

Abstract
Despite many clinical trials conducted with oncolytic viruses, the exact tumor-level mechanisms affecting therapeutic efficacy have not been established. Currently there are no biomarkers available that would predict the clinical outcome to any oncolytic virus. To assess the baseline immunological phenotype and find potential prognostic biomarkers, we monitored mRNA expression levels in 31 tumor biopsy or fluid samples from 27 patients treated with oncolytic adenovirus. Additionally, protein expression was studied from 19 biopsies using immunohistochemical staining. We found highly significant changes in several signaling pathways and genes associated with immune responses, such as B-cell receptor signaling (P < 0.001), granulocyte macrophage colony-stimulating factor (GM-CSF) signaling (P < 0.001), and leukocyte extravasation signaling (P < 0.001), in patients surviving a shorter time than their controls. In immunohistochemical analysis, markers CD4 and CD163 were significantly elevated (P = 0.020 and P = 0.016 respectively), in patients with shorter than expected survival. Interestingly, T-cell exhaustion marker TIM-3 was also found to be significantly upregulated (P = 0.006) in patients with poor prognosis. Collectively, these data suggest that activation of several functions of the innate immunity before treatment is associated with inferior survival in patients treated with oncolytic adenovirus. Conversely, lack of chronic innate inflammation at baseline may predict improved treatment outcome, as suggested by good overall prognosis.
Introduction
Numerous clinical trials have been conducted to assess the safety and efficacy of oncolytic viruses in treatment of human cancer.1 Although in early trials the emphasis is always on safety, there have also been reports of treatment benefits in many studies. However, there is a considerable amount of variation in clinical outcomes in different patients.2,3 In the context of immunotherapy, molecular characterization of the tumors has yielded promising results in identifying factors that contribute to clinical outcome.4 Optimally, only patients likely to benefit would receive any given treatment. A key realization has been that clinical factors, or the organ of origin are rarely able to identify such patients. Instead, most available biomarkers are based on characteristics of the tumor.
Characterization of tumors can be achieved by compiling different types of information by high-throughput analysis, which can incorporate, for example, gene expression analysis and conventional histopathologic evaluation.5,6 Several analyses of cell-surface receptors and/or mutations in specific oncogenes are already in clinical practice.7,8 Technological advances in the fields of molecular diagnostics and genome scale analysis are creating even more opportunities for information-driven approaches and provide a basis for individualized medicine.9,10 For novel technologies, which may be highly potent but could also be expensive once approved, finding biomarkers is a key goal. It has been proposed that a biomarker should accompany every new cancer drug.11,12
Human data has revealed that oncolytic viruses exert many of their effects through the antitumor immune response they cause, instead of through mere lytic effects.3,13 Adenoviruses are known to be immunogenic14 and their efficacy is reported to be dependent on the immunological status of the tumor.15 Accordingly, understanding the immunological environment of the cancer being treated is of paramount importance in the context of oncolytic virotherapy. Studies with oncolytic adenoviruses have shown that they have stimulating effects on both innate and adaptive immunity.13,16,17 There are also reports indicating that baseline activation of innate immunity and interferon pathways could produce an antiviral state in some cancers, consequently blocking the therapeutic efficacy of the viruses.18,19 However, at the moment, there are no available immunological biomarkers that predict the clinical outcome to an adenovirus-based therapy in human cancer patients.
The aim of this study was to assess the baseline cell level characteristics of tumors in patients treated with oncolytic adenovirus. We monitored mRNA expression levels in 31 biopsy or fluid samples from altogether 27 patients using RNA microarrays, and quantified immunohistochemical (IHC) stainings of 19 biopsies. Differentially expressed genes between different clinical categories were then identified from the microarray data. Biological functions and pathways present in the tumors were estimated based the on gene expression levels. IHC staining results were compared between patients with longer and shorter than expected overall survivals. Additionally, to identify possible antiviral phenotypes in tumors, we evaluated the protein-level expression of a major interferon-inducible gene Myxovirus resistance protein A (MxA) in an extension cohort of 10 patients. Finally, we evaluated individual genes as candidate predictive and prognostic markers for oncolytic adenovirus treatment.
Results
Genes are differentially expressed in pretreatment samples between several clinical outcome categories
To evaluate gene expression in tumors of patients prior to treatment with an oncolytic adenovirus, mRNA levels were quantified in pretreatment samples. Microarray analysis was performed on 31 pretreatment samples from a total of 27 patients (Supplementary Table S1). Out of the 31 samples, 16 samples represented tumor biopsies, 8 ascites samples and 7 pleural fluid samples.
Differentially expressed genes between clinical subgroups were determined according to sample material (biopsy, ascites, or pleural fluid), or by using all samples together, with or without sample-type specific normalization that takes into account expression differences between the liquid and solid compartments (Table 1). Gene expression profiles from ascites and pleural samples were similar while a large number of genes were differentially expressed between biopsies and fluid samples. Differences between separate cancer types were relatively small, especially after sample-type–specific normalization.
Table 1. Number of genes found to be differentially expressed in the baseline sample-set.
