

Summary
FtsN is the last known essential protein component to be recruited to the Escherichia coli divisome, and has several special properties. Here we report the isolation of suppressor mutants of ftsA that allow viability in the absence of ftsN. Cells producing the FtsA suppressors exhibited a mild cell division deficiency in the absence of FtsN, and no obvious phenotype in its presence. Remarkably, these altered FtsA proteins also could partially suppress a deletion of ftsK or zipA, were less toxic than wild-type FtsA when in excess, and conferred resistance to excess MinC, indicating that they share some properties with the previously isolated FtsA* suppressor mutant, and bypass the need for ftsN by increasing the integrity of the Z ring. TolA, which normally requires FtsN for its recruitment to the divisome, localized proficiently in the suppressed ftsN null strain, strongly suggesting that FtsN does not recruit the Tol–Pal complex directly. Therefore, despite its classification as a core divisome component, FtsN has no unique essential function but instead promotes overall Z ring integrity. The results strongly suggest that FtsA is conformationally flexible, and this flexibility is a key modulator of divisome function at all stages.
Introduction
Many complex cellular tasks are performed by protein machines. Cell division in Escherichia coli is no exception, with at least 10 proteins known to be required for cytokinesis that colocalize to the site of division at midcell. One of these, FtsZ, is a tubulin homologue that assembles into a structure, called the Z ring, which encircles the inner side of the cytoplasmic membrane. Once the Z ring forms, it recruits other proteins to the divisome complex (Weiss, 2004). FtsA and ZipA associate first, and are required for tethering the ring to the membrane (Hale and de Boer, 1997; Pichoff and Lutkenhaus, 2005). The rest of the complex, consisting of mostly integral membrane proteins, assembles in a second stage (Aarsman et al., 2005). Although some information is available about the protein–protein interactions among these membrane components, only FtsI, a transpeptidase, has a well-defined biochemical function in cell division.
One of the hallmarks of this protein machine is an apparent linear dependency order of recruitment to the Z ring (Goehring and Beckwith, 2005). FtsN, for example, cannot localize to the Z ring until all of the other essential components are present (Addinall et al., 1997; Chen and Beckwith, 2001), while FtsZ is first because it can localize in the absence of any one of the later proteins. The dependency order is FtsZ-ZipA/FtsA-FtsEX-FtsK-FtsQ-(FtsB-FtsL)-FtsW-FtsI-FtsN, with the pairs of proteins separated by a slash exhibiting mutually independent localization, while those in parentheses show co-dependence. However, recent evidence suggests that this order is misleading. First, FtsZ can localize in the absence of either FtsA or ZipA, but cannot form Z rings in the absence of both (Pichoff and Lutkenhaus, 2002). Second, many Z rings fail to localize to potential division sites when FtsN is depleted from cells (Addinall et al., 1997; Chen and Beck-with, 2001). Third, multiple interactions among these proteins exist that are outside the normal dependency order (Di Lallo et al., 2003; Corbin et al., 2004; Karimova et al., 2005). Fourth, the linear dependency order can be bypassed under certain conditions (Geissler et al., 2003; Geissler and Margolin, 2005; Goehring et al., 2005; 2007a). Fifth, overproduction of some cell division proteins can efficiently suppress the requirement for others, indicating functional redundancy, at least under laboratory conditions (Dai et al., 1993; Geissler and Margolin, 2005). These results strongly suggest that the divisome assembles not solely by sequential binary interactions but rather via a web of protein–protein contacts that may also include sequential interactions (Goehring and Beckwith, 2005; Vicente et al., 2006).
FtsN was originally discovered as a multicopy suppressor of an ftsA(ts) mutant, and when overproduced can partially suppress other fts mutants, including ftsQ, ftsI (Dai et al., 1993) and ftsK (Draper et al., 1998; Geissler and Margolin, 2005; Goehring et al., 2007b). This property of FtsN suggested that it had a potentially unique regulatory role. Its late localization was difficult to reconcile with its ability to affect the function of early proteins, such as FtsA. Nevertheless, because it is essential for viability, FtsN has been thought to be part of the core divisome machinery despite the lack of conservation of FtsN outside the enteric bacteria. In support of the importance of FtsN in late divisome functions, the recruitment of non-essential components of the divisome such as AmiC and the Tol–Pal complex to division sites requires FtsN (Bernhardt and de Boer, 2003; Gerding et al., 2007).
Despite its classification as a late divisome protein, recent molecular evidence suggests that FtsN may interact directly with early divisome components such as FtsA. When FtsA or subdomain 1c of FtsA was ectopically localized to cell poles, GFP-FtsN was re-localized from midcell to the poles (Corbin et al., 2004). GFP-FtsI also was recruited to poles, but other divisome proteins were not, suggesting that FtsI was back-recruited by FtsN. Bacterial two-hybrid assays also revealed an interaction between FtsA and FtsI/FtsN (Di Lallo et al., 2003; Karimova et al., 2005), although, as with the polar recruitment assays, a direct protein–protein interaction could not be proven. More recently, premature targeting of FtsQ to midcell in the absence of functional FtsA resulted in the recruitment of all downstream proteins to the septum, except for FtsN (Goehring et al., 2005). Although FtsN is a bitopic protein that has only a small segment in the cytoplasm (Dai et al., 1996; Yang et al., 2004), these results suggest that septal targeting of FtsN has a special requirement, potentially for FtsA.
