

Abstract
Infections with Plasmodium falciparum, the most pathogenic of the Plasmodium species affecting man, have been reduced in part due to artemisinin-based combination therapies. However, artemisinin resistant parasites have recently emerged in South-East Asia. Novel intervention strategies are therefore urgently needed to maintain the current momentum for control and elimination of this disease. In the present study we characterize the phenotypic and genetic properties of the multi drug resistant (MDR) P. falciparum Thai C2A parasite strain in the non-human Aotus primate model, and across multiple passages. Aotus infections with C2A failed to clear upon oral artesunate and mefloquine treatment alone or in combination, and ex vivo drug assays demonstrated reduction in drug susceptibility profiles in later Aotus passages. Further analysis revealed mutations in the pfcrt and pfdhfr loci and increased parasite multiplication rate (PMR) across passages, despite elevated pfmdr1 copy number. Altogether our experiments suggest alterations in parasite population structure and increased fitness during Aotus adaptation. We also present data of early treatment failures with an oral artemisinin combination therapy in a pre-artemisinin resistant P. falciparum Thai isolate in this animal model.
Antimalarial drug resistance is one of the greatest threats to the current malaria eradication agenda1. Oral artemisinin-based combination therapy (ACT) is the standard of care for uncomplicated malaria, while parenteral intravenous (i.v.) treatment is used in severe cases. Artemisinin (QHS) is the fast acting component of ACTs that accelerates clearing of young ring stage parasites by the spleen2. In 2009 decreased susceptibility to AS was first observed in western Cambodia3, and has since been detected in the rest of Southeast Asia, including recent reports of clinical treatment failure of ACTs such as artesunate/mefloquine (AS/MQ) or artemether/lumefantrine (AL or Coartem)4,5. In vitro selection experiments followed by genome-wide association studies have meanwhile identified mutations in the PF3D7_1343700 (PF13_0238) genomic locus encoding a Kelch protein that are strongly correlated with slow clearing parasites6. Such mutations appear to occur in the context of a specific genetic background in Southeast Asia but so far have not been detected in Africa7,8.
The TM90C2A (C2A) parasite is a multidrug resistant P. falciparum strain originally isolated from a patient in Thailand in 1992, prior to the observation of altered susceptibility to QHS in Southeast Asia9. Initial genotyping demonstrated presence of quadruple-mutations in the dhfr-thymidylate synthase gene10 and increased pfmdr1 copy number11. This combination of mutations and pfmdr1 copy number variation (CNV) has been associated in field isolates with higher in vitro inhibitory concentrations to MQ, quinine (QN), halofantrine and QHS, and with failure of MQ monotherapy and AS/MQ combination therapy at the Thai-Cambodian border12,13. Notably, AS/MQ was deployed in 1994 in Thailand and adopted as standard treatment by the Thai health authorities in 2005. However, QHS monotherapy had been used in western Cambodia since the late 1970’s and MQ was introduced in Thailand in 19843.
Recently, we reported the adaptation of the Thai C2A clone to splenectomized Aotus l. lemurinus monkeys (Panamanian Owl monkey). Plasmodium infection in Aotus was first reported in Panama at the Gorgas Memorial Laboratory in 1966 and has since been established as a major non-human primate model to host-parasite-vector interactions, immunology and pathophysiology and evaluation of drugs and vaccines in P. falciparum and P. vivax14. During the adaptation process of passaging the parasite in Aotus, we observed decreasing susceptibility of the parasites to oral MQ or i.v. AS alone or in combination15. To determine the basis for the altered drug sensitivity phenotype during Aotus adaptation, we tested the antimalarial drug efficacy to MQ and artemisinin derivatives in vivo and in vitro in C2A, and correlated these parameters with growth rates, genotypic changes and mutations in antimalarial drug resistance genes.
Results
Artesunate treatment failure with the C2A P. falciparum strain in the Aotus model
We previously demonstrated treatment failures upon AS treatment with the multidrug resistant C2A in the Aotus monkey model15. Specifically, we observed recrudescence in monkey passage VII and suppression in passage VIII, in each case after treatment with AS at 20 mg/Kg i.v. for three days15. To systematically investigate this drug susceptibility phenotype we performed a controlled experiment in Aotus and followed parasitemia upon oral AS treatment alone, or in combination with MQ. Malaria naive and splenectomized male and female Aotus lemurinus lemurinus monkeys were inoculated with 5 × 106 parasites of the C2A clone on its IX serial passage from a donor Aotus and further divided into three oral treatment groups of two monkeys each (Fig. 1, Table 1): one group was treated with a single dose of 40 mg/Kg of oral MQ; a second group was treated with daily doses of 33 mg/Kg of oral AS for three days; and a third group was treated with a combination of a single dose of oral MQ and three daily doses of oral AS. As a control a spleen intact animal was also infected and treated with the same dose regimen as the third group. Parasitemia was patent in all animals between days 1–2 post infection (PI), reaching peak parasitemia of >100,000 parasites/μL in groups 1–3 by day 9–11 PI. In contrast, a peak parasitemia of 340 parasites/μL on day 16 PI was reached in the spleen intact animal. On the first day >100,000 parasites/μL were reached treatment was initiated (Day 0 in Fig. 1).
Figure 1. Antimalarial drug responses of Plasmodium falciparum C2A infected Aotus monkeys.

