

Abstract
Inactivation of the RB protein is one of the most fundamental events in cancer. Coming to a molecular understanding of its function in normal cells and how it impedes cancer development has been challenging. Historically, the ability of RB to regulate the cell cycle placed it in a central role in proliferative control, and research focused on RB regulation of the E2F family of transcription factors. Remarkably, several recent studies have found additional tumour-suppressor functions of RB, including alternative roles in the cell cycle, maintenance of genome stability and apoptosis. These advances and new structural studies are combining to define the multifunctionality of RB.
The RB gene was cloned more than 25 years ago1. Since that time, its encoded protein has been identified as a universal cell cycle regulator with a central role in controlling the commitment of a cell to initiate DNA replication and divide2. Eliminating RB function allows unregulated cell cycle progression and promotes tumour growth. The prominent role that RB has in blocking proliferation has created confusion in understanding its biochemical function. Because of this key function, most upstream events that influence cell proliferation ultimately also affect RB function. However, it is often difficult to distinguish signalling mechanisms that directly impinge on events in the G1 phase of the cell cycle to arrest proliferation from those that act during other cell cycle phases and indirectly cause a G1 arrest. In the context of physiological events that have input into the cell cycle, such as cellular differentiation, cell senescence and the response to DNA damage, it can be unclear how directly signalling pathways actually regulate RB. For these reasons, the ongoing debate on RB function ranges between two extreme views. One view is that RB is a multifunctional protein acting in response to numerous stimuli to create at least as many potential outcomes. The alternative viewpoint is that signals from many different stimuli are ultimately channelled through cyclin-dependent kinase (CDK) regulation of RB phosphorylation, which in turn controls the activity of E2F transcription factors, a family of transcriptional regulators that stimulate proliferation. Both views have merit, and ultimately most models of RB function fall somewhere in-between, as researchers work to pinpoint its precise function.
Most cancers find a way to impair RB function, either through direct mutation of the RB gene or, more commonly, through the altered expression of RB regulators, which include cyclin D, CDK4 and CDK6, and their principal inhibitor, p16 (REF. 3). Given that loss of RB function is frequent in cancer, understanding its mechanism of action is expected to offer crucial insight into the most fundamental properties of cancer cells. Furthermore, loss of RB function in insects and mammals alike leads to overproliferation of cells and defects in numerous stages of organism development. From this perspective, knowledge of RB function promises important advances in our understanding of cancer, as well as of how proliferative control is coordinated in development. These promises can only be realized through a mechanistic understanding of the intricacies of RB function itself.
Much of our molecular knowledge of RB was established using cancer cell lines with aberrant RB pathway function, or was developed before the advent of RNA interference technologies that allow for loss‐of‐function analyses. The tools to address outstanding questions about RB structure and function in normal cells have only recently emerged. Our goal in this Review is to highlight these exciting recent advances in the biochemical understanding of RB. We describe the structure of RB and how its function is regulated. In particular, we discuss the role of key interacting protein partners that can be placed in the context of a few regulatory pathways for which detailed biochemical information is available. Readers who are interested in how these mechanisms are connected to broader questions of development and homeostasis2,4, as well as cancer initiation, progression5,6 and therapy7,8, are directed to other recent reviews specializing in these topics.
RB is a platform for multiple protein contacts
Although recent research has revealed that RB functions in diverse cellular pathways, such as apoptosis and the cell cycle, it has also become clear that RB regulates these pathways through the stimulation or inhibition of the activity of interacting proteins. Therefore, an important starting point for understanding RB function is its structure, which acts as a scaffold for these multiple protein interactions (FIG. 1).
Figure 1. RB is a multidomain protein with several distinct protein-binding surfaces.
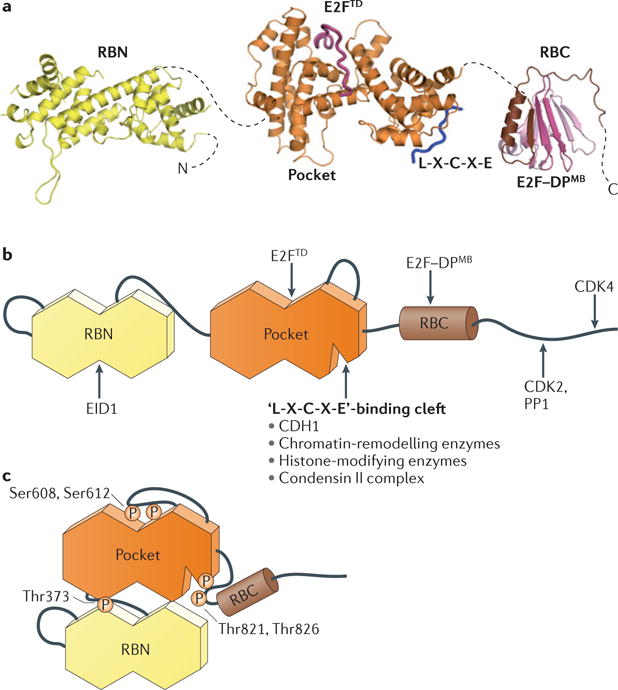
a | Model of active and complexes with E2F and an ‘L-X-C-X-E’ peptide (Protein Data Bank (PDB) codes: 2QDJ, 1GUX, 1N4M and 2AZE). The dashed lines indicate flexible interdomain linkers. b | Schematic diagram of the domain structure and location of known binding sites for protein partners. c | Schematic structure of RB in its inactivated, phosphorylated conformation. Thr373 phosphorylation drives interdomain docking of the RB amino-terminal domain (RBN) and the ‘pocket’, whereas Ser608 and Ser612 and Thr821 and Thr826 phosphorylation induce binding of the pocket loop and the RB carboxy-terminal domain (RBC), respectively, to the pocket domain. These different conformational changes inhibit specific RB–protein interactions. CDH1, CDC20 homologue 1; CDK, cyclin-dependent kinase; DPMB, differentiation-related polypeptide marked box; E2FTD, E2F transactivation domain; EID1, E1A-like inhibitor of differentiation 1; PP1, protein phosphatase 1.
Human RB contains 928 amino acids and is commonly described as having three domains. The central domain was identified as the minimal region necessary to bind viral oncoproteins, such as adenovirus E1A, SV40 TAg and human papilloma virus E7, and it was named the ‘pocket’ (Ref. 9). The pocket comprises two subdomains, A and B, each resembling a cyclin fold with three additional helices10 (FIG. 1a). The A and B subdomains interact with each other through an extensive non‐covalent interface such that the pocket folds into a single structural unit. Two additional cyclin folds constitute the structured amino‐terminal domain (RBN), which resembles the pocket structure except for a few subtle differences11. Approximately the last 150 residues of RB form the carboxy‐terminal domain (RBC), which is intrinsically disordered12. There are several other sequences in RB that similarly lack structure, including insertion loops in the RBN and the pocket, as well as a linker that flexibly tethers these two structured domains (FIG. 1b). The linker sequences are notable because they contain CDK‐dependent phosphorylation sites (BOX 1) that have a critical role for the inactivation of RB13,14. RB inactivation through phosphorylation and reorganization of its structure is depicted in FIG. 1c (see below). The domain structure of the RB family members p107 (also known as RBL1) and p130 (also known as RBL2) is similar, which is consistent with their analogous functions in regulating growth, the cell cycle and E2F function (BOX 2).