These intriguing results prompted us to take the next logical step: remove FtsN entirely and ask whether the cells can compensate for its loss. This would provide important insights into whether FtsN has a unique or redundant function, and the nature of the compensatory changes would suggest how FtsN normally functions. Such an approach has been valuable in identifying redundant functions of ZipA and FtsK that are compensated by alterations in FtsA, as well as in understanding the function of FtsA itself. Here, we show that FtsN can indeed be removed, provided there are once again compensatory changes in FtsA. These compensatory changes are distinct from the FtsA mutation that is able to bypass ZipA, but share some overlapping functions.
Results
Isolation of ftsN bypass suppressors that map to a cloned ftsA
Previous studies implicated an interaction between FtsA and FtsN, and an altered FtsA (R286W) could compensate for the loss of ZipA or FtsK. Therefore, we hypothesized that an altered FtsA might suppress the loss of FtsN. As a first step, we constructed an FtsN depletion strain (WM2355), which contains a cat cassette in place of a complete deletion of chromosomal ftsN, and an ApR plasmid with a thermosensitive replication origin that expresses ftsN. At the permissive temperature of 30°C, WM2355 cells (carrying a control plasmid pBAD18kan, see below) were not as short as cells of the parent strain W3110 (Fig. 1A), probably because the levels of ftsN expressed from the plasmid are suboptimal. However, when shifted to 42°C, which causes dilution-mediated depletion of FtsN, the cells became highly filamentous (Fig. 1B) and lost viability (see below), consistent with the requirement of FtsN for cell division. Whereas cells of this strain had Z rings at most potential division sites at 30°C, many potential division sites in the filamentous cells at 42°C lacked rings (data not shown). This absence of Z rings at many division sites after FtsN depletion has been reported previously (Addinall et al., 1997; Chen and Beckwith, 2001). These results support the idea that although FtsN is a late recruit to the septum, it also has a role in enhancing the integrity or activity of the Z ring.
Fig. 1. The cloned ftsA suppressor mutant (ftsA sup) permits cell division and colony formation in the absence of FtsN or FtsA.

A–F. DIC micrographs of FtsN depletion (WM2355) derivatives containing pBAD18kan (A and B), pBAD18kan-FtsA (C and D) or pBAD18kan-FtsAsup (E and F) grown at either 30°C (A, C and E) or 42°C to deplete FtsN (B, D and F) for 4 h after initial growth for 2 h at 30°C (no arabinose added). Inset in (A) shows cells of the parent W3110 strain.
G–I. P1 transduction of ΔftsN::cat into W3110 containing pBAD18kan (G), pBAD18kan-FtsA (H) or pBAD18kan-FtsAsup (I), selecting for CmR and KmR at 37°C (no arabinose added).
J. Complementation of the FtsA depletion strain WM1281 by FtsA* or FtsAsup. WM1281 cells containing the indicated plasmids were either grown at 30°C or induced at 42°C for 2 h prior to spotting on plates, which were then incubated overnight at the indicated temperatures.
K. Anti-FtsN immunoblot of lysates from strains WM2417, containing the chromosomal E124A suppressor and ΔftsN::cat, or parent strain W3110. Markers to the left are in kDa. The position of the FtsN protein band is shown to the right.
If an altered FtsA could allow survival in the absence of FtsN, then we reasoned that depletion of FtsN in the presence of mutagenized FtsA might allow rare colonies to survive. The ftsA gene was mutagenized using error-prone PCR, then cloned into pET28a, a KanR plasmid which expresses a histidine-tagged ftsA at basal levels sufficient to allow growth of an ftsA12(ts) mutant at the non-permissive temperature (Geissler et al., 2003). This plasmid was then introduced into the FtsN depletion strain, and the strain was then grown at 42°C for many generations to remove the ftsN expression plasmid and to select for variants that could survive in the absence of the plasmid. Several colonies were obtained, and the absence of the ftsN plasmid was confirmed by loss of ApR and by PCR analysis (data not shown).
The ftsA genes in these survivor strains were confirmed to carry the suppressor locus, as they could confer survival at 42°C after re-cloning into other plasmids and introducing them into the parent WM2355 FtsN depletion strain. Moreover, this indicated that the plasmid-borne suppressor was sufficient to bypass the requirement for FtsN, and that no secondary suppressors were involved. Sequencing of several of the cloned ftsA genes from suppressor strains yielded identical mutations (see below), so one isolate was chosen for further analysis. The complete absence of FtsN protein in suppressor strains containing ΔftsN::cat was confirmed by PCR (data not shown) and immunoblotting (Fig. 1K). The altered ftsA, which we will call ftsA sup, is a true gain of function allele, because it complemented an FtsA depletion strain (Fig. 1J) or an ftsA12(ts) mutant (data not shown).
Suppressed cells lacking FtsN have a mild cell division defect
Having confirmed that FtsN was dispensable for colony formation in the presence of a suppressor, we then tested the effectiveness of the suppressor in restoring cell division in the absence of FtsN. In contrast to cells of the FtsN depletion strain containing the pBAD18kan plasmid vector (Fig. 1A and B), cells containing pBAD18kan with the cloned ftsA suppressor were relatively short both at 30°C and at 42°C, although the average length of suppressed cells lacking ftsN was higher than those containing ftsN (Fig. 1E and F). As with pET28a, basal levels of expression of the histidine-tagged ftsA sup gene cloned into pBAD18kan were sufficient for the ftsN bypass. Production of wild-type (WT) FtsA from pBAD18kan did not significantly suppress the filamentation caused by FtsN depletion (Fig. 1C and D), confirming that FtsAsup was required for the bypass.