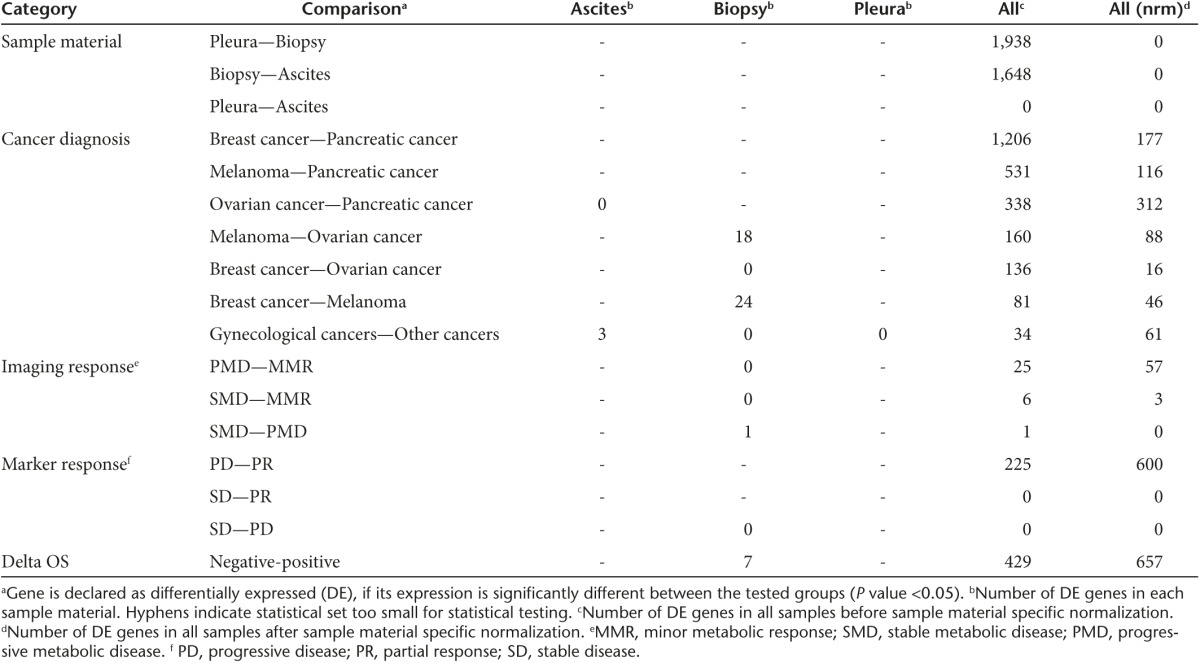
Grouping of samples based on various clinical outcomes and subsequent differential expression analyses of these clinical sample groups revealed various genes with a significantly altered expression profile, especially in comparisons between categories representing the best and the worst outcomes. Most diversity was found in outcome categories defined by imaging response, tumor marker response and deviation from expected overall survival (deltaOS), which was determined by comparison to matched control patients.20
Pathway enrichment analysis reveals distinct biological functions activated in tumors of patients with longer and shorter than expected survival
Differential expression of most relevant biological functions used in pathway analyses were identified and compared between different clinical groups (Supplementary Figure S1). A higher degree of pre-existing immune response was observed in groups with worse outcomes. In contrast, these groups displayed less cancer-related gene expression. Statistically most significant differences were seen in comparisons with samples from positive and negative deltaOS patients, in part due to the largest available dataset.
Most significantly altered biological functions were determined by comparing gene expression between patients having shorter or longer than expected OS (Supplementary Figure S2). Top categories of differently expressed functions were related to cellular growth and proliferation, hematological system development and function, cellular movement, tissue and cellular development, cell to cell signaling, cancer, and importantly, immune cell trafficking, infection and inflammatory responses. Most of the annotated functions associated with immune responses, such as proliferation and quantity of lymphocytes and mononuclear leukocytes at baseline, were increased among patients with shorter than expected OS (Supplementary Table S2).
Pathways associated with inflammatory responses are significantly altered between patients surviving shorter and longer than their matched controls
Pathways that were most significantly changed between patients with longer and shorter than expected survival were identified (Figure 1). Many of the differentially expressed pathways belonged to inflammatory responses. These pathways were all primarily upregulated in patients with shorter than expected OS and included B cell receptor signaling, PI3K signaling in B lymphocytes, GM-CSF signaling, leukocyte extravasation and signaling, role of nuclear factor of activated T-cells (NFAT) in regulation of the immune response, HMGB1 signaling, complement system and pattern recognition receptors (Supplementary Figures S3–S6). Interestingly, in negative deltaOS patients, many pattern-recognition and complement receptors, such as C3aR (P = 0.06), C5aR (P = 0.11), TLR1 (P = 0.12), TLR4 (P = 0.07), TLR6 (P = 0.20), TLR7 (P = 0.07), TLR8 (P = 0.02), CD79A (P = 0.13), and CD79B (P = 0.15), were upregulated, although many of them did not reach statistical significance when analyzed individually.
Figure 1.

Graphical presentation of top 20 most significantly changed signaling pathways between patients with negative or positive deltaOS. All of the presented pathways were primarily upregulated in the deltaOS-negative patients compared to deltaOS-positive patients.
Helper T-cell marker CD4 and macrophage marker CD163 are elevated in patients with worse than expected overall survival
In order to study the phenotypic relevance of the array findings, we performed protein level analyses of immunological variables (Figure 2). In addition to clear trends in several markers, T-cell marker CD4 (P = 0.020) and macrophage lineage marker CD163 (P = 0.016) were significantly differentially expressed between short and long surviving patients (Figure 2a,b,d). The other evaluated macrophage marker CD68 was also elevated in deltaOS negative patients, yet not significantly, further suggesting the presence of tumor-associated macrophages in patients with worse than expected survival.
Figure 2.

Comparison of immunohistochemical scores between different deltaOS groups. (a) CD68 and CD163 biopsy stainings for baseline biopsies taken from two patients O340 (positive outcome in all clinical parameters) and I398 (negative outcome in all clinical parameters). (b–d) Quantitative immunohistochemical analyses of different immunomarkers in tumor biopsies. Biopsy scores were compared between negative and positive deltaOS patients, and they were significantly different for CD4 (P = 0.020) and CD163 (P = 0.016). Error bars are shown as mean + SEM. *P < 0.05
Correlation analysis for coexpression of stained markers was performed separately for deltaOS-negative and -positive patients (Supplementary Figure S7). Coexpression of CD3, CD4, and CD8 T-cell markers was highly significant in deltaOS-negative patients. In addition, significant coexpression of the T-cell activation marker CD25 with several types of T-cells was seen in these patients.