Box 1. RB post-translational modifications.
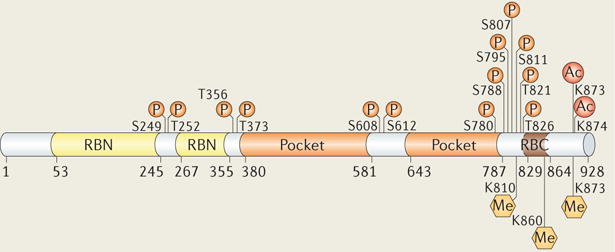
Post-translational modifications have an important role in the regulation of RB function. With a few exceptions, RB phosphorylation (P) results in inactivation, transcriptional derepression and cell cycle progression30. RB is phosphorylated by several different kinases, including cyclin-dependent kinases (CDKs) and checkpoint kinase 2 (CHK2)37. Phosphorylation controls RB interactions with other proteins. This modification typically occurs outside structured domains (see the figure) and promotes conformational transitions from disordered to ordered RB structures that mask protein-binding surfaces12–14 (FIG. 1). Different kinases show preferences for particular phosphorylation sites, and discrete phosphorylation events induce specific structural changes. However, it remains uncertain whether, and in what context, differentially phosphorylated isoforms of RB exist in the cell.
Acetylation (Ac) and methylation (Me) sites have been identified in disordered sequences towards the RB carboxy-terminal domain (RBC)37. In contrast to phosphorylation, these modifications occur in response to signals, such as DNA damage and differentiation, which correlate with RB activation and repression of gene expression82–86. Acetylation occurs on Lys873 and Lys874, which are located within the cyclin-docking sequence, and results in reduced phosphorylation, probably through kinase inhibition83,84. Methylation on Lys873 and Lys810 by SET-domain methyltransferases similarly results in RB hypophosphorylation82,85. SET and MYND domain-containing 2 (SMYD2) methylates Lys860, which results in the recruitment of the transcriptional repressor lethal(3)malignant brain tumour-like 1 (L3MBTL1)86. The reader is referred to other reviews for more details on these and other emerging post-translational modifications on RB and their roles in the regulation of function33,37.
RBN, RB amino-terminal domain.
Box 2. The RB family of proteins.
RB, p107 and p130 are collectively known as the ‘pocket protein’ family. RB shares approximately 25% sequence identity with both homologues, whereas p107 and p130 share approximately 54% identity with each other. Structural data characterizing p107 and p130 are limited; however, sequence analysis suggests domains comparable to those in RB are present (see the figure). Both proteins contain predicted amino-terminal domains (107N and 130N), pocket domains and carboxy-terminal domains (107C and 130C), with analogous secondary and tertiary structural elements. Consistent with these parallel structural features, a number of common molecular functions have been identified87,88. All three proteins negatively regulate the cell cycle, and for each protein this effect has been tied to its ability to associate with E2F transcription factor family members and influence E2F-mediated gene expression. The pocket domains of all homologues are predicted to contain L-X-C-X-E-binding clefts, which bind viral proteins and probably partially overlapping sets of cellular proteins. Pocket proteins are all inactivated by cyclin-dependent kinases (CDKs), and sequence analysis suggests several common structural effects of phosphorylation.
Genetic and cellular investigations have revealed various key functional differences between pocket proteins. RB knockout is embryonic lethal in mice, whereas knockout of p107 or p130 does not have a phenotype in a mixed genetic background89–91. Examination of mice lacking different combinations of pocket protein genes suggests that p107 and p130 have an overlapping role in development that is distinct from that of RB91. Importantly, the tumour suppressor properties of the RB gene are significantly stronger than those of p107 and p130, and only RB mutations are commonly found in human cancer. Consistent with these genetic differences, pocket proteins have been observed to control distinct E2F target genes and arrest cells in different cell cycle phases47. In a recent striking example, a genome-wide screen of gene repression in fibroblast cells revealed a unique role for RB in promoting senescence42. Further structural and biochemical analysis is needed to understand the molecular basis for these functional differences in pocket proteins, although distinct protein interactions have already been identified. For example, pocket proteins show preferences for different E2F family members, and p107 and p130 bind and inhibit CDKs, whereas RB exclusively forms a stable complex with protein phosphatase 1 (PP1)28,87,88.

RBC, RB carboxy-terminal domain; RBN, RB amino-terminal domain.
Evolutionarily conserved interaction surfaces on RB
The modular domain organization of RB, like that of many signalling proteins, allows for independent binding and regulation of multiple protein interactors (FIG. 1). However, unlike typical signal transducers, RB does not contain canonical protein–protein interaction domains (for example, Src homology 2 and phosphotyrosine‐binding domains) and instead uses the cyclin folds and the disordered RBC to form binding interfaces. To inhibit transactivation, RB binds the E2F transactivation domain (E2FTD) using a highly conserved region of the pocket domain, which lies at the interface between the A and B cyclin folds15,16. Also in the pocket domain, an ‘L‐X‐C‐X‐E‐binding cleft’ (in which X represents any amino acid) formed by three helices of the second cyclin fold binds an L‐X‐C‐X‐E sequence found in viral proteins such as TAg and E7 (REFS 17,18). This interaction is required for the cellular transforming activity of these proteins. The conservation of this surface in RB orthologues from other species, as well as in its related family members p107 and p130, suggests a crucial cellular role (BOX 2). Importantly, a number of RB–protein interactions have been mapped to the L‐X‐C‐X‐E‐binding cleft19,20. Although it has been common to use the L‐X‐C‐X‐E sequence motif as a consensus for identifying cellular RB‐interacting proteins, it has become clear from structural studies with viral and cellular proteins that there are other determinants of high‐affinity interactions with the pocket cleft21. Furthermore, several proteins that contact this region of RB do not contain a clear L‐X‐C‐X‐E motif22,23, and there is little evidence that these or other L‐X‐C‐X‐E‐containing cellular proteins bind RB directly. Together, these observations indicate that RB interactions with cellular proteins at the L‐X‐C‐X‐E‐binding cleft are probably more complex than understood from the crystal structures of RB bound to viral proteins. In light of the recent discovery of small molecule therapeutics that can target the cleft24, further structural studies to understand these subtleties are required.