(A) Parasitemia response plots of Aotus monkeys infected with the P. falciparum C2A clone passage X. Animals were inoculated with 5 × 106 infected red blood cells from a donor monkey (MN26006, passage IX) and treated when parasitemia reached >105 parasites × μL of blood. Panel (A) shows group 1 animals treated with MQ at 40 mg/Kg orally once, group 2 animals treated with AS at 33 mg/Kg orally × three days and group 3 animals treated with MQ and AS at 33 mg/Kg × three days and MQ at 40 mg/Kg orally once. Panel (B) shows a spleen intact control treated with AS at 33 mg/Kg × three days +MQ at 40 mg/Kg orally once on day 16 of infection. Panel (C) shows parasitemia responses of MN24058 and MN26008 inoculated with PfC2A passages III and IV respectively and treated with AS at 33 mg/Kg × three days i.v. +MQ at 40 mg/Kg orally once.
Table 1. Parasitological treatment responses of Aotus monkeys infected with a Plasmodium falciparum C2A clone.
| Group | ID | Aotus | Weight g. | Parasitemia | Day of peak | Group | Regimen mg/Kg | Route | Treatment | Day of treatment | Day rescue | Results primary |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Peak 103 × μL | Duration days | ||||||||||
| 1 | 26058 | 2 | 829 | 104.60 | 11 | MQ | 40 | p.o. | 1 | 11 | 14 | Failed |
| 1 | 26033 | 1 | 758 | 272.74 | 9 | MQ | 40 | p.o. | 1 | 9 | 5 | Failed |
| 2 | 22028 | 2 | 764 | 449.73 | 9 | AS | 33 | p.o. | 3 | 9 | 10 | Failed |
| 2 | 26045 | 1 | 789 | 110.58 | 11 | AS | 33 | p.o. | 3 | 11 | 8 | Failed |
| 3 | 26063 | 1 | 782 | 177.84 | 9 | AS + MQ | 33 + 40 | p.o. | 3 + 1 | 9 | 16 | Failed |
| 3 | 26025 | 2 | 785 | 106.02 | 10 | AS + MQ | 33 + 40 | p.o. | 3 + 1 | 10 | None | Failed |
| 4 | 23042 λ | 1 | 724 | 0.340 | 16 | AS + MQ | 33 + 40 | p.o. | 3 + 1 | 16 | None | Cured |
Male = 1; Female = 2.
MQ = mefloquine; AS = artesunate.
λ = spleen intact.
p.o. = per os.
In each of the three treatment regimes, the animals remained parasitemic with 10–100 parasites/μL for >10 days (Fig. 1A, Table 1 and S3). In the spleen intact animal, parasitemia persisted at low levels until treatment on day 16 PI and was cleared by day 3 of treatment (Fig. 1B). In contrast, when splenectomized animals were infected with parasites from Aotus passages III and IV and treated i.v. with AS at 33 mg/Kg for 3 days and MQ at 40 mg/Kg once at peak parasitemia (<100,000 parasites/μL), parasites were cleared on average on day 5 following initiation of treatment (Fig. 1C, Table S3). Similar clearance times are found in human cohorts: in one study 25% of patients with parasitemia >4% (>100,000 parasites/μL) were positive beyond day 3 of treatment, but only 5% with parasitemia <4%16. Notably, the i.v. AS dosage used in our study is about eight-fold higher than what is commonly given to humans for oral treatment of uncomplicated17,18 or severe malaria (33 mg/Kg versus 4 mg/kg)19. Converting the Aotus animal dose of AS used in this study (33 mg/Kg) to a human equivalent dose based on body surface area20 would result in ~11 mg/Kg, approximately three times the human standard dose of 4 mg/Kg. Together these experiments demonstrate that infections with the MDR C2A isolate in the Aotus/P. falciparum non-human primate model show characteristics of MQ resistance and reduced AS susceptibility, including parasite suppression upon i.v. or oral AS at 33 mg/Kg × three days, or in combination with MQ at 40 mg/Kg once (Fig. 1).
C2A shows altered drug susceptibility profiles ex vivo
To further investigate the drug resistance profile of the C2A strain we performed ex vivo assays of C2A from several Aotus passages with a series of antimalarial compounds. Specifically, we determined the ex vivo IC50 values for Aotus passages II and X and the original in vitro-adapted TM90C2A isolate against MQ, CQ, atovaquone (ATV), QHA, DHA and AS (Fig. 2). As a control we also measured the IC50 of the sensitive D6 strain (CQ and MQ susceptible). These experiments confirmed that TM90C2A is indeed resistant to MQ, CQ and ATV (Fig. 2A), as observed in our previous experiments in the Aotus model15. MQ IC50s are >30 nM, a concentration that has been associated with treatment failures in infected patients21,22. Interestingly we also observed reduced susceptibility with C2A to the three different artemisinin derivatives, QHA, DHA and AS, and compared to D6 (Fig. 2B). IC50 concentrations were above the previously defined thresholds of 12 nM21 for DHA (C2A passage X), and 20 nM21,23 for AS (C2A passages II and X). Interestingly, parasites showed a significant decrease in susceptibility against MQ, QHS and DHA between in vitro-adapted TM90C2A and C2A passages II and X. In contrast, CQ IC50 was not significantly different across C2A passages. To further evaluate possible reduction in artemisinin susceptibility across C2A passages, we performed the recently established ring stage assay (RSA)24. This experiment indeed demonstrated some reduced susceptibility for ex vivo C2A passages II and X compared to the in vitro-adapted original TM90C2A isolate and the D6 control (Fig. 2C), although still below the defined threshold of 1% ring stage survival for resistance24. Altogether these data demonstrated phenotypic changes in C2A across Aotus passages such as altered susceptibility to QHS, DHA, and MQ. Interestingly the in vitro-adapted TM90C2A did not show any signs of altered artemisinin susceptibility while being equally resistant to MQ and CQ compared to the Aotus passages.