Low MxA protein expression on pretreatment tumor biopsies trends for improved overall survival
Since several different innate immune receptors were upregulated in patients with shorter than expected survival, we evaluated interferon-stimulated gene expression to address whether activated innate immune phenotype in cancer cells reflects outcome after oncolytic immunotherapy as proposed in preclinical studies.19 Therefore we analyzed major interferon-inducible gene Myxovirus resistance protein A (MxA) by IHC in an extension patient cohort with available pretreatment tissue samples (Figure 3a). Patients with lower MxA score of +1 to +2 showed a trend for improved overall survival (P = 0.054) over patients with the highest MxA score of +3 in this extension cohort, (Figure 3b). The average deltaOS was also lower in the group with high MxA score (−220 versus 177 days) (Figure 3c).
Figure 3.

MxA immunohistochemistry on pretreatment tumor biopsies. Low interferon-inducible MxA protein expression on baseline tumor biopsies correlates with improved overall survival after oncolytic adenovirus therapy. Ten patients with available baseline biopsy material were grouped based on MxA protein expression on pretreatment biopsy samples. (a) Staining panels of the biopsies are grouped in columns based on the assessed MxA score. Asterisk indicates strong stromal staining, instead of strong staining at the tumor. Patients C261 and G322 were not involved in the microarray analyses. Patient C261 was a 46-year-old male, who was diagnosed with colorectal cancer. He received treatment with CGTG-401 and had an overall survival of 123 days. Imaging and marker data were not available. Patient G322 was a 49-year-old female. She was diagnosed with gastric cancer and received treatment with CGTG-102. She had an overall survival of 71 days and a progressive disease marker response. Imaging response was not available. (b) Patients with the highest MxA score +3 showed shorter overall survival than with score +1 or +2, which was found borderline significant (P = 0.054). (c) Average DeltaOS in patients with different MxA scores. The P values were not considered significant. Error bars are shown as mean + SEM.
Genes related to innate immunity are upregulated at baseline in patients with worse clinical outcomes
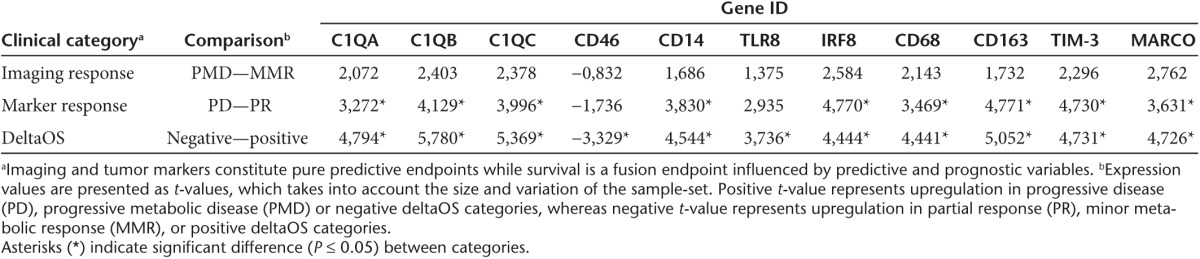
In total, the differentially expressed genes used for pathway analyses contained 1,496 upregulated genes in patients with better than expected OS and 1,296 upregulated genes in patients with worse than expected OS. We examined the 50 most upregulated genes in both groups (Supplementary Tables S3 and S4). The most highly upregulated genes in patients with worse than expected OS included many genes associated with the innate immune system, for example, macrophage markers CD68 (P = 0.009) and CD163 (P = 0.004), macrophage receptor MARCO (P = 0.006), TLR4 coreceptor CD14 (P = 0.008), and complement system subcomponent C1q (subunits A, B, and C, P = 0.001–0.005). Interestingly, we also found T-cell exhaustion marker TIM-3 (P = 0.006) to be significantly upregulated in deltaOS-negative patients, alongside with several other nonsignificantly upregulated T-cell exhaustion markers (Figure 4). We compared these genes in more detail between various clinical categories (Supplementary Table S5) and found significant differences in expression values in comparisons between deltaOS and marker response categories (Table 2).
Figure 4.

Graphical presentation of genes associated with T-cell exhaustion. Positive t-value indicates upregulation in deltaOS-negative patients compared to deltaOS-positive patients, whereas negative value indicates upregulation in positive deltaOS group. Dashed line indicates threshold for significant t-value. Upregulation of TIM-3 in deltaOS-negative patients was significant (P = 0.006). *P < 0.05
Table 2. Selected genes with potential predictive or prognostic value and their expression values between imaging response, marker response and deltaOS comparisons.
Discussion
In this study, we have evaluated gene expression patterns in pretreatment samples of patients subsequently treated with an oncolytic adenovirus. To verify some of the key immunological findings on the protein level, IHC stainings were also performed. Genes were mapped to pathways, allowing detection of highly significant changes in several biological functions and signaling pathways between patients with long or short survival. Due to relatively small sample size, statistical significance was reached only between extremities of the clinical classification. However, the findings were consistent across all tested clinical categories. Interestingly, many of the most significant results were associated with different immune functions, especially in patients with shorter than expected OS.
Two of the most significantly altered pathways in patients with shorter than expected OS were axonal guidance signaling and B-cell receptor (BCR) signaling. Axonal guidance signaling has been found to be associated with migration and survival of cancer cells and the development of tumor vasculature.21,22 Previously, it has also been reported that axonal guidance signaling is activated in T-cell-dependent germinal center B-cells.23 The presence of activated B-cells in patients with worse than expected survival is further suggested by increased BCR signaling, required for the initial antigen recognition and activation of the B-cells. Another factor affecting B-cell activation is signaling through Toll-like receptors (TLRs),24,25 which we found upregulated. Although there were more B-cells in tumors of short surviving patients than long surviving patients (average biopsy scores 0.0017 and 0.0007, respectively), it was not possible to stain for activated B-cells and thus this remains a hypothesis for future studies.