Interactions through intrinsically disordered regions
RB binds several proteins using sequences in the RBN and RBC. The RBN binds E1A‐like inhibitor of differentiation 1 (EID1), along with several other proteins, although data to map protein interactions in detail are limited for this domain of RB. A second RB–E2F interaction is made between the RBC and the ‘marked box’ domains of E2F and its heterodimerization partner differentiation-related polypeptide (DP)12 (FIG. 1). Although the RBC is intrinsically disordered, approximately 30 residues adopt a strand–turn–helix conformation on binding the marked box region of E2F–DP. At least part of this second interaction seems to be specific between RB and E2F1 in vivo, and it may be important for the function and regulation of an E2F1‐specific activity, such as apoptosis25,26, which is discussed below.
The RBC is also the location of kinase and phosphatase docking sites27–29. These enzymes bind short, linear peptide sequences distal to their target RB phosphorylation sites, and this increases enzyme efficiency by a stronger substrate interaction. Distinct binding sites in the RBC are accessed by cyclin A–CDK2, cyclin D–CDK4 and cyclin D–CDK6, although the implications of this difference for CDK‐site preference by these enzymes is not clear27,29. A recent structural study of protein phosphatase 1 (PP1) bound to the RBC demonstrated that the phosphatase and cyclin A binding sites overlap and that PP1 can inhibit CDK activity towards RB through a docking competition mechanism28. RB binds PP1 in a manner that resembles other PP1 regulatory subunits, and it will be interesting to learn whether RB also functions in targeting PP1 to specific substrates for dephosphorylation.
Cell cycle control through E2F regulation
Initial studies of RB focused on its inhibition of E2F transcription before the G1 to S phase transition, as the observed negative regulation of the cell cycle offered a powerful explanation for why RB is a tumour suppressor2,30. These early experiments correlated RB function with its capacity to physically associate with E2F family proteins and thereby inhibit E2F‐dependent activation of genes that stimulate DNA synthesis and cell cycle advancement. When RB is in a hypophosphorylated state, pocket–E2FTD contacts and interactions involving the RBC are required for E2F inhibition31,32. CDK‐dependent phosphorylation, which occurs on approximately 13 conserved consensus sites in vivo33, inactivates RB‐dependent E2F repression by dissociating the RB–E2F complex30 (BOX 1).
Regulation of RB–E2F binding
Recent studies have characterized the structural changes in RB that result in binding inhibition12–14 (FIG. 1c). Phosphorylation induces interdomain associations that occlude, or allosterically disrupt, the E2FTD‐binding site in the pocket and the interaction between the RBC and the E2F marked box. An important result of this is that discrete phosphorylation events result in distinct RB structures. Ser608 and Ser612 phosphorylation stabilizes the association of the pocket loop with the E2FTD‐binding site within the pocket domain. Thr373 phosphorylation induces interdomain docking between the RBN and the pocket, which allosterically inhibits E2FTD and directly inhibits binding at the L‐X‐C‐X‐E‐binding cleft. Finally, Thr821 and Thr826 phosphorylation induces RBC binding to the pocket, which excludes the E2F marked box and L‐X‐C‐X‐E interactions. It is possible that these phosphorylation‐dependent conformational changes can control RB interactions with other proteins as well as E2F. Mutational analysis of CDK phosphorylation sites on RB suggests that a large number of sites need to be phosphorylated for RB inactivation and cell cycle advancement34,35. However, there are few data available to indicate the quantity of sites that are phosphorylated on each individual RB molecule during G1 to S phase transition. For this reason, models such as the one depicted in FIG. 1c require further testing, and it is possible that partial phosphorylation could create different functional outputs depending on which specific sites are phosphorylated.
Unlike many other cell cycle regulatory proteins, RB is not typically degraded upon inactivation but persists until mitosis, when it is dephosphorylated by PP1 to enter the following G1 phase36. This presence throughout the cell cycle allows reactivation by dephosphorylation in response to S phase and other checkpoints. Several recent studies have found that other post‐translational modifications, including methylation and acetylation, can control RB activity by regulating RB phosphorylation levels37 (BOX 1).
Chromatin structure regulation at E2F target promoters
RB interactions with chromatin‐regulating enzymes and histone‐modifying enzymes have been identified and are mediated through the L‐X‐C‐X‐E‐binding cleft19. The physical separation between the E2F and L‐X‐C‐X‐E‐binding surfaces in RB supports a model whereby RB recruits chromatin regulators to E2F‐regulated promoters for repression or activation of target genes (FIG. 2). CDK phosphorylation disrupts RB interactions with these proteins by inducing conformational changes that inhibit binding at the L‐X‐C‐X‐E‐binding cleft and E2Fs12,14,38,39. Mutations in the L‐X‐C‐X‐E‐binding cleft do not impair the ability of RB to regulate the G1 to S phase transition and cell growth40, so the significance of chromatin‐modifying enzymes in RB regulation of E2F‐controlled gene expression in cycling cells is not clear. The action of these proteins may be more relevant for changes in chromatin structure associated with permanent gene silencing observed in cell cycle exit. Supporting this idea, a crucial role for RB–L‐X‐C‐X‐E interactions has been observed in the formation of heterochromatin in senescence41, and RB and its homologues p107 and p130 contribute to the response of upstream signals that induce senescence and maintenance of cell cycle exit42–44. Phenotypically, these results indicate an important role for RB in recruiting chromatin regulators; however, biochemical demonstration of these complexes remains rare. Recent work has now demonstrated the RB‐dependent recruitment of histone deacetylase enzymes using its L‐X‐C‐X‐E‐binding cleft at E2F target genes by chromatin immunoprecipitation assays45.
Figure 2. Regulation of E2Fs by RB.
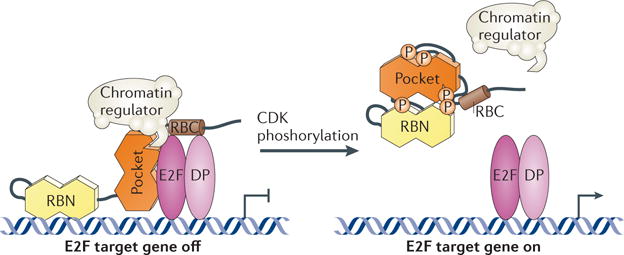
In the G1 phase of the cell cycle, RB is phosphorylated at low levels and associates with E2F transcription factors, which are dimeric proteins containing E2F and differentiation-related polypeptide (DP) subunits. RB also recruits enzymes that regulate chromatin structure to these complexes. Transcription of these genes is repressed until cyclin-dependent kinases (CDKs) phosphorylate (P) RB and prevent binding of E2Fs and chromatin regulators. E2F transcription factors then transcribe genes necessary for S phase and the cell cycle advances. RBC, RB carboxy-terminal domain; RBN, RB amino-terminal domain.