The effectiveness of FtsAsup in bypassing ftsN was also assessed independently by introducing the ΔftsN::cat allele by transduction into W3110 recipient strains containing either pBAD18kan, pBAD18kan-ftsA or pBAD18kan-ftsA sup. Consistent with the above data, only the strain expressing the cloned ftsA sup allele allowed efficient transduction of the ΔftsN::cat allele (~200 transductants, Fig. 1I, see also below). This was consistently well above the typical background of 0–10 colonies per plate for the negative controls (Fig. 1G and H); it is possible that these background colonies represent some type of rearrangement, as a few that were tested were CmR but positive for ftsN (data not shown). We cannot rule out the possibility that some of these result from acquisition of suppressors.
To characterize the suppression more thoroughly, we compared cell lengths and FtsZ-GFP rings in an FtsN+ strain (EC448) versus the isogenic strain lacking FtsN and containing ftsA sup (WM2600). Cells of the control EC448 strain averaged 3.0 ± 0.7 μm in length, with 88% having a single Z ring at midcell. The calculated cell length per Z ring was 3.4 μm (Fig. 2A). In contrast, cells of WM2600 were about three times longer, averaging 9.7 ± 4.8 μm, and Z rings were sparser, with 5.6 μm per ring (Fig. 2B). DAPI staining indicated that the longer cells had four or more nucleoids (data not shown), suggesting that cell division is delayed under these conditions.
Fig. 2. Z rings and relative viability of ftsN + cells with the suppressor and suppressed cells lacking ftsN.

A and B. Representative fluorescence/DIC overlays of logarithmically growing cells of EC448 (FtsZ-GFP) (A) or of WM2600, which is EC448 plus pBAD18kan-FtsAsup and ΔftsN::cat (B).
C and D. Colony viabilities of EC448 carrying derivatives of pBAD18kan (C) or W3110 derivatives (also containing leu::Tn5) with or without the chromosomal E124A allele (D), in the presence or absence of ftsN.
We then asked how the viability of suppressed ΔftsN cells compared with ftsN+ cells with or without expression of ftsA sup. Mid-exponential cultures of WM2600 or EC448 carrying pBAD18kan derivatives at equivalent densities were spotted onto LB plates to measure colony growth. Consistent with its moderate increase in cell length, strain WM2600 formed colonies with a slightly lower (approximately twofold) efficiency compared with the equivalent ftsN+ strains (Fig. 2C, compare rows 1 and 2).
One possibility was that if ftsA sup encoded an activated form of FtsA, perhaps it would be deleterious in the presence of FtsN. However, the colony viabilities of EC448/pBAD18kan-FtsAsup and EC448/pBAD18kan were essentially identical (Fig. 2C, compare rows 2 and 4). Moreover, other plasmid derivatives expressing ftsA sup in W3110 showed no differences from W3110 alone either in cell morphology or in colony viability (data not shown). Finally, an equivalent ftsA suppressor mutant in the absence of WT ftsA also had no detectable phenotype on its own (see below). These results indicate that despite their dramatic suppression of ΔftsN::cat, the ftsA suppressor alleles themselves have no obvious phenotype on their own.
FtsAsup is a triple missense mutant
Sequence analysis of several of the plasmids conferring the ftsN bypass phenotype revealed that they all expressed ftsA genes encoding three altered amino acids: K48R, K117R and E124G. Interestingly, the latter two of these mutations map to subdomain 1c, the region of FtsA sufficient to interact with GFP-FtsN in a polar recruitment assay (Corbin et al., 2004). Figure 3 (bottom)
Fig. 3. Conservation of ftsA suppressor mutations in FtsA.

Top. An alignment of segments comprising the K48, K117 and E124 residues involved in suppression. Conserved residues at these three positions, denoted by numbers at the top, are shown in bold. Species containing a likely homologue of FtsN, defined as an E-value < 0.001 after a BLAST search with E. coli FtsN, are underlined.
Bottom. Positions of the three FtsAsup mutations and R286W (FtsA*) in the corresponding crystal structure of Thermotoga maritima FtsA. shows the positions of the three mutations mapped onto the crystal structure of FtsA from Thermotoga maritima.
An alignment of divergent FtsA sequences show that aspartate or glutamate residues are quite conserved at position 124 among diverse species of bacteria (Fig. 3, top). Nevertheless, the ability of the suppressor mutant to function normally in the absence of WT ftsA indicates that an acidic residue at this position must not be critical for normal function. Moreover, as can be seen in the alignment, the next most common residue at this position is glycine, which is present in several alpha-proteobacteria but also is present in Haemophilus influenzae, a close relative of E. coli that contains an ftsN homologue (Dai et al., 1996). Lysines (or arginines) at position 48 are also quite well conserved, although basic residues at position 117 are less well conserved (Fig. 3, top).
The E124G mutation is sufficient for the ftsN bypass but less efficient than the triple mutant
To identify which of the three mutations were crucial for bypassing the requirement for FtsN, they were separated into single and double combinations, and then tested them for their ability to confer survival of WM2355, the FtsN depletion strain. We cloned these ftsA derivatives into plasmid pCSB1, containing the weakened Trc99 promoter, so their expression levels could be regulated by isopropyl-β-D-thiogalactopyranoside (IPTG).