Figure 2. Ex vivo/in vitro antimalarial drug susceptibility and Pfmdr1 copy number variation of PfC2A across Aotus passages.

(A,B) IC50 antimalarial drug nM concentration bar plots of reference strains D6 (white bar), in vitro culture adapted TM90C2A (blue bar) and pfC2A Aotus adapted passage levels II (black bar) and X (red bar). Drug IC50 nM resistance threshold (black line). Mean ± sample SEM (see also Table S1). (C) DHA drug susceptibility based on RSA. Survival percentage = DHA treated parasites/DMSO treated parasites × 100. Red bar represents survival of artemisinin resistant positive control strain KH001-029, D6 is included as negative reference strain. Significant survival threshold (black line) is set at 1% as in Witkowski et al.24. (D) Passage level pfmdr1 copy number fold change of a Plasmodium falciparum C2A clone during adaption to Aotus monkeys. P. falciparum strains with two pfmdr1 gene copies are considered resistant to MQ. Mean ± sample SEM. Dashed line indicates threshold for pfmdr1 CN level indicative of MQ resistance. Dotted line indicates threshold for pfmdr1 CN level indicative of MQ sensitivity. MQ = Mefloquine; CQ = Chloroquine; ATV = Atovaquone; AS = Artesunate; QHS = Artemisinin; DHA = Dihydroartemisinin. p = Mann-Whitney U significance t test unpaired samples. ***p < 0.005; **p < 0.05.
Genetic analysis of C2A during Aotus passages and in vitro adaptation
To track possible changes in the genetic composition of C2A across Aotus passages and during in vitro adaptation we first analyzed copy number variation (CNV) of pfmdr1 (Fig. 2D). These experiments demonstrated variation in pfmdr1 copy number between 2 and 2.5 across all passages, and copy number of 2 in the in vitro-adapted line, suggesting presence of mixed genotypes across Aotus passages.
Given the pfmdr1 CNV data and the observed drug resistant profiles with C2A, we performed detailed genetic analysis of C2A across passages and with the in vitro-adapted TM90C2A. To determine whether C2A represented a single genotype across passages, despite the observed pfmdr1 CNV, we performed genotyping using a molecular barcode assay that assesses the parasite genotype based on a combination of 24 neutral loci across the 14 P. falciparum chromosomes25. Genotyping by molecular barcode assay demonstrated that C2A parasites across all passages from TM90C2A to passage X show identical or very closely related major haplotypes (Fig. 3A). Genetic analysis of drug resistance loci revealed resistance mutations across all passages including a mutation in the in vitro-adapted TM90C2A within the pfcrt (M74I, N75E, N76T, A220S, N326S, I356T) and pfdhfr (N51I, C59R, I164L, S108N) loci (Fig. 3B). No mutations were found in the Kelch K13 locus across TM90C2A and passage II, III and X. However, compared to the 3D7 reference sequence we found a tandem asparagine insert (NN) after codon 142 in the propeller domain in passages II, III and X as well as in the in vitro-adapted TM90C2A. This mutation has been observed in other parasites and is not related to an artemisinin resistance phenotype26. Interestingly specific mutations in pfmdr1 (Y184F), pfdhps (A437G, A581G) and pfATPase (L263A) were only found in passages II and III (Fig. 3B). Similarly distinct sets of mutations were found in the in vitro-adapted TM90C2A in pfdhfr (N51Y, C59M) and pfATPase (L263I, A623R, I431L, S769A/N), and these parasites also harbor unique mutations in pfcytb (Y268E, M270S). Given the molecular barcode data these observations suggest that the original C2A strain was a mixture of closely related parasite genotypes from which at least two were selected during subsequent Aotus passages (represented by passages II/III and passages IV-X, respectively) and another genotype was selected during in vitro adaptation (TM90C2A).
Figure 3. Molecular barcode and antimalarial drug resistance loci across C2A passages.

(A) Plasmodium falciparum barcode. (B) Drug resistance genotyping profiles. nd = not determined; λ = spleen intact.