In addition to B-cell-related pathways, GM-CSF signaling was significantly increased in patients with worse than expected survival. This finding is important considering that GM-CSF is used as a transgene in many advanced oncolytic virus constructs.17,26,27 In a recent report by Kanerva et al.,20 certain tumor types were characterized as GM-CSF sensitive or insensitive based on clinical findings, and a significant improvement in overall survival was seen in the GM-CSF-sensitive group. Elevated GM-CSF production at baseline could explain a decreased effect of GM-CSF coding viruses on non-GM-CSF-sensitive tumors.
In our data, several constituents of the complement system were upregulated in patients with short survival. These included all soluble C1q subcomponents, C8 and receptors for C3 and C5A. The complement system is traditionally recognized as a key player in innate immunity, which defends host against microbes by using opsonization of pathogens and direct killing by lysis.28 The complement system also acts as a bridge between the innate immune response and the subsequent activation of adaptive immunity. Complement activation has also been identified in patients with different types of cancers.28 Moreover, previous findings suggest that complement components may contribute to the immune surveillance of malignant tumors and may also promote tumor growth.28
Despite the activity of complement, tumor cells seem to be resistant to its effects.29 This resistance may be due to the expression of membrane complement regulatory proteins (mCRPs), which include, for example, SerpinG1, CD35 (complement receptor type 1 or CR1), CD46 (membrane cofactor protein or MCP), CD55 (decay-accelerating factor or DAF), and CD59. Previous studies demonstrate that various tumor cells express at least one mCRP, most often CD46, CD55, or CD59 (refs. 30,31). Interestingly, our data show that many mCRPs including SerpinG1, CR1 and complement factor D are upregulated in patients surviving a shorter time than their controls, indicating resistance to complement. These findings are also compatible with the notion that, even if tumor cells are protected against an elevated activation status of complement, oncolytic viruses are not. As a consequence, this could increase viral clearance at the tumor.
In contrast to other mCRPs, CD46 (or MCP) was upregulated in patients with positive deltaOS. Interestingly, CD46 acts also as a receptor for serotype 3 adenovirus.32 Many of the viruses used in this study were based on the chimeric Ad5/3 backbone, which includes the Ad3 knob. Accordingly, it is interesting to hypothesize, that baseline upregulation of CD46 would lead to more robust virus infection and replication in patients with longer than expected survival. It should be noted, however, that it is not clear if CD46 is one of the receptors for 5/3 chimeric adenoviruses.33
Another factor affecting the prognosis of adenovirus treated patients could be the presence of tumor-associated macrophages (TAMs). In previous reports, these cells have been shown to promote tumor angiogenesis, invasion, intravasation, and metastasis in animal models.34 There are also several reports indicating CD68+ or CD68+CD163+ TAMs to be associated with worse clinical outcomes in different human cancers.35,36 In our data, the presence of TAMs and their effect on the prognosis is supported by the list of the most upregulated genes in patients with worse than expected overall survival, which contains many macrophage-associated genes, such as CD14, CD68, and CD163. We also found TAM-associated genes34 CD34, CCR2, and CSFR1 to be more expressed in this patient subset. Moreover, in IHC analysis, the TAM protein markers CD68 and CD163 were increased in the biopsy samples of these patients (Figure 2a,d).
The main endpoint used in this study was overall survival as compared to matched controls (“deltaOS”). Survival is primarily a prognostic endpoint, although if treatment increases survival, it becomes also a predictive endpoint. Therefore, we consider survival a mixed predictive/prognostic endpoint. In contrast, imaging and tumor markers constitute pure predictive endpoints. We identified possible predictive markers from the baseline gene expression data (Table 2). These candidates included macrophage markers, antigen receptors, components of the complement and the interferon regulatory factor 8 (IRF8). We found significant differences in expression of these genes in tumor marker response categories. Although the findings are preliminary, partly due to the limited availability of imaging and marker response data, and because of the confounding effect tumor inflammation has on size based imaging evaluation,37 these genes again point toward the importance of innate immunity with regard to tumor response or lack thereof.
MxA protein expression has been shown to be an accurate indicator of the antiviral response induced by IFN signaling.38 Upregulation of MxA expression in tumors has been previously found to correlate with virus resistance in animal models.19 Although post-treatment IFN response is potentially important in boosting the development of anti-tumor immunity and combating tumor immunosuppression,39,40 pretreatment IFN signaling marked by MxA expression was associated with shorter overall survival in our data (Figure 3). Also, significant upregulation of IRF8 in patients with worse than expected survival and patients with worse post-treatment marker response suggests that IFN signaling is activated at baseline in patients with worse outcome. High baseline IFN signaling could indicate that the tumor has already been detected by the immune system41 and, possibly, an extensive immunosuppression has been developed. Another possible mechanism contributing to inferior prognosis is the antiviral response created by IFN signaling.42
Overall, our data is in accord with the notion that a heightened baseline immune state indicates that the tumor has progressed further immunologically, and has consequently developed an immunosuppressive micro-environment.43 Several of our findings suggest that one aspect contributing to the immunosuppression in tumors of short surviving patients could be the activation of regulatory B-cells (Bregs). In addition to the BCR and TLR signaling described earlier, the differentiation of regulatory Bregs is dependent on IL10 (ref. 44). Accordingly, we found the IL10 receptor to be upregulated in this patient subset. Bregs promote immunosuppression through several mechanisms, such as complement mediated chronic inflammation45 and regulation of macrophage phenotype.46 Correspondingly, complement activation and macrophage infiltration were seen in patients with worse than expected survival. Therefore it is tempting to speculate that the activation of regulatory B-cells could represent a pivotal step in the development of tumor immunosuppression with a decisive impact on the efficacy of oncolytic virotherapy. This could be studied by evaluation of this cellular subset with flow cytometry, which would enable further characterization of B-cell activation markers in addition to CD19 staining seen in IHC. While specimens for flow cytometry were not available in this study, they could be collected in future trials.