Transcriptional repression by other RB-family proteins in G1
RB belongs to a family comprising three homologous proteins that are referred to as pocket proteins (BOX 2). p130 is the most abundant pocket protein in stable cell cycle arrest events such as quiescence and senescence46,47. When bound to E2F4, it represses genes required for cell cycle re‐entry under both conditions48,49. DREAM, a complex containing p130, E2F4–DP and the highly conserved MuvB complex, was recently shown to facilitate this p130‐dependent activity49,50. In human cells, chromatin immunoprecipitation assays showed that MuvB proteins are located at most E2F‐regulated promoters, and gene expression analysis has implicated DREAM as a repressor of cell cycle genes. Few details are known about the biochemical function of MuvB, with the exception that it acts as a scaffold for the recruitment of different transcription factors. DREAM is regulated by dual specificity Tyr‐phosphorylation‐regulated kinase 1A (DYRK1A) and CDK phosphorylation51. Upon cell cycle entry MuvB dissociates from p130 and binds Myb‐related protein B (MYBB; also known as MYBL2) and forkhead box M1 (FOXM1)52,53. These two transcription factors, which are upregulated in many cancers, then activate late‐stage cell cycle genes53.
Putting RB–E2F regulation into perspective
The canonical model of RB tumour suppression is based on the negative regulation of E2F transcription54. However, p107 and p130 also repress E2F transcription and inhibit the cell cycle, yet only RB is commonly mutated in cancer8. For this reason, some cancers may retain the ability to restrict E2F activation. Two recent studies have also shown that activator E2Fs (E2F1, E2F2 and E2F3) are dispensable for proliferation in vivo55,56. From this perspective, it is difficult to envision how RB inhibition of these particular E2Fs can arrest proliferation when they are not required for proliferation. These results suggest that transcriptional regulation of E2Fs may not be the only way by wayRB can act as a tumour suppressor. A number of novel RB activities have emerged, and they serve to illustrate the multifunctional nature of this protein.
Cell cycle control through CDK inhibition
An E2F‐independent mechanism for RB‐induced cell cycle arrest has recently been uncovered. Ectopic expression of RB in an RB‐deficient cell line induces a G1 arrest before E2F target genes become repressed57. Thus, progression through G1 can be blocked, and CDKs can be inhibited, in an RB‐dependent fashion, without altering the transcription of E2F gene targets. Two mechanisms, which are not mutually exclusive, have emerged to explain this observation (FIG. 3). In the first case, the CDK inhibitor p27 (also known as CDKN1B) is stabilized through the antagonism of its cognate F box protein, S phase kinase-associated protein 2 (SKP2). In this model, RB physically binds SKP2 through its unstructured RBC domain. This interaction prevents SKP2 from recognizing p27 and targeting it for degradation via the ubiquitin–proteasome system (FIG. 3a,b). Consequently, p27 is stabilized, causing CDK inhibition and cell cycle arrest57. In the second mechanism, RB interacts simultaneously with SKP2 and the APC/C (anaphase‐promoting complex; also known as the cyclosome)22. This interaction targets SKP2 for ubiquitylation and degradation, leading to the stabilization of p27, CDK inhibition and cell cycle arrest. In this model, again, RB acts as a scaffold, binding SKP2 at the RBC and the APC subunit CDC20 homologue 1 (CDH1) through the pocket (FIG. 3c). Although structural details of these interactions have not been elucidated, RB mutagenesis studies suggest that CDH1 binding involves the L‐X‐C‐X‐E‐binding cleft in the pocket22. The RB–CDH1 interaction was further destabilized in an RB mutant that included mutations in both the L‐X‐C‐X‐E‐binding cleft and the E2F‐binding site in the pocket. An interesting possibility is that the same RB molecule cannot regulate E2F and APC–SKP2–p27 activities simultaneously. Studies suggest that APC and E2F binding to RB may be competitive, although the complexity of RB–E2F1 interactions described below suggests that this requires further investigation58. Together, this mechanism of function and its apparent independence from E2F strongly suggest that RB is a multifunctional protein. Future structural studies will be needed to fully appreciate this mechanism and its regulation.
Figure 3. Transcription-independent regulation of cyclin-dependent kinases by RB.
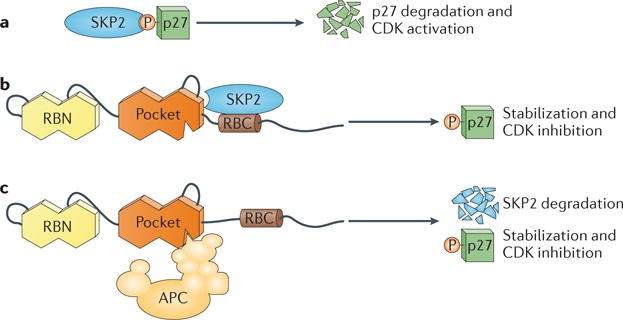
a | Schematic depiction of the F box protein S phase kinase-associated protein 2 (SKP2) recognizing phosphorylated p27. Binding results in ubiquitylation and degradation of p27 and the activation of cyclin-dependent kinases (CDKs). b | SKP2 is bound by the RB carboxy-terminal domain (RBC) and the ‘pocket’ domain, which competes with SKP2 for interaction with phosphorylated p27. RB thereby prevents p27 ubiquitylation, resulting in inhibition of CDK activity. c | The RBC region also binds SKP2 to recruit it to the APC/C (anaphase-promoting complex; also known as the cyclosome) E3 ubiquitin ligase complex. Ubiquitylated SKP2 (not shown) is targeted for degradation, leading to the increased expression of p27 and inhibition of CDKs. RBN, RB amino-terminal domain.
An E2F-independent paradigm of tumour suppression
To test the relevance of RB regulation of p27 in vivo, SKP2‐deficient mice were crossed with animals heterozygous for Rb, and SKP2 loss was found to suppress cancer susceptibility of the Rb+/− genotype59. Surprisingly, it was discovered that inactivation of SKP2 in RB‐deficient cells did not inhibit proliferation, but rather induced a p27‐dependent apoptotic programme. Apoptosis leads to loss of the intermediate lobe of the pituitary, which is where tumours generally arise in Rb+/− mice. It is noteworthy that in these mice, cells deficient in both RB and SKP2 exhibited deregulated E2F expression but not deregulated proliferation. Again, this result suggests that RB‐dependent regulation of non‐E2F pathways is crucial. The existence of such pathways is more consistent with a multifunctional model of RB and is less supportive of an E2F‐centric mechanism of function.
Heterochromatin and chromosome stability
RB has been shown to have an impact on genome stability through various distinct mechanisms.