Serial passaging of C2A in Aotus selects for increased growth phenotype
As in other biological systems P. falciparum adaptation (in vitro and in animals) selects for the genotypes with maximal growth in the absence of any other selection (i.e., drug pressure)27,28,29. Increased growth rates typically result in decreased drug susceptibility profiles as demonstrated in the rodent malaria model30. We therefore wanted to quantify the altered parasite growth rates across Aotus passages, as a possible contributor to the observed changes in drug susceptibility. We analyzed growth rates during Aotus adaptation based on the recorded parasitemia from each of the 10 passage experiments (Fig. 4A), considering the time (Fig. 4B) and peak parasitemia (Fig. 4C). Specifically we estimated parasite multiplication rate (PMR, Fig. 4D) from the median of all increases in parasitemia at 48-hour intervals averaged across monkeys in the same passage. These data demonstrated that parasite growth increased significantly from a PMR just above 1 in passage I to PMR of above 7 in passage X. Interestingly increased growth rates were not linked to genomic deletions on chromosomes 2 and 9 (Fig. S1) that occur frequently during in vitro parasite adaptation31 and have been observed phenotypically in a splenectomized monkey model32.
Figure 4. Plasmodium falciparum C2A growth across Aotus passages.

(A) Parasitemia plots from individual monkeys (colored lines) with time of treatment (black dotted lines) across each passage. Parasitemia was followed for up to 100 days post inoculation and frequently fell below detectability (disappearance of colored lines). (B) Day of peak parasitemia trend. The day of peak parasitemia is the time since inoculation when peak parasitemia was achieved, averaged across monkeys for each passage. (C) Peak parasitemia trend. The value of peak parasitemia is the maximum parasitemia reached over the course of infection, averaged across monkeys for each passage. (D) Parasite multiplication rate (PMR). The PMR is the median of parasitemia increases across 48-hr periods over the course of an infection, averaged across monkeys for each passage. The data is modeled with linear regression.
Discussion
In a previous study we observed MQ and AS treatment failures with the Thai multi-drug resistant PfC2A in Aotus monkeys15. Here we systematically investigate the antimalarial drug responses of this isolate and its changes during host adaptation.
Artemisinin resistance in humans is defined as reduced parasite clearance rate or persistence of microscopically detectable parasites on the third day of ACT therapy33. We observed suppression and persistence of microscopically detectable parasites beyond the third day of oral AS alone or in combination with MQ, indicating resistance of the Aotus-adapted PfC2A isolate based on the above definition. The observed phenotype is unlikely due to limited AS and/or MQ bioavailability by the oral delivery route as our previous experiments in Aotus with the FVO P. falciparum strain have efficiently cleared parasites at day 4 when AS was administered i.v. or orally at 8 mg/Kg for three days34. Similarly MQ treatment as a single dose of 20 mg/Kg cleared parasites on day 5 following initiation of treatment in Aotus infected with FVO35. However, we cannot exclude that an inoculum effect36 may have contributed to the observed relative lower efficiency of the oral route at high parasitemia densities (≥100 × 103/μL) compared to the i.v. route at low parasitemia (<1.0 × 103/μL). Finally, splenectomy may have resulted in reduced efficiency of artemisinin-mediated parasite clearance as discussed previously35.
In vitro and ex vivo experiments demonstrated reduced DHA susceptibility at later Aotus passages, although remaining below the standard resistance threshold of 1% ring stage survival. Genotyping experiments demonstrated presence of wild type Kelch K13 locus across all C2A passages, therefore ruling out involvement of this major resistance locus in the observed in vivo and in vitro or ex vivo drug susceptibility phenotypes. We also demonstrated treatment failure upon MQ treatment in Aotus and MQ drug resistant phenotype in vitro and ex vivo. Pfmdr1 copy number fluctuated between 2 to 2.5 during Aotus adaptation while MQ susceptibility decreased at later passages. These observations contrast with in vitro studies where pfmdr1 de-amplification results in increased MQ susceptibility. For example, in an artelinic acid (AL)-resistant line of P. falciparum (W2AL80) and clones originating from it, pfmdr1 de-amplification resulted in partial reversal of resistance to AL and increased susceptibility to MQ, even in the absence of drug pressure37,38.
It is possible that both, reduced artemisinin susceptibility and MQ resistance during Aotus adaptation are due to increased growth rates. For example, treatment of neurosyphilis with the zoonotic malaria parasite P. knowlesi was eventually abandoned due to increased virulence of this species after multiple passages in humans39. We modeled PfC2A growth rates during Aotus adaptation and indeed demonstrated a significant increase in parasite multiplication rate from near 1 (passage I) to above 7 (passage X). It is therefore possible that the increased growth (i.e., parasite fitness) during serial passaging in Aotus resulted in altered drug susceptibility phenotypes. Similar observations have been made in the mouse malaria model, where increased fitness in P. chabaudi correlated with reduced susceptibility to artemisinin30.
In conclusion, we present a comprehensive study on Aotus adaptation of the multidrug resistant P. falciparum C2A strain. Combined phenotypic and molecular analysis of parasites across Aotus passages demonstrates that host adaptation has co-selected for reduced drug susceptibility in this non-human primate model. Our data also suggest that this model may be used for the evaluation of anti-malarial drugs against the recently detected Artemisinin resistant strains emerging from South-East Asia.
Materials and Methods
Plasmodium falciparum parasite strains
The original P. falciparum TM90C2A strain and subsequent passages in Aotus (Fig. 5)15, as well as ex vivo cultures thereof, were used in this study. In addition, the Aotus adapted strain P. falciparum FVO34,40 and in vitro adapted strains, D6, D10, CS2 and W2mef, were used as control parasites for these studies.
Figure 5. Genealogy of Plasmodium. falciparum C2A adaptation to Aotus lemurinus lemurinus monkeys.