Together with B-cell-mediated immunosuppression, the exhaustion of antitumor T-cells could contribute to the impaired effects of adenovirus therapy.47 We found T-cell exhaustion marker TIM-3 to be significantly upregulated in patients with worse than expected survival. Additionally, several other markers associated with T-cell exhaustion, such as PD-L1, PD-L2, CTLA-4, LAG-3, GAL-9, CD244, CD69, and CD160 were upregulated, although not significantly, in this patient group. It should be noted, however, that the exact role of CD69 in T-cell function remains to be fully clarified.48 Nonetheless, this observation implies that the local accumulation of CD4+ cells, which was seen in the IHC analysis, could actually represent an exhausted helper T-cell population. Since CD4+ T-cells are central for the development of antitumor immunity,49 their dysfunction could indicate an unfavorable environment for immunostimulatory virotherapy. More specifically, as the immune response caused by oncolytic adenoviruses is probably at least partly mediated by T-cells,17 exhaustion at baseline could effectively diminish the therapeutic effect of the viruses. Without immunostimulatory capability, oncolytic adenoviruses would cause chiefly local oncolytic effects at tumors, which might compromise optimal antitumor effects. This indicates potential benefits in combining oncolytic adenoviruses with checkpoint inhibitors.50,51,52
The link between immunosuppression and activation of pathways related to humoral and innate immunity, such as GM-CSF, IFN and complement signaling, requires further investigation. Future experiments could also shed light on the complex roles of B cells, T-cell exhaustion and tumor infiltrating macrophages in the context of virotherapy. Additionally, it would be important to study the different tumor infiltrating T-cell populations, such as regulatory T-cells, in more detail. The expression of PD-1 was not measured due to technical reasons, but this information would be highly interesting to study in the context of possible T-cell exhaustion. The data presented here comprises analyses from baseline biopsy samples. Thus, further studies with follow-up biopsy samples and larger patient materials would be useful in investigating what happens to the identified variables during oncolytic virus treatment. Importantly, our data provide the first comprehensive assessment of biopsies from tumors subsequently treated with oncolytic adenoviruses. The hypotheses generated herein could eventually lead to analyses clarifying the mechanism of action of oncolytic viruses in different tumors. Most importantly, we have identified candidate prognostic and predictive markers, which can be used as a test set for confirmatory studies, possibly resulting in biomarkers for selection of patients for oncolytic virotherapy.
In conclusion, the data presented here show that several functions of innate and humoral immunity are associated with inferior survival of patients treated with oncolytic adenoviruses. Further dissection of these functions, which include B-cell activation and differentiation, complement, TAMs, and interferon signaling, could yield potential prognostic and predictive biomarkers that could help in optimizing the viral treatments. Overall, our data indicate that high baseline activity of the immune system at the tumor is disadvantageous for oncolytic immunotherapy, because it leaves little room for the stimulatory effects of the treatment. Tumors featuring high immunological activation at baseline probably represent tumors that were rather immunogenic at earlier stages of carcinogenesis, and therefore were immunoedited and developed a high degree of immunosuppression. For such tumors, targeting immunosuppression should be incorporated in trial design to produce more relevant results, which might help in transforming oncolytic virotherapy from an experimental therapy into a conventional, clinically available drug. Moreover, from the point of view of tumor immunotherapy, our data indicates that there are two types of tumors: (i) Initially immunogenic tumors which have developed strong countermeasures including significant immunosuppression, and (ii) tumors that are not as immunoedited or immunosuppressive. In the former group, “releasing of the brake” with, e.g., checkpoint inhibiting antibodies may be required for immunogenic immunotherapy (such as oncolytic viruses) to work optimally. “Pressing on the gas” may be sufficient in the latter group, allowing effective single agent oncolytic therapy.
Materials and Methods
Patient samples. We monitored mRNA expression levels from 31 samples collected with ethics committee consent from patients treated with several oncolytic adenoviruses in the Advanced Therapy Access Program.53 Advanced Therapy Access Program was not a clinical trial, and the decisions regarding the therapy were made individually for each patient, based on the characteristics of the patient and his or her cancer. Samples were collected before any viral therapies, and contained eight ascites fluid, sixteen biopsies, and seven pleural fluid samples for microarray analyses (Supplementary Table S1). All biopsies were histologically verified by an experienced pathologist and biopsies containing only normal tissue were removed from the data analyses. The biopsy study was positively evaluated by the Helsinki University Central Hospital operational Ethics committee (Dnro 368/13/03/02/2009) and samples were obtained under written informed consent. Some of the other analyses presented here were positively evaluated by the Helsinki University Central Hospital Operative Ethics Committee (HUS 62/13/03/02/2013).
Oncolytic adenoviruses. The viruses used in this study have been published previously.54,55,56,57,58,59 All of these viruses are replicating oncolytic adenoviruses, which have been modified for tumor selectivity. CGTG-103 (ref. 54) includes Ad5 serotype capsid, whereas viruses CGTG-401 (ref. 55) and CGTG-201 (ref. 56) have Ad3 capsid. Viruses CGTG-102 (ref. 57), CGTG-602 (ref. 58), and CGTG-301 (ref. 59) have chimeric 5/3 serotype capsid. Transgenes that have been inserted in the virus constructs are listed in Supplementary Table S1. The treatments were predominantly administered intratumorally (i.t.). Four patients received all of the treatment dose i.t., whereas one patient received the dose completely intravenously and 1 patient completely intraperitoneally. Remaining patients were treated using two or three administration routes. However, majority of these patients received most of the treatment i.t.