E2F-dependent mechanisms
Overexpression of the E2F target gene mitotic arrest deficient 2 (MAD2) as a consequence of RB loss causes lagging chromosomes, in which chromosome arms become fused leading to double‐strand DNA breaks or missegregation. These aberrations can result either from an overactive spindle assembly checkpoint60 or from an inability to resolve merotelic microtubule attachments to sister chromatids61. Furthermore, deregulation of E2F transcription due to loss of RB function can lead to abnormally low nucleotide pools and replication stress62. This stress leads to double‐strand DNA breaks and abnormal firing of replication origins and, ultimately, causes aneuploidy. Thus, loss of RB function has several negative effects on chromosome stability that are mediated by increased E2F activity, and the mechanistic involvement of RB in genome maintenance in these scenarios is probably best described by the classic mechanisms of E2F regulation described above. These paradigms of RB function exemplify the idea that regulation of E2F is paramount to RB, and that its diverse effects can ultimately be traced back to E2F control.
E2F-independent regulation of heterochromatin
RB and its family members have emerged as regulators of heterochromatin domains that surround the centromere and have a key role in chromosome structure and the attachment to spindle microtubules. Cells deficient for all RB family proteins display decondensed pericentromeric heterochromatin with reduced trimethylation of Lys20 on histone H4 (H4K20me3) as well as reduced DNA methylation63. This defect leads to tangled chromosomes at metaphase, missegregation and aneuploidy. Many of these characteristics are observed in cells from a mouse strain bearing an RB mutation that impairs its L‐X‐C‐X‐E‐binding cleft but not its E2F interactions, indicating that this chromosomal phenotype is caused by loss of just one aspect of RB function40 (FIG. 4). The root cause of this phenotype remains uncertain. For example, loss of the histone tail modification H4K20me3 caused by deletion of the methyl transferases SUV420H1 (suppressor of variegation 4–20 homologue 1) and SUV420H2 similarly depletes this modification from pericentromeric regions but does not result in the chromosomal fusions seen in RB‐deficient cells64.
Figure 4. Regulation of pericentromeric heterochromatin by RB.
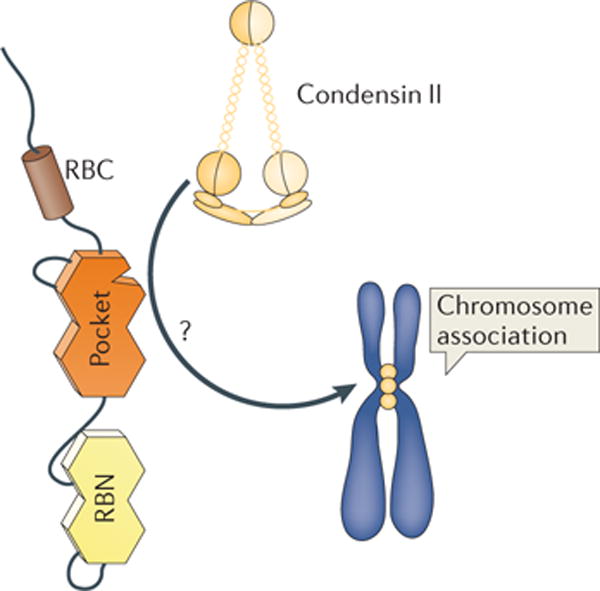
The condensin II complex is enriched at pericentromeric and centromeric regions of mitotic chromosomes. Loading this complex onto chromatin is dependent on L-X-C-X-E-type interactions with RB. How this directs the condensin II complex to this genomic location is not known (indicated by a question mark), but it is suggested that additional proteins are involved. Similarly, the localization of cohesins to the centromere and pericentromere remains to be elucidated. Given the prominence of genome instability in mice deficient for L-X-C-X-E-type interactions, RB function in chromosome structure is key for the well-conserved L-X-C-X-E binding site. RBC, RB carboxy-terminal domain; RBN, RB amino-terminal domain.
A series of recent reports have provided further insight into this aspect of RB function and narrowed down the possibilities for how this mechanism works. First, examination of the fruit fly orthologue of RB, called Rbf1, and comparison with fly mutations in components of the condensin II complex revealed similar phenotypes of chromatin decondensation, not only in mitotic chromosomes but also under growth arrest conditions23. This report demonstrated a conserved interaction between RB and the condensin II complex in human and fly cells. Furthermore, loss of RB function leads to defective loading of condensin II and cohesin, specifically at the centromeric region of mitotic chromosomes65–67. Phenotypically, this is identifiable in mitosis as defective chromosome congression, reduced cohesion and misaligned centromeres.
Beyond defects in cohesion and condensation of mitotic chromosomes, RB‐deficient cells exhibit chromosome missegregation and aneuploidy. In addition to these mitosis‐specific molecular defects, spontaneous double‐strand breaks are seen in cells deficient for all RB family proteins67. These defects reveal the means by which RB loss can influence genome stability independently of E2F transcriptional regulation. The effect of RB‐mediated genome stability in cancer was investigated by crossing RB mutant mice that are viable but defective for L‐X‐C‐X‐E inter actions with p53‐deficient mice66. In this way, compound mutant animals, and p53 knockout‐only mice have similar G1 arrest defects. Compound mutant animals obtained from this cross succumbed to cancer significantly earlier than their p53‐knockout controls. Furthermore, tumours were more metastatic in mice deficient in both RB and p53, and analysis of DNA copy number variation indicated that these mice had more alterations. Thus, maintenance of genome stability through the regulation of pericentromeric heterochromatin contributes to RB tumour suppressor activity.
Perspectives on RB regulation of chromosome stability
First, it should be noted that loss of RB does not universally cause chromosome instability68. Studies of primary retinoblastoma tumours propagated in mice indicate that, in this tumour type, Rb‐null mutations do not contribute to chromosome instability. However, given the relevance of the role of RB in cancer in a genomically unstable mouse model, understanding the mechanism of RB function in establishing heterochromatin at pericentromeric regions is vital. To this end, many fundamental questions remain (FIG. 3). Defects in condensin II and cohesin loading are central to the effect of RB on chromatin structure, but there are no data suggesting whether RB targets them to pericentromeric locations directly or regulates their loading by another means. Furthermore, phosphorylation by CDKs in late G1 typically inactivates RB by disrupting protein interactions2, so it is difficult to envision how RB facilitates cohesion and condensation in M phase. Likewise, this aspect of RB function is dependent on interactions at the L‐X‐C‐X‐E‐binding cleft, but the identity of the protein (or proteins) that it associates with is unknown. The condensin II subunit chromosome‐associated protein D3 (CAPD3) has been proposed as a candidate23, but there is little evidence that it directly contacts RB. RB could also bind other proteins in the condensin II ring or a separate bridging factor. Clearly, much work remains for us to understand this newly emerging aspect of RB function. However, its independence from E2F transcriptional regulation is well established and contributes to a growing body of literature that is revealing the multifunctional nature of RB.