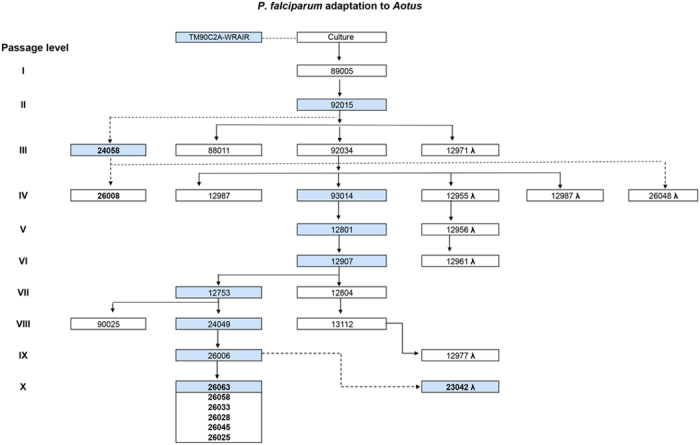
Blue highlighted squares indicate samples from which gDNA was extracted for genetic studies. Aotus monkey numbers in bold mark those that have been used in this study.
Animals
Monkeys of the species Aotus l. lemurinus (Panamanian Owl monkey) Karyotype VIII & IX41 were used in this study. Animals were housed at Gorgas Memorial Institute of Health Studies (ICGES) in Panama, and cared and maintained as described42. Briefly, the animals were kept in stainless steel 4 unit quads cages (Lab Products Inc., Seaford, DE) with dimensions of 27 × 23.5 × 29.5 inches. Each cage was fitted with a 3⁄4-inch-diameter PVC pipe perch placed across 2/3 of the length of the cage and a 6-inch-diameter × 14.5 inches long PVC T pipe nest-box attached to the roof and back of the cage with cable zip ties. Cages were routinely cleaned and sterilized at 180° F at weekly intervals in a cage washing machine (Steris®, Erie, PA). During experimental infections the animals did not receive analgesics.
The experimental protocol was approved by the ICGES Institutional Laboratory Animal Care and Use Committee (CIUCAL) in accordance with procedures described in the “Guide for the Care and Use of Laboratory Animals,” 1996; protocol approval number 2006/02. All experiments described herein were performed in accordance with the approved guidelines.
Drug efficacy studies in Aotus monkeys
To determine drug efficacy using the commonly used delivery route in humans, MQ and AS were applied orally alone or in combination. Six splenectomized male and female laboratory bred Aotus weighing between 758–829 g and one spleen intact control were inoculated intravenously (i.v.) in the saphenous vein with 5 × 106 parasitized erythrocytes of the C2A strain from a donor monkey (passage IX). The animals were splenectomized ~30 days prior to inoculation as described43 (Fig. 5, Table 1). Parasite density was determined daily by Giemsa stained thick blood smears using the method described by Earle and Perez44, with 50 μL of blood obtained with a lancet prick from the marginal ear vein. When parasitemia reached ~100,000 infected RBCs/μL, animals were assigned to one of the three treatment arms (groups) by weight and sex and treated as shown in Table S3. Drug doses were calculated on a milligram (mg) base per kilogram (kg) of body weight basis45. Oral administration of drugs was by gastric intubation with a 14 French red rubber catheter, while for i.v. administration a 25g butterfly needle was used. Response to treatment was categorized as no effect, parasite suppression without clearance, parasite clearance and recrudescence, or parasite clearance and cure. The day of parasite clearance was defined as the first of three consecutive days in which the thick blood films were parasite negative. The day of recrudescence was defined as the first of 3 consecutive days of positive thick blood films after a period of clearance. Parasite suppression was defined as a transient decrease in parasite density post-treatment without clearance15. Animals were considered cured if no recrudescence was observed after parasite clearance and 100 days of follow up with twice a week negative thick smears.
Before initiation of treatment, citrated whole blood was collected from each animal, plasma removed, and cryopreserved with Glycerolyte® in liquid nitrogen and labeled passage level X for further in vitro IC50s determination, Ring Susceptibility Assay (RSA) and genotypic studies. Rescue treatment with MQ at 40mg/Kg orally once and AS at 20 mg/Kg i.v. for three days was triggered by any of the following: Hematocrit (HTO) below 50% of baseline, thrombocytopenia (<50 × 103/μL) or signs of depression or anorexia as determined by the attending veterinarian. Two animals were also included in this study as historical references representing infections with C2A passages III (MN24058) and IV (MN26008) shown in Fig. 5 and Table S1.
In vitro drug assays
In vitro IC50 values were obtained using the hypoxanthine incorporation assay46. IC50 cutoff values indicative of resistance were adopted from the literature as follows: >100 nM for CQ21,22,23,47, 30 nM for MQ21,22,47, 20 nM for AS21,23, and 12 nM for DHA21. Resistance cutoff points for QHS and ATV were not available from the literature. Ex vivo parasite passages were cultured for up to 8 serial passages in RPMI media containing 25% human AB + serum and 4% packed red blood cells. Thin smears were stained with Giemsa for morphological studies and detection of gametocytes. In vitro ring stage 0–3 hours survival assays (RSA) were carried out as described24 and the threshold for resistance was established at 1% survival.