Response evaluation. The clinical status of patients was classified before and after virus treatments. These evaluations allowed us to divide samples into clinical categories that were used for statistical evaluations and to divide patients to subgroups showing clinical response or lack of response after adenoviral treatment. The clinical measurements used in this study were cancer diagnosis, imaging response evaluated by positron emission tomography with CT (F18-FDG-PET-CT), marker response from patient's serum and deltaOS. Previously described PET criteria37 were used for the PET-CT imaging results. DeltaOS was measured as overall survival normalized with control patients' cancer type–specific median survival time.20 Patients were divided into negative (survived less than median of matched controls) and positive deltaOS categories. The rationale for using deltaOS was to convert overall survival statistics to a binary end-point, which was needed for pathway enrichment analysis. Additionally, deltaOS allowed normalization of survival times in the presence of various tumor types, with big differences in prognosis.20
RNA microarrays. Ascites and pleural fluids were collected and kept on ice. Then, cells were pelleted from 10–50 ml of fluid by centrifugation 900 rpm for 8' at +4 °C. The pellet was snap-frozen in liquid nitrogen and stored at −80 °C until use. Biopsies were collected with biopsy gun and stored in RNALater (Life Technologies, Carlsbad, CA) until RNA extraction.
Total RNA was extracted from the samples using TRIZOL Reagent (Life Technologies) and sequentially purified with the RNAEAsy Mini kit (Qiagen, Hilden, Germany) according standard procedures. Finally, RNA was eluated to 30 µl of RNase-free water (Thermo Fisher Scientific, Waltham, MA). RNA quantity was evaluated spectrophotometrically by using Nanodrop (Thermo Fisher Scientific), and the quality was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Normally, samples with no evidence for RNA degradation (RNA integrity number >8) were kept for further experiments.
Genome-wide gene expression profiling of RNA samples was done by hybridizing the RNA to the Illumina HumanHT-12 v4 Expression BeadChips arrays (Illumina, San Diego, CA). The labeling and hybridization was performed with TotalPrep RNA Labeling Kit (Illumina) according to manufactureŕs instructions. BeadChips were washed, blocked and stained with streptavidin-Cy3 and scanned with Illumina iScan (Illumina) by using manufacturer provided protocols. Genome Studio software (Illumina) was used to control the quality of the data. The statistical analysis of the microarray data is detailed in Supplementary Materials and methods.
Immunohistochemistry. Pre- and post-treatment core needle biopsies were collected with written informed consent from patients undergoing oncolytic virus treatment. Biopsies were taken in ultrasound or visual guidance depending on the location of the tumor. Tissue blocks were sectioned using conventional histological techniques. Serial sections (3.5 µm) were taken and mounted on electrically charged glass slides (SuperFrost Plus, Menzel-Gläser, Germany). The first set of sections was stained with hematoxylin and eosin and further sets were used for immunohistochemistry stainings with CD3, CD4, CD8, CD11c, CD19, CD25, CD68, and CD163 antibodies were performed according to standard protocols using 3,3'-diaminobenzidine as detection agent. Collection of biopsies from patients undergoing treatment in Advanced Therapy Access Program received a positive evaluation by the Helsinki University Central Hospital operational Ethics committee (Dnro 368/13/03/02/2009).
IHC analysis. A color information based image processing methodology was applied to quantify the IHC stainings. The samples were first digitized with an automated whole-slide scanner (Pannoramic 250 FLASH, 3DHISTECH, Budapest, Hungary) using a Plan-Apochromat 20× objective (numerical aperture 0.8) and a VCC-F52U25CL camera (CIS, Tokyo, Japan) equipped with three 1,224 × 1,624 pixel Charge Coupled Device (CCD) sensors. After digitization, samples were annotated for tumorous regions by an experienced pathologist and then automatically analyzed. Based on the standard color deconvolution,60 a monochrome channel (CDAB) was extracted, identifying the image pixels stained with 3,3'-diaminobenzidine and their staining intensities. A threshold value was defined for the CDAB monochrome image to further detect exclusively positively stained cellular regions and to filter out possible unspecific staining. The IHC samples were quantified by calculating a fraction of positively stained cellular region in the whole region-of-interest, i.e., tumor region. The image-processing pipeline was implemented in matrix laboratory (MATLAB, version R2012b; MathWorks, Natick, MA) numerical computing environment.
MxA protein analyses. MxA protein expression was analyzed from an extension cohort of 10 patients with available pretreatment tumor biopsy samples. Similarly in this cohort, the patients received oncolytic adenovirus treatments after biopsies, and overall survival was recorded starting on the day of the first virus treatment. The baseline tumor biopsies were assessed for MxA by immunohistochemistry, and the MxA stainings were scored by an independent pathologist from scale of 0 to +3.
Statistical analysis of immunohistological data. Statistical analysis for MxA and other IHC stainings was performed with SPSS Statistics (International Business Machines Corporation, Armonk, NY), Microsoft Excel (Microsoft Corporation, Redmond, WA) and GraphPad Prism (GraphPad Software, La Jolla, CA). Data from immunohistochemistry were analyzed with two-tailed t-test. Correlations between different immunomarkers were analyzed using Pearson's correlation coefficient. For MxA results, differences between staining intensity groups were analyzed using Mann–Whitney U-test and overall survival data with log rank test. P values <0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Table S1. Baseline samples before viral treatments. Table S2. List of the top 50 biological functions differently activated between deltaOS negative and positive patients. Table S3. List of the 50 most up-regulated genes in patients with shorter than expected overall survival. Table S4. List of the most up-regulated 50 genes in patients with longer than expected survival. Table S5. Genes that may have value as predictive or prognostic markers and their expression values (t-value) between different types of cancers and used clinical categories. Figure S1. Heat-map presentations for biological function variation in different clinical comparisons. Figure S2. Graphical presentation of 20 top biological function categories significantly changed between patients with negative or positive deltaOS. Figure S3. Graphical presentation of B Cell Receptor Signaling pathway. Figure S4. Graphical presentation of GM-CSF Signaling pathway. Figure S5. Graphical presentation of Leukocyte Extravasation Signaling pathway. Figure S6. Graphical presentation of Role of Pattern Recognization Receptors in Recognization of Bacteria and Viruses pathway. Figure S7. Correlations of different immunomarkers at tumor hotspot across all patients with positive and negative deltaOS. Supplementary Materials and Methods. Microarray data analysis.