Regulation of apoptosis
Largely through the study of E2F overexpression in cultured mammalian cells and deregulated E2Fs in RB‐knockout mice, it was discovered that E2Fs could induce apoptosis69. More recently, it has been found that apoptotic phenotypes in Rb‐null mice are secondary to defective placental development70, and hence are not exclusively caused by overactive E2Fs. Almost simultaneously, new data have emerged describing a number of post‐translational modification changes on E2F1 that allow it to respond to DNA damage and induce cell death37. Specifically, E2F1 is phosphorylated by ataxia‐telangiectasia mutated (ATM) and checkpoint kinase 2 (CHK2), two kinases that are activated by double‐strand DNA breaks. E2F1 is also acetylated by p300/CBP‐associated factor (PCAF; also known as KAT2B), an acetyltransferase that has been implicated in transcriptional control. In addition, several basic residues on E2F1 are also demethylated in response to DNA damage signalling. This level of regulation is unique to E2F1 as these modifications are not shared by other E2F family members. In general, the ability of RB to bind and inhibit E2F1‐dependent transcription has been interpreted as a means to block apoptosis, and this is consistent with RB–E2F1 interactions ultimately mediating the effects of RB on cell viability. However, recent mechanistic advances in our understanding of RB and E2F1 in apoptosis now suggest multifunctional roles for these proteins.
DNA damage-induced alterations to RB–E2F1 function
A recent, transformative development in efforts to understand the role of RB in apoptotic regulation has been the discovery that it can have a pro‐apoptotic role71. DNA damage induces the recruitment of PCAF to RB and E2F1, resulting in histone acetylation and transcription at specific pro‐apoptotic promoters. To understand how RB can regulate both apoptosis and the cell cycle in response to different stimuli, it is important to consider how it regulates E2F1 in normal proliferating cells and how E2F1 control is altered in response to DNA damage.
The biochemical mechanism of E2F1 interactions with RB is different from that of other E2Fs. E2F1 was the E2F protein that was used to define the association between the RB pocket and the transactivation domain of E2F72. However, E2F1 has an additional interaction with RB that is unique among E2F family members25 (FIG. 5a). The best evidence for the unique RB–E2F1 interaction is the demonstration that the adenoviral E1A protein can compete with and disrupt the common RB–E2F interaction but not the E2F1‐specific interaction73. Likewise, RB–E2F1 association persists when RB is phosphorylated74. The structural determinants for the specific RB–E2F1 interaction have been mapped to the RBC26. However, it is unclear to what extent the crystal structure of the RBC complex with the E2F1–DP1 marked box domains represents the E2F1‐specific interaction12,74.
Figure 5. Differential regulation of E2F1 in apoptosis.
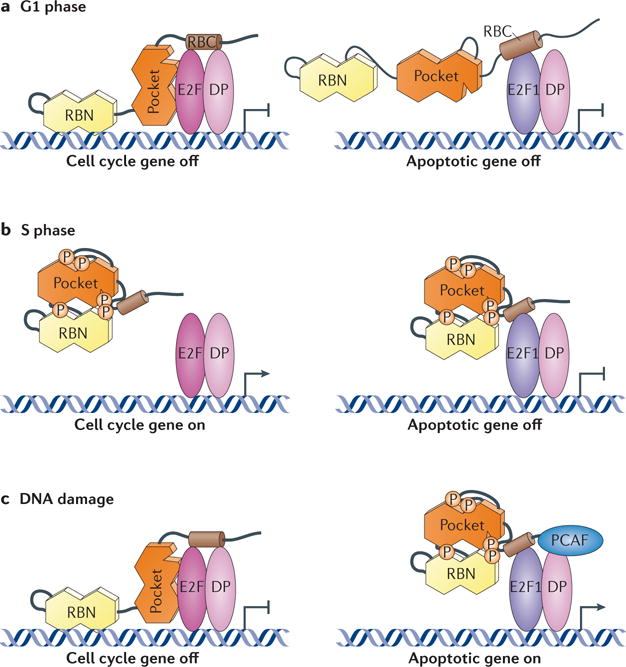
a | RB associates with E2F transcription factors and differentiation-related polypeptide (DP) heterodimers in G1 to repress transcription of cell cycle genes. RB can also form a unique interaction with E2F1 involving sequences in the RB carboxy-terminal domain (RBC). b | In S phase, RB is phosphorylated (P) and unable to bind to E2Fs that are bound the promoters of cell cycle genes, allowing the cell cycle to advance. Phosphorylated RB can still interact with E2F1, and this complex can repress the expression of apoptotic target genes. c | In response to DNA damage, RB is dephosphorylated and regains the ability to repress E2F transcription at cell cycle promoters. Simultaneously, phosphorylated RB remains in contact with E2F1 transcription factors, and the recruitment of p300/CBP-associated factor (PCAF) through unknown signals leads to histone acetylation (not shown) and activation of pro-apoptotic target genes. RBN, RB amino-terminal domain.
The picture that has emerged from this differential regulation of E2F1 by RB is that CDK phosphorylation of RB releases most E2Fs from its control at the G1 to S phase boundary to stimulate transcription of cell cycle target genes, but at least some E2F1 remains bound to RB through the unique interaction of these proteins in S phase (FIG. 5b). This model explains a number of paradoxical reports that previously suggested RB–E2F1 interactions persist in S phase, even in the face of CDK phosphorylation75–77. The unique RB–E2F1 interaction seems to negatively regulate transcription at pro‐apoptotic promoters, such as TAp73, in reporter assays74. In response to DNA damage, both RB and E2F1 undergo extensive changes in post‐translational modifications37. RB is dephosphorylated at CDK target sites but is phosphorylated by CHK2, as well as acetylated and methylated. E2F1 is phosphorylated and acetylated as described above. The cumulative effect results in repression of the transcription of E2F‐dependent cell cycle genes and the activation of pro‐apoptotic genes (FIG. 5c). How these modifications lead to persistence of only the specific RB–E2F1 complex is unknown; however, two reports now demonstrate that a complex containing phosphorylated RB and E2F1 has pro‐apoptotic activity and is found at the TAp73 promoter coincident with its activation71,78.
Further experimentation to understand the molecular mechanism of RB–E2F1 transcriptional regulation will continue to be a challenging endeavour. It represents an area in which RB is again emerging as more than just an E2F regulator that is controlled by CDK phosphorylation. A deeper molecular understanding of RB–E2F1 interactions and regulation will undoubtedly clarify its role in apoptosis and the DNA‐damage response and offer further evidence for the multifunctional nature of RB.