As shown in Fig. 4A, neither pCSB1 plasmid vector alone (row 1) nor WT FtsA (row 2) could permit significant growth after FtsN depletion at 42°C. However, induction of expression of the triple mutant with different levels of IPTG after FtsN depletion resulted in efficient colony formation (row 10, columns 2–4). On the other hand, no suppression occurred in the absence of IPTG (row 10, column 1). IPTG induction from this plasmid system also complemented ftsA mutants (data not shown). The levels of WT or mutant FtsA produced at 0, 0.1 and 1 mM IPTG at 30°C were measured by immunoblotting with anti-FtsA antibodies (Fig. 4B). Although all ftsA derivatives in pCSB1 are under identical transcriptional and translational controls, FtsA protein levels were somewhat variable from plasmid to plasmid; nonetheless, levels consistently increased with IPTG as expected.
Fig. 4. Effects of various ftsA mutants on bypassing the requirement for FtsN and inhibiting colony formation.

A. FtsN depletion strains (WM2355) containing pCSB1 plasmid derivatives expressing IPTG-inducible alleles of ftsA shown at left were tested for colony viability on plates at 30°C (permissive) or 42°C (to deplete FtsN). IPTG was present in the plates at 0, 0.1, 0.5 or 1 mM, as shown at the top.
B. Immunoblots of representative cultures used for the 30°C spots were probed with polyclonal anti-FtsA antibodies. Both blots show cultures expressing WT FtsA as controls.
Importantly, the single E124G mutation was sufficient to permit significant growth in the absence of FtsN, although the efficiency was not as high as with the triple mutant (Fig. 4A, compare rows 6 and 10). When this was tested independently by transducing the ΔftsN::cat allele into W3110 containing IPTG-induced pCSB1 derivatives containing the triple mutant and the E124G mutant, similar results were observed: the transductant colonies were more numerous and larger for the triple mutant than for E124G (data not shown).
The effects of the other two mutations were more subtle but reproducible. The K117R or K48R single mutants were unable to bypass the need for FtsN, and neither could the K48R K117R double mutant (Fig. 4A, rows 4, 5, 7). However, K48R enhanced the suppression ability of E124G, resulting in colony-forming efficiency similar to that of the triple mutant (row 8). K117R, on the other hand, had no significant effect on E124G (row 9). Therefore, we can conclude that E124G is sufficient to bypass partially the requirement for FtsN, and K48R enhances this activity to the point that the efficiency of colony formation at 42°C is similar to that at 30°C.
E124A is an efficient chromosomal bypass suppressor of ftsN and equivalent to ftsA sup
We also undertook an unbiased selection for spontaneous ftsN bypass suppressors by selecting for rare surviving colonies of a WM2355 derivative after long-term FtsN depletion. We have characterized one suppressor in detail. Genetic mapping and sequencing revealed that the suppressor mutation mapped to ftsA, and is a single missense mutation that results in an E124A change (strain WM2417). When ftsA(E124A) was cloned into pCSB1 and tested for the efficiency of transduction of ΔftsN::cat, E124A and FtsAsup were indistinguishable in their ability to bypass ftsN, and both were significantly more efficient than E124G (Fig. 4, compare lane 6 with lanes 10 and 11; Fig. 5A; and data not shown). E124A was similar to FtsAsup by other criteria, including requirement of IPTG for efficient ftsN bypass activity when in pCSB1, lack of overproduction toxicity upon IPTG induction from the same plasmid (Fig. 4, row 11), and ability to suppress other cell division mutations (see below). We conclude that a single amino acid change in FtsA can bypass the requirement for FtsN. Interestingly, whereas a glycine was often present at this position in some bacteria, alanine was not (Fig. 3).
Fig. 5. Suppressor mutants of ftsA can suppress the loss of ftsK or zipA in addition to the loss of ftsN, with varying efficiencies.

A. The ΔzipA::kan, ΔftsK::kan or ΔftsN::cat alleles were introduced by P1 transduction into W3110 carrying the plasmids indicated, and plated on selective plates containing 0.1 mM IPTG to show the frequency and size of the initial transductants.
B–E. Colony viabilities of various combinations of ftsA suppressor mutants and cell division gene mutants are shown in each row. The ftsA suppressor mutants were expressed either from the native chromosomal locus in the absence of WT ftsA (C and D), or from plasmid pRR48 derivatives (supplemented with 0.1 mM IPTG) in addition to the WT ftsA in the chromosome (B and E).
Because E124A in WM2417 replaced the WT version of ftsA in the chromosome, we asked whether this allele of ftsA was as efficient a suppressor as when expressed in a merodiploid. Like cells of ΔftsN::cat strains expressing E124A (or ftsA sup) from a plasmid, such as WM2600 (see above), cells of strain WM2417 were longer on average than the corresponding ftsN+ strain WM2886 (7.5 ± 2.6 μm versus 4.1 ± 1.0 μm, at 37°C). Length per Z ring (6.9 μm) was also longer than that for the ftsN+ parent strain (4.4 μm). Furthermore, as shown in Fig. 2D, colony growth of WM2417 was similar to that of its ftsN+ counterparts, indicating that the chromosomal E124A allele is an efficient suppressor of ΔftsN::cat.
We also asked whether W3110 containing E124A in place of WT ftsA displayed any noticeable phenotype compared with W3110 itself. The average lengths of WM2886 (W3110 ftsA E124A) and WM2887 (W3110) were the same (4.1 ± 1.0 μm versus 4.0 ± 0.8 μm, at 37°C). FtsZ-GFP localization (in WM2891 and WM2892) and colony growth phenotypes of the two strains were identical (Fig. 2D and data not shown). Growth rates of strains WM2886, WM2887 and WM2417 were essentially the same (data not shown). These three strains were also grown to stationary phase (18 h) and plating revealed that viability was equivalent among all three strains, indicating that the suppression was effective in stationary phase (data not shown). Therefore, we conclude that despite its dramatic suppression of cell division defects in cells lacking ftsN, the E124A mutant of FtsA has no obvious effect on growth and division of E. coli when in an otherwise WT context.