Genetic analysis of parasite strains
Molecular barcode and drug resistance genotyping
Genomic DNA was obtained from blood samples using phenol extraction-ethanol precipitation protocol for purification and concentration of DNA as described48. DNA concentrations of the samples were measured using a NanoDrop® spectrophotometer ND1000. Extracted DNA was pre-amplified and genotyped across 24 genomic loci with both high-resolution melting (HRM) and TaqMan technologies (Life Technologies, Grand Island, NY) as previously described25,49. Samples were diluted 1:20 and 5 μL of each pre-amplified product used for the molecular barcode. TaqMan barcoding assays were run on a ViiA system (Applied Biosystems) and genotypes called using the pre-installed analysis software. To confirm haplotypes and determine drug resistance genotypes of the C2A Aotus passages, we used a high resolution melting (HRM) analysis as described50.
Pfmdr1 copy number variation (CNV)
To determine CNV in the pfmdr1 gene across C2A Aotus-adapted passages, gDNA was extracted and used in a qPCR assay, as described13. Pfmdr1 fold changes between C2A passage levels III-X and reference strain FVO were calculated as described51. Fold change results were rounded to the next significant integer for the purpose of determining copy number. Primer sequences are included in Table S2.
Gene sequencing
To determine single nucleotide polymorphisms (SNPs) in the K13 propeller domain, PCR sequencing was carried out. The full ORF of K13 was PCR amplified using Phusion HF DNA Polymerase kit and primers 1F and 1R. The resulting PCR product of ~2.2kb was purified by gel extraction (QIAquick Gel Extraction Kit, Qiagen) and sequenced at Genewiz using primers 1F, 2F, 3F, 1R, 2R and 3R. Primer sequences are included in Table S2.
Chromosomal deletions
To determine whether common genomic deletions in the subtelomeric regions of either chromosome 2 or 9 occurred during C2A passages in Aotus, several loci were analyzed by PCR. For this purpose genomic DNA was extracted using Qiagen™ DNA kit across P. falciparum C2A Aotus-adapted passages, as well as P. falciparum reference strains FVO, D10 and 3D7. PCR amplification of genes PF3D7_0201500 (PFB0075c) and PF3D7_0202000 (PFB0100c) on chromosome 2, as well as PF3D7_0935400 (PFI1710w), PF3D7_0936300 (PFI1755c), PF3D7_0936800 (PFI1780w) on chromosome 9 were carried out in a BioRad™ PCR machine using the following amplification program: 95o Celsius (C) × 5 minutes, 95 °C × 30 minutes, 51.4 °C × 30 minutes, 61.0 °C × 3 minutes for 35 cycles, plus 72 °C × 10 minutes. Amplicons were loaded onto a 1% agarose gel and subjected to electrophoresis at 150 Volts for 30 minutes, stained with ethidium bromide and photo-documentation done with a UV light reader. Primer sequences are included in Table S2; primers for KAHRP (PFB0100c) amplification were published previously52.
Statistical analysis
Prism 5 Graph Pad® Software was used to plot and calculate the mean and standard error of the mean (SEM), and the JMP® Pro 10.0.0 statistical program (SAS Institute Inc, Middleton, MA) was used for the Mann-Whitney U significance test between unpaired IC50 values for TM90C2A and passage levels II and X. Matlab R2013a was used to plot parasitemia curves and calculate parasite multiplication rate (PMR). PMR is modeled via polynomial regression using the polynomial degree corresponding to the lowest Akaike Information Criteria.
Additional Information
How to cite this article: Obaldía, N. et al. Altered drug susceptibility during host adaptation of a Plasmodium falciparum strain in a non-human primate model. Sci. Rep. 6, 21216; doi: 10.1038/srep21216 (2016).
Supplementary Material
Acknowledgments
The authors wish to thank the Directors of the Gorgas Memorial Institute in Panama City, Drs. Jorge Motta and Nestor Sosa; Gines Sanchez and Gladys Calvino at Tropical Medicine Research in Panama City for administrative support; Maritza Brewer for secretarial assistance; Camilo Marin, Temistocles Gonzales and the animal care takers for their assistance with animal handling and care. We thank Katelyn Durfee for help with the molecular barcoding and drug resistance marker typing and Courtney Edison for help with the kelch13 propeller domain sequencing. This work was supported in part by a USAMDA contract W81XWH-07-C-044, a SENACYT-IFHARU Panama doctoral fellowship to Nicanor Obaldia III, the Sistema Nacional de Investigacion of Panama (SNI) and the Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, Boston, MA, USA. LC and COB were supported by award number U54GM088558 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
Author Contributions N.O., G.S.D., W.O. and D.K. planned and executed Aotus experiments; N.O., M.M., W.O. and G.O.D. collected and analyzed Aotus monkey experimental data. N.O., L.G., S.K.V., A.M. and M.M. planned, executed and analyzed in vitro drug susceptibility assays. N.O., S.K.V., M.M., R.D., N.B., A.M. and P.Y.M. planned, executed and analyzed molecular biology and genotyping studies. N.O., L.M.C., C.B. and M.M. planned, analyzed and developed growth phenotype models. N.O., M.M., S.K.V., M.T.D. and D.F.W. designed the study. N.O. and M.M. wrote the paper with input from all coauthors.