Reassessing the uses of RB function in cancer
Tumour specimens are often graded on proliferative indexes, and high levels of proliferating cells are generally correlated with poor outcomes. Recent advances in genomic analyses of tumours have established expression signatures that are indicative of deregulation of E2F activity79,80. However, no markers are available that predict the ability of cancer cells to activate RB and restrict proliferation, or that predict its potential to stimulate apoptosis and eliminate tumours. Given the direct effect of genotoxic chemotherapeutics and novel kinase‐inhibitor drugs for growth and survival pathways that affect RB and its downstream effectors, more rigorous classification of the functional state of RB in tumours will be beneficial for clinical outcomes. Furthermore, the key role of RB–E2F1 in regulating TAp73 expression in response to DNA damage offers tremendous potential for the development of therapeutics, as TAp73 expression has been shown to control sensitivity to genotoxic therapeutics81. A clearer structural picture of RB and E2F1 in their various interaction configurations will create the opportunity for therapeutic manipulation.
Conclusions and perspectives
Recent structural and functional data highlighted here are beginning to provide evidence that RB is a truly multifunctional protein. Current data also suggest that some novel functions of RB are potentially tumour suppressive. The RB–SKP2–p27 regulatory pathway is, seemingly, an E2F‐ independent proliferative control mechanism. Initial experiments demonstrate that CDH1 and E2F compete for RB association58, and this type of biochemical insight defines RB as multifunctional. It is not clear whether CDK regulation through the RB–SKP2–p27 pathway is equivalent in importance with E2F transcriptional repression. The best available data suggest that both pathways affect the pituitary, but it is not yet possible to determine whether one pathway has a relatively more important role in a particular tissue. It is also possible that the RB–E2F and RB–SKP2–p27 pathways are crucial under distinct physiological circumstances that have yet to be determined.
A similar comparative discussion is possible for each of the different RB‐dependent pathways described in this Review, and others that do not yet have an extensive structural understanding. Can RB engage in each one simultaneously as a higher order complex, or are their interactions mutually exclusive, forcing cellular RB to be rationed among its different roles? Given the growing support for multifunctionality of RB, the next challenges in cell cycle and RB research will be to assign priority to these functions. Does RB rely more strongly on one function compared with others to act as a tumour suppressor? Only through the understanding and comparison of all functions in relation to one another will we appreciate the tumour suppressive role of RB in biochemical detail.
Acknowledgments
Research in the authors laboratories is supported by awards from the Canadian Institutes of Health Research (MOP89765 and MOP64253) and the Canadian Cancer Society Research Institute (2011-700720) to F.A.D, and the US National Institutes of Health (R01CA132685) and the American Cancer Society (RSG-12-131-01-CCG) to S.M.R. F.A.D is The Wolfe Senior Fellow in Tumour Suppressor Genes.
Glossary
- Cyclin
A family of proteins that activate cyclin-dependent kinases and whose stability is cell-cycle regulated.
- CDK
(Cyclin-dependent kinase). A family of kinases that are activated by cyclins.
- E2F
(E2-binding factor). A family of cell-cycle regulated transcription factors.
A region in RB-family proteins that was originally determined to bind to viral oncoproteins such as SV40 TAg
- Differentiation-related polypeptide
(DP). The E2F dimerization partner.
- Pocket proteins
RB-family proteins defined by their possession of the central ‘pocket’ domain
- p27
An inhibitor of cyclin-dependent kinase activity.
- F box protein
A protein containing the F box domain an approximately 50-amino acid motif that facilitates protein–protein interactions
- S phase kinase-associated protein 2
(SKP2). An adaptor protein that recruits p27 to the SKP–cullin–F box E3 ligase complex.
- APC/C
(Anaphase-promoting complex also known as the cyclosome). An E3 ubiquitin ligase.
- Merotelic
When multiple microtubules emanating from opposite spindle poles, simultaneously bind to a single kinetochore
- Aneuploidy
An abnormal number of chromosomes in a cell.
- Centromere
A constricted region of a chromosome that interacts with kinetochores and is the attachment point for spindle microtubules.
- Condensin II
A protein complex made up of seven protein subunits that create a ring structure to link and supercoil DNA strands.
- Cohesin
A ring-structured protein complex similar to the condensins that creates cohesion between replicated homologous DNA strands and regulates their separation during cell division
- Pericentromere
A repetitive chromosomal region adjacent to the centromere.
Footnotes
Note added in proof
A recent paper also describes a phosphorylation‐dependent change in RB conformation that is relevant for understanding how RB interactions with other proteins are regulated92.
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Frederick A. Dick, Email: fdick@uwo.ca.
Seth M. Rubin, Email: srubin@ucsc.edu.
References
- 1.Friend SH, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 2.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nature Rev Cancer. 2002;2:910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 4.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nature Rev Mol Cell Biol. 2008;9:713–724. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- 5.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nature Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nature Rev Cancer. 2012;12:20–226. doi: 10.1038/nrc3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacPherson D, Dyer MA. Retinoblastoma: from the two-hit hypothesis to targeted chemotherapy. Cancer Res. 2007;67:7547–7550. doi: 10.1158/0008-5472.CAN-07-0276. [DOI] [PubMed] [Google Scholar]
- 8.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nature Rev Cancer. 2008;8:714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 10.Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 11.Hassler M, et al. Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol Cell. 2007;28:371–385. doi: 10.1016/j.molcel.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the Rb C-terminal domain bound to E2F1–DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123:1093–1106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 13.Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem. 2010;285:16286–16293. doi: 10.1074/jbc.M110.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012;26:1156–1166. doi: 10.1101/gad.189837.112. Examines molecular contacts of phosphates on RB and how they influence the structure of the pocket domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao B, et al. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc Natl Acad Sci USA. 2003;100:2363–2368. doi: 10.1073/pnas.0436813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HY, Ahn BY, Cho Y. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J. 2001;20:295–304. doi: 10.1093/emboj/20.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 19.Brehm A, Kouzarides T. Retinoblastoma protein meets chromatin. Trends Biochem Sci. 1999;24:142–145. doi: 10.1016/s0968-0004(99)01368-7. [DOI] [PubMed] [Google Scholar]
- 20.Morris EJ, Dyson NJ. Retinoblastoma protein partners. Adv Cancer Res. 2001;82:1–54. doi: 10.1016/s0065-230x(01)82001-7. [DOI] [PubMed] [Google Scholar]
- 21.Singh M, Krajewski M, Mikolajka A, Holak TA. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J Biol Chem. 2005;280:37868–37876. doi: 10.1074/jbc.M504877200. [DOI] [PubMed] [Google Scholar]
- 22.Binne UK, et al. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nature Cell Biol. 2007;9:225–232. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- 23.Longworth MS, Herr A, Ji JY, Dyson NJ. RBF1 promotes chromatin condensation through a conserved interaction with the condensin II protein dCAP-D3. Genes Dev. 2008;22:1011–1024. doi: 10.1101/gad.1631508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fera D, et al. Identification and characterization of small molecule antagonists of pRb inactivation by viral oncoproteins. Chem Biol. 2012;19:518–528. doi: 10.1016/j.chembiol.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell. 2003;12:639–649. doi: 10.1016/s1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- 26.Julian LM, Palander O, Seifried LA, Foster JE, Dick FA. Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene. 2008;27:1572–1579. doi: 10.1038/sj.onc.1210803. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, et al. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin–CDK complexes. Mol Cell Biol. 1999;19:1068–1080. doi: 10.1128/mcb.19.2.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirschi A, et al. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nature Struct Mol Biol. 2010;17:1051–1057. doi: 10.1038/nsmb.1868. Demonstrates that CDKs and phosphatases compete for access to a common binding site on RB to regulate the phosphorylation status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan W, Cox S, Hoess RH, Grafstrom RH. A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 2001;61:2885–2891. [PubMed] [Google Scholar]
