–190. doi: 10.1016/j.mcn.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, Wex H, Langnaese K, Bockmann J, Garner CC, Gundelfinger ED. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J Neurosci. 1999;19:6506–6518. doi: 10.1523/JNEUROSCI.19-15-06506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Beri S, De Agostini C, Novara F, Fichera M, Grillo L, Galesi O, Vetro A, Ciccone R, et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 2011;7:e1002173. doi: 10.1371/journal.pgen.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguière E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340:1243–1246. doi: 10.1126/science.1232380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lacy N, King BH. Revisiting the relationship between autism and schizophrenia: toward an integrated neurobiology. Annu Rev Clin Psychol. 2013;9:555–587. doi: 10.1146/annurev-clinpsy-050212-185627. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euston DR, Gruber AJ, McNaughton BL. The role of medial prefrontal cortex in memory and decision making. Neuron. 2012;76:1057–1070. doi: 10.1016/j.neuron.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk AJ, Rumbaugh G, Harotunian V, McCullumsmith RE, Meador-Woodruff JH. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport. 2009;20:1019–1022. doi: 10.1097/WNR.0b013e32832d30d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Spiegelman D, Piton A, Lafrenière RG, Laurent S, St-Onge J, Lapointe L, Hamdan FF, Cossette P, Mottron L, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Côté M, Noreau A, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci USA. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol. 2014;74:113–122. doi: 10.1002/dneu.22128. [DOI] [PubMed] [Google Scholar]
- Han K, Holder JL, Jr, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503:72–77. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang-Xie LF, Liao HM, Chen CH, Chen YT, Ho SY, Lu DH, Lee LJ, Liou HH, Fu WM, Gau SS. Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol Autism. 2014;5:32. doi: 10.1186/2040-2392-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joober R, Zarate JM, Rouleau GA, Skamene E, Boksa P. Provisional mapping of quantitative trait loci modulating the acoustic startle response and prepulse inhibition of acoustic startle. Neuropsychopharmacology. 2002;27:765–781. doi: 10.1016/S0893-133X(02)00333-0. [DOI] [PubMed] [Google Scholar]
- Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R, Héron D, Salomon D, Glessner J, Restituito S, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. 2014;511:236–240. doi: 10.1038/nature13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, Kelleher E, Ennis S, Tropea D, Anney R, et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2014;19:872–879. doi: 10.1038/mp.2013.127. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- King BH, Lord C. Is schizophrenia on the autism spectrum? Brain Res. 2011;1380:34–41. doi: 10.1016/j.brainres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N, Liu S, Jaramillo TC, Bangash M, Xiao B, et al. Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J Neurosci. 2013;33:18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, State MW. Psychiatric disorders: diagnosis to therapy. Cell. 2014;157:201–214. doi: 10.1016/j.cell.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, Witte JS, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E. Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem. 1999;274:29510–29518. doi: 10.1074/jbc.274.41.29510. [DOI] [PubMed] [Google Scholar]
- Lin D, Boyle MP, Dollar P, Lee H, Lein ES, Perona P, Anderson DJ. Functional identification of an aggression locus in the mouse hypothalamus. Nature. 2011;470:221–226. doi: 10.1038/nature09736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N. Autism spectrum disorders and childhood-onset schizophrenia: clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry. 2009;48:10–18. doi: 10.1097/CHI.0b013e31818b1c63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci. 2005;25:3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Lapish CC, Durstewitz D. Comparing the prefrontal cortex of rats and primates: insights from electrophysiology. Neurotox Res. 2008;14:249–262. doi: 10.1007/BF03033814. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3:3. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, Shmelkov E, Kushner JS, Baljevic M, Dincheva I, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16:598–602. doi: 10.1038/nm.2125. 1p, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biol Psychiatry. 1993;33:298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Unenge Hallerbäck M, Lugnegård T, Gillberg C. Is autism spectrum disorder common in schizophrenia? Psychiatry Res. 2012;198:12–17. doi: 10.1016/j.psychres.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Uylings HB, Groenewegen HJ, Kolb B. Do rats have a prefrontal cortex? Behav Brain Res. 2003;146:3–17. doi: 10.1016/j.bbr.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Wang F, Zhu J, Zhu H, Zhang Q, Lin Z, Hu H. Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science. 2011a;334:693–697. doi: 10.1126/science.1209951. [DOI] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011b;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Kessels HW, Hu H. The mouse that roared: neural mechanisms of social hierarchy. Trends Neurosci. 2014a;37:674–682. doi: 10.1016/j.tins.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Wang X, Xu Q, Bey AL, Lee Y, Jiang YH. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol Autism. 2014b;5:30. doi: 10.1186/2040-2392-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, Newell KA, Pellen D, Huang XF, Catts SV, Weickert TW. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013;18:1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.