Gain of function ftsA mutants are less toxic than WT ftsA when expression is increased
Massive overproduction of FtsA, FtsAsup or FtsA* from the strong araBAD promoter after arabinose induction of the pBAD18kan plasmids blocked cell division and sharply reduced viability (data not shown), consistent with the known negative effects of excess FtsA on cell division (Dai and Lutkenhaus, 1992; Dewar et al., 1992; Geissler et al., 2007). However, during induction of the weak Trc99 promoter on pCSB1 derivatives, we noticed that increased expression of WT ftsA abolished viability as IPTG was increased (Fig. 4A, row 2 at 30°C), but similar increased expression of R286W (ftsA*) (row 3), ftsA sup (row 10) or E124A (row 11) caused no detectable reduction in viability. This lack of toxicity of the mutant proteins was observed despite somewhat higher levels of R286W, FtsAsup or E124A protein relative to WT FtsA protein at a given IPTG concentration, as measured by immunoblotting (Fig. 4B). Interestingly, similar induction of K117R or K48R expression from pCSB1 derivatives caused toxicity similar to that of WT ftsA (Fig. 4A, rows 4 and 5).
It is notable that the degree of toxicity correlated with the inability to bypass FtsN. However, the data indicate that the failure of some of the derivatives to bypass FtsN is not solely because of toxicity. For example, cells overproducing FtsA* (Fig. 4B) had normal viability at 1 mM IPTG, but were only able to suppress the loss of FtsN partially at 0.1 mM IPTG and not at 0.5 or 1 mM IPTG (Fig. 4A, row 3). Likewise, cells overproducing K48R were viable at 0.1 mM IPTG (Fig. 4A, row 4, and Fig. 4B), but were not able to suppress the loss of FtsN at this or any other IPTG concentrations (Fig. 4A, row 4).
As R286W or FtsAsup can localize to the Z ring as GFP fusions (Geissler et al., 2003 and data not shown) and hence must interact with FtsZ, it is not surprising that very high levels of these FtsA derivatives still cause toxicity. Nevertheless, the lower toxicity of the gain of function mutants compared with WT FtsA suggests that the mutant FtsA proteins may resist perturbations in the crucial FtsA:FtsZ ratio by increasing the overall integrity of the Z ring.
FtsAsup or E124A can also partially compensate for the inactivation of ZipA or FtsK
FtsA* (R286W) can suppress multiple cell division gene mutants to varying degrees. We explored whether FtsAsup or E124A had similar gain of function activities. To allow introduction of ΔftsK::kan or ΔzipA::kan alleles, the ftsA*, ftsA sup and ftsA(E124A) mutant genes were cloned into the ApR plasmid pRR48, which allows for their IPTG-dependent expression. Remarkably, W3110 expressing these three ftsA alleles allowed efficient transduction of either ΔzipA::kan or ΔftsK::kan null alleles compared with the vector control (Fig. 5A). In addition, an ftsK44(ts) strain was complemented efficiently at the non-permissive temperature by pBAD18kan-ftsA* or pBAD18kan-ftsA sup (data not shown).
Nonetheless, ftsA sup or E124A expressed from pRR48 were not able to compensate for the loss of ZipA as efficiently as the ftsA* allele R286W, with a 10- to 100-fold loss of viability (Fig. 5B). The same was true for either R286W or E124A alleles replacing WT ftsA in the chromosome (Fig. 5C), further supporting the idea that the E124A suppressor can compensate for the loss of ZipA, but not as effectively as R286W. The ΔftsK::kan allele was suppressed equally well by R286W or E124A, with ftsA sup showing a modest decrease (Fig. 5E); the significance of this is unclear. When the R286W and E124A alleles replaced chromosomal ftsA, ΔftsK::kan transductants were always smaller with R286W, which may reflect the prevalence of long cell chains seen with R286W compared with short filaments with E124A (data not shown).
Ability of FtsA R286W or increased levels of early Z ring components to compensate for the loss of FtsN
The functional overlap between FtsAsup and FtsA R286W prompted us to investigate whether the latter could compensate for the loss of FtsN. Although ΔftsN::cat transductants could be isolated in W3110 strains with R286W, the colonies were small compared with those expressing ftsA sup or E124A (Fig. 5A, right) and had low viability compared with ΔftsN::cat strains containing FtsAsup or E124A (Fig. 4). This was true whether the R286W or E124A alleles were expressed from a plasmid or replaced the native ftsA at the chromosomal locus (Fig. 5D). Therefore, despite their overlapping functions, FtsA R286W and FtsAsup have somewhat different suppression profiles, suggesting that the lack of FtsN imposes different requirements for compensatory changes in FtsA.
Because the loss of zipA or ftsK was suppressed quite efficiently by merely increasing expression of ftsZ, ftsA and ftsQ from the pZAQ plasmid (Geissler et al., 2003), we investigated whether pZAQ might help to suppress the cell division defects in the absence of FtsN. However, when W3110 cells containing pZAQ were transduced with the ΔftsN::cat allele, no transductants were obtained, while several hundred transductants were isolated with ΔzipA::kan in W3110/pZAQ or with ΔftsN::cat in W3110 derivatives expressing ftsA sup or E124A (data not shown). This suggests that the presence of extra early Z ring components cannot compensate for the lack of FtsN, and supports the idea that the ftsN bypass suppression may act via a distinct mechanism.