- 30.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 31.Hiebert SW, Chellappan SP, Horowitz JM, Nevins JR. The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 1992;6:177–185. doi: 10.1101/gad.6.2.177. [DOI] [PubMed] [Google Scholar]
- 32.Qin XQ, Chittenden T, Livingston DM, Kaelin WG., Jr Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 1992;6:953–964. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- 33.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–19. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol. 1999;19:3246–3256. doi: 10.1128/mcb.19.5.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolupaeva V, Janssens V. PP1 and PP2A phosphatases - cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2012;280:627–643. doi: 10.1111/j.1742-4658.2012.08511.x. [DOI] [PubMed] [Google Scholar]
- 37.Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: is there a code of conduct? Oncogene. 2012;31:4343–4352. doi: 10.1038/onc.2011.603. [DOI] [PubMed] [Google Scholar]
- 38.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 39.Harbour J, Luo R, Dei Santi A, Postigo A, Dean D. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 40.Isaac CE, et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol. 2006;26:3659–3671. doi: 10.1128/MCB.26.9.3659-3671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Talluri S, et al. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol. 2010;30:948–960. doi: 10.1128/MCB.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chicas A, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. Identifies precise E2F targets for RB-dependent repression in senescence by chromatin immunoprecipitation followed by sequencing and microarray approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dannenberg J-H, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G1 control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sage J, et al. Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourgo RJ, et al. RB restricts DNA damage-initiated tumorigenesis through an LXCXE-dependent mechanism of transcriptional control. Mol Cell. 2011;43:663–672. doi: 10.1016/j.molcel.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurford RK, Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- 48.Cam H, et al. A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell. 2004;16:399–411. doi: 10.1016/j.molcel.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 49.Litovchick L, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–551. doi: 10.1016/j.molcel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 50.Korenjak M, et al. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell. 2004;119:181–193. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 51.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pilkinton M, Sandoval R, Colamonici OR. Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene. 2007;26:7535–7543. doi: 10.1038/sj.onc.1210562. [DOI] [PubMed] [Google Scholar]
- 53.Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 55.Chen D, et al. Division and apoptosis of E2f-defient retinal progenitors. Nature. 2009;462:925–929. doi: 10.1038/nature08544. Reveals the surprising finding that activator E2F family proteins are dispensable for proliferation in the developing retina. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chong JL, et al. E2f1–3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462:930–934. doi: 10.1038/nature08677. Demonstrates the paradox that activator E2Fs are only required for repression and cell cycle exit in the intestine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ji P, et al. An Rb–Skp2–p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol Cell. 2004;16:47–58. doi: 10.1016/j.molcel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 58.Gao D, et al. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell. 2009;20:3305–3316. doi: 10.1091/mbc.E09-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nature Genet. 2010;42:83–88. doi: 10.1038/ng.498. Determines that SKP2 deficiency stabilizes p27 expression and prevents tumorigenesis in Rb+/− mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kabeche L, Compton DA. Checkpoint-independent stabilization of kinetochore–microtubule attachments by Mad2 in human cells. Curr Biol. 2012;22:638–644. doi: 10.1016/j.cub.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bester AC, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. Demonstrates that loss of RB function leads to nucleotide deficiency and aberrant DNA replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalo S, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nature Cell Biol. 2005;7:420–428. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- 64.Benetti R, et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. 2007;178:925–936. doi: 10.1083/jcb.200703081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24:1364–1376. doi: 10.1101/gad.1917310. Shows that RB controls centromere structure and function and that in its absence mitotic errors lead to aneuploidy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coschi CH, et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24:1351–1363. doi: 10.1101/gad.1917610. Demonstrates, using a knock-in mutant of RB, that defective chromosome condensation contributes to cancer pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Harn T, et al. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010;24:1377–1388. doi: 10.1101/gad.580710. Shows that deletion of all three RB-family proteins causes chromosomal breaks and aneuploidy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nature Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 70.Wu L, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 71.Ianari A, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–194. doi: 10.1016/j.ccr.2009.01.026. Reveals that phosphorylated RB participates in a transcriptional activation mechanism that drives the expression of apoptotic target genes in response to DNA damage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Helin K, et al. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- 73.Seifried LA, et al. pRB–E2F1 complexes are resistant to adenovirus E1A-mediated disruption. J Virol. 2008;82:4511–4520. doi: 10.1128/JVI.02713-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cecchini MJ, Dick FA. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem J. 2011;434:297–308. doi: 10.1042/BJ20101210. [DOI] [PubMed] [Google Scholar]
- 75.Wells J, Yan PS, Cechvala M, Huang T, Farnham PJ. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene. 2003;22:1445–1460. doi: 10.1038/sj.onc.1206264. [DOI] [PubMed] [Google Scholar]
- 76.Avni D, et al. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol Cell. 2003;12:735–746. doi: 10.1016/s1097-2765(03)00355-1. [DOI] [PubMed] [Google Scholar]
- 77.Calbo J, et al. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J Biol Chem. 2002;277:50263–50274. doi: 10.1074/jbc.M209181200. [DOI] [PubMed] [Google Scholar]
- 78.Carnevale J, Palander O, Seifried LA, Dick FA. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol Cell Biol. 2012;32:900–912. doi: 10.1128/MCB.06286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bosco EE, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117:218–228. doi: 10.1172/JCI28803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irwin MS, et al. Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 82.Carr SM, Munro S, Kessler B, Oppermann U, La Thangue NB. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 2011;30:317–327. doi: 10.1038/emboj.2010.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nature Genet. 2001;3:667–674. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 84.Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006;7:192–198. doi: 10.1038/sj.embor.7400591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Munro S, Khaire N, Inche A, Carr S, La Thangue NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29:2357–2367. doi: 10.1038/onc.2009.511. [DOI] [PubMed] [Google Scholar]
- 86.Saddic LA, et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 2010;285:37733–37740. doi: 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 89.Jacks T, et al. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 90.Lee MH, et al. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- 91.Cobrinik D, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 92.Lamber EP, et al. Structural insights into the mechanism of phosphoregulation of the retinoblastoma protein. PLoS ONE. 2013;8:e58463. doi: 10.1371/journal.pone.0058463. [DOI] [PMC free article] [PubMed] [Google Scholar]