References
- Alonso P. L. et al. A research agenda to underpin malaria eradication. PLoS medicine 8, e1000406, doi: 10.1371/journal.pmed.1000406 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley C. H. Understanding artemisinin resistance. Science 347, 373–374 (2015). [DOI] [PubMed] [Google Scholar]
- Price R. N. et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364, 438–447, doi: 10.1016/S0140-6736(04)16767-6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M. et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361, 455–467, doi: 10.1056/NEJMoa0808859 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takala-Harrison S. et al. Independent Emergence of Artemisinin Resistance Mutations Among Plasmodium falciparum in Southeast Asia. J Infect Dis, doi: 10.1093/infdis/jiu491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55, doi: 10.1038/nature12876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet, doi: 10.1038/ng.3189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga C. et al. Plasmodium falciparum founder populations in western Cambodia have reduced artemisinin sensitivity in vitro. Antimicrob Agents Chemother 58, 4935–4937, doi: 10.1128/AAC.03055-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C. A. et al. Effects of body size and gender on the population pharmacokinetics of artesunate and its active metabolite dihydroartemisinin in pediatric malaria patients. Antimicrob Agents Chemother 57, 5889–5900, doi: 10.1128/AAC.00635-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edstein M. D. et al. Antimalarial pharmacodynamics and pharmacokinetics of a third-generation antifolate—JPC2056—in cynomolgus monkeys using an in vivo in vitro model. J Antimicrob Chemother 60, 811–818, doi: 10.1093/jac/dkm280 (2007). [DOI] [PubMed] [Google Scholar]
- Looareesuwan S. et al. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54, 62–66 (1996). [DOI] [PubMed] [Google Scholar]
- Woodrow C. J. & Bustamante L. Y. Mechanisms of artemisinin action and resistance: wider focus is needed. Trends Parasitol 27, 2-3; author reply 3–4, doi: 10.1016/j.pt.2010.10.002 (2011). [DOI] [PubMed] [Google Scholar]
- Chavchich M. et al. Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob Agents Chemother 54, 2455–2464, doi: 10.1128/AAC.00947-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M. D., Porter J. A. Jr. & Johnson C. M. Plasmodium vivax transmitted from man to monkey to man. Science 153, 1006–1007 (1966). [DOI] [PubMed] [Google Scholar]
- Obaldia N. 3rd, Milhous W. & Kyle D. Adaptation of a Thai multidrug-resistant C2A clone of Plasmodium falciparum to Aotus monkeys and its preliminary in vivo antimalarial drug efficacy-resistance profile. Am J Trop Med Hyg 81, 587–594, doi: 10.4269/ajtmh.2009.08-0445 (2009). [DOI] [PubMed] [Google Scholar]
- Stepniewska K. et al. In vivo parasitological measures of artemisinin susceptibility. J Infect Dis 201, 570–579, doi: 10.1086/650301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilairatana P. et al. An open randomized clinical trial of Artecom vs artesunate-mefloquine in the treatment of acute uncomplicated falciparum malaria in Thailand. Southeast Asian J Trop Med Public Health 33, 519–524 (2002). [PubMed] [Google Scholar]
- Vijaykadga S. et al. Delayed Plasmodium falciparum clearance following artesunate-mefloquine combination therapy in Thailand, 1997-2007. Malar J 11, 296, doi: 10.1186/1475-2875-11-296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M., Davidson R., Brent A. & Wilkinson R. Oxford Handbook of Tropical Medicine. 56–61 (Oxford Universtiy Press Inc., 2008). [Google Scholar]
- Reagan-Shaw S., Nihal M. & Ahmad N. Dose translation from animal to human studies revisited. Faseb J 22, 659–661, doi: 10.1096/fj.07-9574LSF (2008). [DOI] [PubMed] [Google Scholar]
- Quashie N. B. et al. A SYBR Green 1-based in vitro test of susceptibility of Ghanaian Plasmodium falciparum clinical isolates to a panel of anti-malarial drugs. Malar J 12, 450, doi: 10.1186/1475-2875-12-450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basco L. Field application of in vitro assays for the sensitivity of human malaria parasites to antimalarial drugs. (WHO, 2007). [Google Scholar]
- Toure A. O. et al. [In vitro susceptibility of P. falciparum isolates from Abidjan (Cote d’Ivoire) to quinine, artesunate and chloroquine]. Sante 18, 43–47, doi: 10.1684/san.2008.0103 (2008). [DOI] [PubMed] [Google Scholar]
- Witkowski B. et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13, 1043–1049, doi: 10.1016/S1473-3099(13)70252-4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R. et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J 7, 223, doi: 10.1186/1475-2875-7-223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrentino-Madamet M. et al. Limited polymorphisms in k13 gene in Plasmodium falciparum isolates from Dakar, Senegal in 2012-2013. Malar J 13, 472, doi: 10.1186/1475-2875-13-472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walliker D., Hunt P. & Babiker H. Fitness of drug-resistant malaria parasites. Acta Trop 94, 251–259, doi: 10.1016/j.actatropica.2005.04.005 (2005). [DOI] [PubMed] [Google Scholar]