The E124A mutation in ftsA increases resistance to the MinC division inhibitor
One way to test whether E124A or FtsAsup might increase the integrity of the Z ring was to determine if these mutant FtsA proteins, like R286W, could resist the inhibitory effects of MinC, a key inhibitor of FtsZ assembly. As we have shown that E124A and FtsAsup are essentially equivalent, we used WM2886, which contains the E124A allele in place of the WT ftsA gene on the chromosome. A plasmid expressing an IPTG-inducible functional flag–minC fusion (pWM2801) was introduced into this strain and WM2887, the isogenic strain containing WT ftsA.
Without IPTG, cells of both strains containing the flag–minC fusions were short and divided normally, although WM2887 cells were slightly longer (Fig. 6A and B). As expected, WM2887 cells filamented after several hours of FLAG–MinC overproduction upon IPTG induction (Fig. 6D). WM2886 cells, on the other hand, remained fairly short, with only occasional filamentation (Fig. 6C). FLAG–MinC levels in both strains were equivalent at a given IPTG concentration (Fig. 6E), indicating that the E124A allele in WM2886 conferred resistance to Z ring disassembly caused by overproduction of MinC. Viability tests confirmed a ~10-fold decrease in colony-forming units in IPTG-induced WM2887 versus WM2886 at the time the micrographs were taken (data not shown). Therefore, despite having no visible phenotype in otherwise WT strains, including no observed minicells, the mutant FtsA subtly increases in the integrity of the Z ring.
Fig. 6. The E124A allele of FtsA is resistant to inhibition caused by overproduction of MinC. WM2886 (ftsA E124A) or WM2887 (WT ftsA) containing plasmid pWM2801, which expresses flag–minC under control of IPTG, were grown at 37°C with either 0 mM or 1 mM IPTG for 5 h. Cells were then harvested for microscopy and for immunoblotting.

A–D. WM2886/flag–minC without (A) or with IPTG (B); WM2887/flag–minC without (C) or with IPTG (D).
E. Immunoblot of cell extracts from (A)–(D), probed with anti-FLAG antibody, showing the overproduction of FLAG–MinC after IPTG addition. Equal cell density equivalents were loaded in each lane.
GFP-FtsN localizes normally in cells with the E124A mutant of FtsA
In WT cells, FtsN is the last essential cell division protein to localize, and its septal localization depends on all other known essential cell division proteins. However, the ability of FtsN to be bypassed by an altered FtsA prompted us to test whether FtsN could still be targeted to the Z ring in the presence of the altered FtsA. One possible scenario was that the FtsA suppressor promotes a bypass of the normal divisome protein assembly pathway. As FtsN depends on prior localization of other divisome components, this model predicts that FtsN would fail to localize to the divisome in an E124A mutant (WM2886).
To test this idea, we introduced a plasmid expressing gfp-ftsN into WM2886, WM2887 and WM2417, which contains both ΔftsN::cat and ftsA (E124A). GFP-FtsN localized to the septum in approximately half of the cells of WM2887 and WM2886, and no other differences between the two strains were detected (Fig. 7A and B). GFP-FtsN also localized to division septa in WM2417 cells (Fig. 7C). Therefore, we conclude that FtsN, at least in the form of GFP-FtsN, retains the ability to localize to division septa in the presence of E124A. This suggests that the normal recruitment properties of the divisome, at least involving FtsN, are unchanged.
Fig. 7.

GFP-FtsN localizes normally in cells with the E124A suppressor. Shown are representative fluorescence micrographs of (A) WM2886 (ftsA E124A), (B) WM2887 (WT ftsA) or (C) WM2417 (ftsA E124A, ΔftsN::cat) producing GFP-FtsN from pWM1152. Cells were grown in LB without IPTG for 4 h at 37°C to mid-exponential phase.
The requirement of FtsN for localization of the Tol–Pal complex to the cell division site is indirect
Recently, Gerding et al. (2007) showed that the Tol–Pal complex localizes to division sites and helps to co-ordinate invagination of the outer membrane with division septum formation. Using GFP fusions to several components of the complex, including TolA, they demonstrated these fusions failed to localize after FtsN depletion, indicating that FtsN is required for the localization of this complex to the divisome. This suggests two possible models. The first is that FtsN directly recruits the Tol–Pal complex, either via direct protein–protein interactions or via an intermediate protein(s). If this model were correct, FtsN should be indispensable for recruitment of Tol–Pal. The second is that FtsN does not directly recruit Tol–Pal components, but instead FtsN activity is indirectly required for Tol–Pal to localize to the divisome. If this model were correct, FtsN activity could theoretically be replaced by another compensatory activity.
The FtsN bypass suppressors allowed us to test these models directly. We constructed a GFP–TolA fusion and introduced it into WM2886 (E124A), WM2887 (WT) and WM2417 (E124A ΔftsN::cat). Importantly, GFP–TolA localized to the divisome in all three strains (Fig. 8). This rules out the first model and supports the second model, in which FtsN does not directly recruit Tol–Pal but instead increases the integrity of the Z ring, which then becomes competent to recruit Tol–Pal. To provide additional support for this idea, we grew WM2417 in LB with no added salt, conditions that normally cause chaining of cells in tol mutants (Gerding et al., 2007). As expected, WM2417 was unchanged from its normal phenotype observed in regular LB medium, with no cell chains detected (data not shown). This result suggests that as long as the appropriate ftsA suppressor is present to compensate, the lack of FtsN does not inhibit the function of the Tol–Pal complex in cell–cell separation.