Acknowledgments
We thank Elina Haavisto and Aila Karioja-Kallio for expert assistance. This study was supported by Institute for Molecular Medicine Finland, Oncos Therapeutics Ltd, ASCO Foundation, HUCH Research Funds (EVO), Sigrid Juselius Foundation, Biocentrum Helsinki, Biocenter Finland, Finnish Cancer Organizations, University of Helsinki. A.H. is Jane and Aatos Erkko Professor of Oncology at the University of Helsinki. A.H. is shareholder in Oncos Therapeutics, Ltd. A.H. is employee and shareholder in TILT Biotherapeutics Ltd.
Supplementary Material
References
- Pol, J, Bloy, N, Obrist, F, Eggermont, A, Galon, J, Cremer, I et al. (2014). Trial Watch:: Oncolytic viruses for cancer therapy. Oncoimmunology 3: e28694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichty, BD, Breitbach, CJ, Stojdl, DF and Bell, JC (2014). Going viral with cancer immunotherapy. Nat Rev Cancer 14: 559–567. [DOI] [PubMed] [Google Scholar]
- Le, DT, Uram, JN, Wang, H, Bartlett, BR, Kemberling, H, Eyring, AD et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, K, Soda, M, Togashi, Y, Suzuki, R, Sakata, S, Hatano, S et al. (2012). RET, ROS1 and ALK fusions in lung cancer. Nat Med 18: 378–381. [DOI] [PubMed] [Google Scholar]
- Doucette, T, Rao, G, Rao, A, Shen, L, Aldape, K, Wei, J et al. (2013). Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res 1: 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott, U and Settleman, J (2009). Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol 27: 5650–5659. [DOI] [PubMed] [Google Scholar]
- Chapman, PB, Hauschild, A, Robert, C, Haanen, JB, Ascierto, P, Larkin, J et al.; BRIM-3 Study Group. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff, DD, Stephenson, JJ Jr, Rosen, P, Loesch, DM, Borad, MJ, Anthony, S et al. (2010). Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 28: 4877–4883. [DOI] [PubMed] [Google Scholar]
- Tsimberidou, AM, Iskander, NG, Hong, DS, Wheler, JJ, Falchook, GS, Fu, S et al. (2012). Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 18: 6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, K, Masuda, S and Kimura, H (2013). Impact of biomarker usage on oncology drug development. J Clin Pharm Ther 38: 62–67. [DOI] [PubMed] [Google Scholar]
- Smith, AD, Roda, D and Yap, TA (2014). Strategies for modern biomarker and drug development in oncology. J Hematol Oncol 7: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu, I, Cerullo, V, Hirvinen, ML, Escutenaire, S, Ugolini, M, Pesonen, SK et al. (2012). Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res 72: 2327–2338. [DOI] [PubMed] [Google Scholar]
- Tuve, S, Liu, Y, Tragoolpua, K, Jacobs, JD, Yumul, RC, Li, ZY et al. (2009). In situ adenovirus vaccination engages T effector cells against cancer. Vaccine 27: 4225–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, AW, Senzer, N, Cerullo, V, Templeton, NS, Hemminki, A and Nemunaitis, J (2012). Oncolytic viruses for induction of anti-tumor immunity. Curr Pharm Biotechnol 13: 1750–1760. [DOI] [PubMed] [Google Scholar]
- Cerullo, V, Seiler, MP, Mane, V, Brunetti-Pierri, N, Clarke, C, Bertin, TK et al. (2007). Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther 15: 378–385. [DOI] [PubMed] [Google Scholar]
- Kanerva, A, Nokisalmi, P, Diaconu, I, Koski, A, Cerullo, V, Liikanen, I et al. (2013). Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res 19: 2734–2744. [DOI] [PubMed] [Google Scholar]
- Monsurrò, V, Beghelli, S, Wang, R, Barbi, S, Coin, S, Di Pasquale, G et al. (2010). Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: potential relevance for adenoviral gene therapy. J Transl Med 8: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liikanen, I, Monsurrò, V, Ahtiainen, L, Raki, M, Hakkarainen, T, Diaconu, I et al. (2011). Induction of interferon pathways mediates in vivo resistance to oncolytic adenovirus. Mol Ther 19: 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva, A, Koski, A, Liikanen, I, Oksanen, M, Joensuu, T, Hemminki, O et al. (2015). Case-control estimation of the impact of oncolytic adenovirus on the survival of patients with refractory solid tumors. Mol Ther 23: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédotal, A, Kerjan, G and Moreau-Fauvarque, C (2005). The brain within the tumor: new roles for axon guidance molecules in cancers. Cell Death Differ 12: 1044–1056. [DOI] [PubMed] [Google Scholar]
- Mehlen, P, Delloye-Bourgeois, C and Chédotal, A (2011). Novel roles for Slits and netrins: axon guidance cues as anticancer targets? Nat Rev Cancer 11: 188–197. [DOI] [PubMed] [Google Scholar]
- Yu, D, Cook, MC, Shin, DM, Silva, DG, Marshall, J, Toellner, KM et al. (2008). Axon growth and guidance genes identify T-dependent germinal centre B cells. Immunol Cell Biol 86: 3–14. [DOI] [PubMed] [Google Scholar]
- Browne, EP (2012). Regulation of B-cell responses by Toll-like receptors. Immunology 136: 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, Z and Hou, B (2013). TLR signaling in B-cell development and activation. Cell Mol Immunol 10: 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andtbacka, RH, Kaufman, HL, Collichio, F, Amatruda, T, Senzer, N, Chesney, J et al. (2015). Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 33: 2780–2788. [DOI] [PubMed] [Google Scholar]