- Gimode W. R. et al. Fitness cost of resistance for lumefantrine and piperaquine-resistant Plasmodium berghei in a mouse model. Malar J 14, 38, doi: 10.1186/s12936-015-0550-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eber D. In Evolutionary Biology of Host-Parasite Relationship: Theory Meets Reality (eds Poulin R., Morand S. & Skorping A.) 163–184 (Elsevier Science B.V., 2000). [Google Scholar]
- Schneider P. et al. Virulence, drug sensitivity and transmission success in the rodent malaria, Plasmodium chabaudi. Proc Biol Sci 279, 4677–4685, doi: 10.1098/rspb.2012.1792 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carret C. K. et al. Microarray-based comparative genomic analyses of the human malaria parasite Plasmodium falciparum using Affymetrix arrays. Mol Biochem Parasitol 144, 177–186 (2005). [DOI] [PubMed] [Google Scholar]
- David P. H., Hommel M., Miller L. H., Udeinya I. J. & Oligino L. D. Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proceedings of the National Academy of Sciences of the United States of America 80, 5075–5079 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S. & Kremsner P. G. Antidogmatic approaches to artemisinin resistance: reappraisal as treatment failure with artemisinin combination therapy. Trends Parasitol 29, 313–317, doi: 10.1016/j.pt.2013.04.001 (2013). [DOI] [PubMed] [Google Scholar]
- Ohrt C. et al. Efficacy of intravenous methylene blue, intravenous artesunate, and their combination in preclinical models of malaria. Malar J 13, 415, doi: 10.1186/1475-2875-13-415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffet P. A. et al. Ex vivo perfusion of human spleens maintains clearing and processing functions. Blood 107, 3745–3752, doi: 10.1182/blood-2005-10-4094 (2006). [DOI] [PubMed] [Google Scholar]
- Duraisingh M. T. et al. Inoculum effect leads to overestimation of in vitro resistance for artemisinin derivatives and standard antimalarials: a Gambian field study. Parasitology 119 (Pt 5), 435–440 (1999). [DOI] [PubMed] [Google Scholar]
- Chen N. et al. Deamplification of pfmdr1-containing amplicon on chromosome 5 in Plasmodium falciparum is associated with reduced resistance to artelinic acid in vitro. Antimicrob Agents Chemother 54, 3395–3401, doi: 10.1128/AAC.01421-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. S., Mutka T., Sparks K., Patel J. & Kyle D. E. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother 56, 302–314, doi: 10.1128/AAC.05540-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatney G. R., Collins W. E., Warren M. & Contacos P. G. The Primate Malarias. (US Government Print Off, 1971). [Google Scholar]
- Obaldia N. 3rd et al. Evaluation of artemisone combinations in Aotus monkeys infected with Plasmodium falciparum. Antimicrob Agents Chemother 53, 3592–3594, doi: 10.1128/AAC.00471-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N. S. et al. Banding patterns of the chromosomes of two new karyotypes of the owl monkey, Aotus, captured in Panama. J Med Primatol 7, 146–155 (1978). [DOI] [PubMed] [Google Scholar]
- Obaldia N. 3rd, Otero W., Marin C., Aparicio J. & Cisneros G. Long-term effect of a simple nest-box on the reproductive efficiency and other life traits of an Aotus lemurinus lemurinus monkey colony: an animal model for malaria research. J Med Primatol 40, 383–391, doi: 10.1111/j.1600-0684.2011.00489.x (2011). [DOI] [PubMed] [Google Scholar]
- Gramzinski R. A. et al. Susceptibility of Panamanian Aotus lemurinus lemurinus to sporozoite-induced Plasmodium falciparum (Santa Lucia) infection. Am J Trop Med Hyg 61, 19–25 (1999). [DOI] [PubMed] [Google Scholar]
- Earle W. & Perez M. Enumeration of parasites in the blood of malaria patients. Journal of Laboratory and Clinical Medicine 17, 1124–1130 (1932). [Google Scholar]
- Rossan R. N. In Antimalarials Drugs I: Biological background, experimental methods, and drug resistane (eds Peters W. & Richards WHG.) Ch. 9, 265–280 (Springer-Verlag, 1984). [Google Scholar]
- Desjardins R. E., Canfield C. J., Haynes J. D. & Chulay J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16, 710–718 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndong J. M. et al. In vitro activity of chloroquine, quinine, mefloquine and halofantrine against Gabonese isolates of Plasmodium falciparum. Trop Med Int Health 8, 25–29 (2003). [DOI] [PubMed] [Google Scholar]
- Moore D. & Dowhan D. Purification and concentration of DNA from aqueous solutions. Curr Protoc Mol Biol Chapter 2, Unit 2 1A, doi: 10.1002/0471142727.mb0201as59 (2002). [DOI] [PubMed] [Google Scholar]
- Mharakurwa S. et al. Pre-amplification methods for tracking low-grade Plasmodium falciparum populations during scaled-up interventions in Southern Zambia. Malar J 13, 89, doi: 10.1186/1475-2875-13-89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R. et al. Rapid, field-deployable method for genotyping and discovery of single-nucleotide polymorphisms associated with drug resistance in Plasmodium falciparum. Antimicrob Agents Chemother 56, 2976–2986, doi: 10.1128/AAC.05737-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J. & Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, doi: 10.1006/meth.2001.1262 (2001). [DOI] [PubMed] [Google Scholar]
- Marti M., Good R. T., Rug M., Knuepfer E. & Cowman A. F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 306, 1930–1933 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.