Fig. 8.
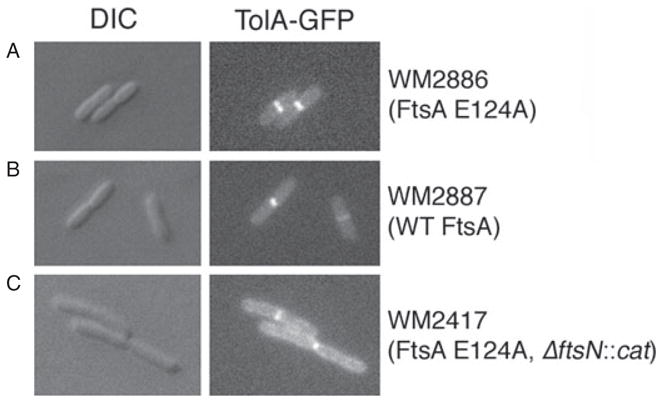
GFP–TolA localizes to division septa of cells lacking FtsN when the E124A suppressor is present. Shown are representative DIC (left) and fluorescence (right) micrographs of GFP–TolA producing cells of (A) WM2886 (ftsA E124A), (B) WM2887 (WT ftsA) or (C) WM2417 (ftsA E124A, ΔftsN::cat). Cells were grown in LB without IPTG for 5 h at 37°C to mid-exponential phase.
Discussion
We have shown that the essential cell division gene ftsN can be bypassed by mutations in another cell division gene, ftsA. The immediate conclusion is that FtsN, while normally essential, cannot be a core component of the divisome. This is a significant finding, as a number of papers have been published about FtsN, but no unique function has been ascribed to it. FtsN is also poorly conserved outside the enteric bacteria, consistent with its revised role as an accessory factor for the divisome.
Despite the robust viability of cells with ftsA sup or ftsA(E124A) but lacking ftsN, there is a modest cost. For example, suppressed cells lacking FtsN are longer than WT cells with more space between Z rings, suggesting that Z rings are less stable or less active, resulting in less efficient septation. On the other hand, without the FtsA suppressor, depletion of FtsN results in formation of long filaments and death, probably because the remaining Z rings are too inactive to divide cells sufficiently for viability. Therefore, the gain-of-function mutations in FtsA reported here probably increase ring integrity or activity to mostly counteract the loss of FtsN. The resistance of the E124A mutant to the toxic effects of MinC overproduction directly supports this idea. Further work will be needed to determine whether additional factors can reduce the cost of not having FtsN. One prediction would be that removal of other Z ring destabilizing factors or activation of ring stabilizing factors might further compensate for its absence. However, it is also possible that FtsN has a unique function that, while not essential for cell division in the presence of a suppressor, increases its efficiency.
The stronger FtsN bypass activity of the E124A mutant FtsA versus the E124G mutant protein suggests that any conformational changes in FtsA resulting from E124A are more dramatic than those resulting from E124G. Because the triple mutation in FtsAsup has FtsN bypass activity equivalent to E124A alone, and K48R combined with E124G are sufficient for this activity, it is likely that K48R induces additional subtle conformational changes in the E124G mutant protein that allow it to function more efficiently to bypass the FtsN requirement. The fact that the gain-of-function mutants of FtsA have no detectable phenotype by themselves and seem to recruit FtsN normally, at least in the form of GFP-FtsN, suggests that the divisome has tolerance for conformational flexibility of the FtsA molecule.
Our results provide additional support for the idea that FtsN enhances the activity of the divisome. The mutations in FtsA that bypass the requirement for FtsN also partially suppress the inactivation of other divisome proteins, including ZipA or FtsK, suggesting that FtsAsup and E124A are not FtsN-specific, but instead have a general role in divisome integrity similar to R286W. Importantly, however, suppression efficiencies suggest that FtsAsup and E124A are biased towards suppressing FtsN, while R286W fails to suppress the loss of FtsN significantly. Consistent with the bias of R286W away from suppressing FtsN, increased levels of FtsQ, FtsA and FtsZ from pZAQ phenocopy R286W and suppress the loss of ZipA and FtsK to some degree, but do not confer viability on a strain lacking FtsN. Therefore, we speculate that different subtle alterations in FtsA function result in overlapping but distinct suppression profiles because different divisome subcomplexes, as outlined in a recent review (Vicente et al., 2006), are influenced differentially by FtsA binding. The recent isolation of an ftsA mutant that suppresses a defective ftsQ allele but that also suppresses defects in some other cell division genes (Goehring et al., 2007a) is consistent with this idea.
This model is supported by additional evidence. First, despite the ability of E124A or FtsAsup to suppress the loss of either ZipA or FtsN, neither allele permitted both ZipA and FtsN to be inactivated simultaneously (T. Hammerstrom and W. Margolin, unpubl. results). This suggests that the effects of ZipA and FtsN on the divisome are synergistic and distinct and cannot be replaced by a single form of FtsA, and is consistent with the partial specificity of suppression that we have observed. We also attempted to delete both zipA and ftsN in a strain expressing ftsA* from the chromosome and ftsA sup from a plasmid, but could only make the single deletions (W. Margolin, unpubl. results). There are many potential explanations for this, but once again it is consistent with a synergistic effects of ZipA and FtsN and a lack of simple additive effects of the suppressors.