- Markiewski, MM and Lambris, JD (2009). Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol 30: 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donin, N, Jurianz, K, Ziporen, L, Schultz, S, Kirschfink, M and Fishelson, Z (2003). Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol 131: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell, S, Geis, N, Rutz, R, Schultz, S, Giese, T and Kirschfink, M (2007). Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol 150: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, NF, Durrant, LG, Madjd, Z, Ellis, IO, Scholefield, JH and Spendlove, I (2006). Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol Immunother 55: 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischli, C, Sirena, D, Lesage, G, Havenga, MJ, Cattaneo, R, Greber, UF et al. (2007). Species B adenovirus serotypes 3, 7, 11 and 35 share similar binding sites on the membrane cofactor protein CD46 receptor. J Gen Virol 88(Pt 11): 2925–2934. [DOI] [PubMed] [Google Scholar]
- Wang, H, Li, ZY, Liu, Y, Persson, J, Beyer, I, Möller, T et al. (2011). Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med 17: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce, JA and Pollard, JW (2009). Microenvironmental regulation of metastasis. Nat Rev Cancer 9: 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud, SM, Lee, AH, Paish, EC, Macmillan, RD, Ellis, IO and Green, AR (2012). Tumour-infiltrating macrophages and clinical outcome in breast cancer. J Clin Pathol 65: 159–163. [DOI] [PubMed] [Google Scholar]
- Tan, KL, Scott, DW, Hong, F, Kahl, BS, Fisher, RI, Bartlett, NL et al. (2012). Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: a correlative study from the E2496 Intergroup trial. Blood 120: 3280–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski, A, Ahtinen, H, Liljenback, H, Roivainen, A, Koskela, A, Oksanen, M et al. (2013). [(18)F]-fluorodeoxyglucose positron emission tomography and computed tomography in response evaluation of oncolytic adenovirus treatments of patients with advanced cancer. Hum Gene Ther 24: 1029–1041. [DOI] [PubMed] [Google Scholar]
- Sadler, AJ and Williams, BR (2008). Interferon-inducible antiviral effectors. Nat Rev Immunol 8: 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, MS, Kinder, M, Matsushita, H, Mashayekhi, M, Dunn, GP, Archambault, JM et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuri, T, Ghosh, A, Kosaka, A, Zhu, J, Ikeura, M, David, M et al. (2014). STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res 2: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, GP, Koebel, CM and Schreiber, RD (2006). Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6: 836–848. [DOI] [PubMed] [Google Scholar]
- Ahtiainen, L, Mirantes, C, Jahkola, T, Escutenaire, S, Diaconu, I, Osterlund, P et al. (2010). Defects in innate immunity render breast cancer initiating cells permissive to oncolytic adenovirus. PLoS One 5: e13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann, JB and Smyth, MJ (2007). Immune surveillance of tumors. J Clin Invest 117: 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill, F, Montfort, A and Capasso, M (2013). B regulatory cells in cancer. Trends Immunol 34: 169–173. [DOI] [PubMed] [Google Scholar]
- He, Y, Qian, H, Liu, Y, Duan, L, Li, Y and Shi, G (2014). The roles of regulatory B cells in cancer. J Immunol Res 2014: 215471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affara, NI, Ruffell, B, Medler, TR, Gunderson, AJ, Johansson, M, Bornstein, S et al. (2014). B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 25: 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry, EJ (2011). T cell exhaustion. Nat Immunol 12: 492–499. [DOI] [PubMed] [Google Scholar]
- González-Amaro, R, Cortés, JR, Sánchez-Madrid, F and Martín, P (2013). Is CD69 an effective brake to control inflammatory diseases? Trends Mol Med 19: 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, HJ and Cantor, H (2014). CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res 2: 91–98. [DOI] [PubMed] [Google Scholar]
- Duraiswamy, J, Freeman, GJ and Coukos, G (2013). Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 73: 6900–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman-Saint Victor, C, Rech, AJ, Maity, A, Rengan, R, Pauken, KE, Stelekati, E et al. (2015). Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520: 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken, KE and Wherry, EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki, A, Oksanen, M and Merisalo-Soikkeli, M (2013). Oncolytic virotherapy trials–letter. Clin Cancer Res 19: 4541–4542. [DOI] [PubMed] [Google Scholar]
- Pesonen, S, Diaconu, I, Cerullo, V, Escutenaire, S, Raki, M, Kangasniemi, L et al. (2012). Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors. Int J Cancer 130: 1937–1947. [DOI] [PubMed] [Google Scholar]
- Pesonen, S, Diaconu, I, Kangasniemi, L, Ranki, T, Kanerva, A, Pesonen, SK et al. (2012). Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res 72: 1621–1631. [DOI] [PubMed] [Google Scholar]
- Hemminki, O, Diaconu, I, Cerullo, V, Pesonen, SK, Kanerva, A, Joensuu, T et al. (2012). Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol Ther 20: 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski, A, Kangasniemi, L, Escutenaire, S, Pesonen, S, Cerullo, V, Diaconu, I et al. (2010). Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 18: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki, O, Parviainen, S, Juhila, J, Turkki, R, Linder, N, Lundin, J et al. (2015). Immunological data from cancer patients treated with Ad5/3-E2F-Δ24-GMCSF suggests utility for tumor immunotherapy. Oncotarget 6: 4467–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajecki, M, Sarparanta, M, Hakkarainen, T, Tenhunen, M, Diaconu, I, Kuhmonen, V et al. (2012). SPECT/CT imaging of hNIS-expression after intravenous delivery of an oncolytic adenovirus and 131I. PLoS One 7: e32871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruifrok, AC and Johnston, DA (2001). Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23: 291–299. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.