Second, recent evidence suggests that FtsA interacts, probably indirectly, with the late divisome proteins FtsN and/or FtsI. In one of these approaches, premature targeting of FtsQ to the Z ring can recruit all known later divisome proteins in the absence of FtsA or FtsK except for FtsN. Therefore, FtsN likely has a direct requirement for FtsA to be stably recruited to the divisome. Third, overproduced FtsN can partially suppress a number of fts mutants much like FtsAsup and E124A, suggesting that the FtsA suppressor mutants allow FtsA to function more efficiently to bypass the FtsN requirement. Finally, as shown here, FtsAsup, E124A and R286W seem to be less toxic than WT FtsA at equivalent levels of IPTG induction. These alleles may alter the requirement for a strict stoichiometric ratio between FtsZ and FtsA, although the mechanistic explanations for this are not clear. The challenge for the future is to understand how the various mediators of divisome integrity and activity function at the molecular level.
An alternative model for FtsN function is that it senses the completion of divisome assembly, triggering Z ring contraction (Corbin et al., 2004). Although it is not required for FtsN function, FtsN binds peptidoglycan (Ursinus et al., 2004), suggesting that part of the putative signal may originate from FtsI-mediated septal peptidoglycan biosynthesis. The known roles of FtsN in stimulating septation under conditions normally unfavourable for septation are consistent with this idea, as is its (probably indirect) interaction with FtsA, which might undergo a conformational change in response to FtsN and transduce the signal to FtsZ. In the absence of FtsN, the sensing pathway would be inactive, but could be compensated by a mutation that would mimic the FtsN-induced conformational change in FtsA. While this model is intriguing, it predicts that the ftsA mutants on their own would display a significant phenotype, which they do not, either when produced from a plasmid or as the sole copy of ftsA in the cell. The absence of ftsN homologues in most species also argues against a special signalling role for FtsN.
Another potential role for FtsN is the recruitment of proteins involved in cell–cell separation, including AmiC and the Tol–Pal complex. These proteins are not recruited to the septum after FtsN depletion, indicating that FtsN is required for their recruitment (Bernhardt and de Boer, 2003; Gerding et al., 2007). However, we found that FtsN is not required for recruitment of a key Tol–Pal component, TolA, in the presence of the E124A suppressor mutant of FtsA. This result is important because it eliminates the most obvious model, which proposes that FtsN directly recruits these proteins to the septum via specific protein–protein interactions or a specific enzymatic activity. Instead, it strongly suggests that FtsN helps to recruit these later proteins by its indirect effects on divisome integrity, which can be replaced by an altered FtsA.
Despite previous evidence strongly suggesting that FtsN is a special component of the divisome (Corbin et al., 2004; Vicente et al., 2006; Goehring et al., 2007a), the present study demonstrates conclusively that FtsN is not a unique divisome component. Instead, FtsN is one of several factors that potentially regulate conformational changes within divisome components such as FtsA, which in turn regulate the activity of the Z ring. The fact that point mutations of FtsA can bypass the requirement for an early acting, FtsZ-binding protein (ZipA), a middle-stage protein (FtsK) or a late-stage protein (FtsN) strongly supports the model that FtsA is conformationally flexible, and can exert subtle effects on the divisome at different stages. In contrast to FtsN, this points to a key and core role for FtsA in regulating divisome function.
Experimental procedures
Growth conditions, microscopy and protein detection
Unless stated otherwise, cells were grown in LB (1% tryptone, 0.5% yeast extract, 0.5% NaCl). SOC medium (2% tryptone, 0.5% yeast extract. 8.6 mM NaCl, 2.5 mM KCl, 20 mM glucose, 20 mM MgSO4) was used for outgrowth of transformed cells. Antibiotics were added to LB at 50 μg ml−1 for ampicillin or kanamycin (kan), and 10 μg ml−1 for tetracycline (tet) or chloramphenicol (cam). Top10 was used as a recipient in transformations of ligations. TOE44, which is ftsK44(ts) and thyA, was supplemented with 50 μg ml−1 thymine. Immunoblotting was performed as described previously (Geissler et al., 2003) with rabbit polyclonal antibodies against purified FtsN or FtsA. Loading of gel lanes was normalized to cell density. FtsN protein was purified for antibody production by overproduction from a pET28-FtsN construct. Microscopy to detect GFP fusions in live cells or in fixed cells with immuofluorescence was performed as described previously (Sun and Margolin, 2001). Cell lengths were measured with Object Image 2.19 freeware (Norbert Vischer).
Plate viability spot assays were performed by spotting 10-fold dilutions of exponentially growing cells that were at equivalent densities, starting with a 10−1 dilution. For assays of plasmid derivatives in the FtsN depletion strain WM2355, cultures were grown overnight in LB kan at 30°C, diluted 1:100 and grown at 30°C for 2 h, then split into 30°C and 42°C cultures and grown for an additional 2 h each prior to spotting on plates with different IPTG concentrations at 30°C or 42°C.
Constructing the FtsN depletion strain
All strains are listed in Table 1. To make a strain with the ΔftsN::cat allele, the GFP-FtsN plasmid pWM1152 (Corbin et al., 2004) was first introduced into strain DY329 (Yu et al., 2000). The ΔftsN::cat allele was assembled by PCR amplification of the cat cassette from the chromosomal ftsK1::cat allele (Diez et al., 1997) and, flanking the cat cassette, 30 bp regions flanking the chromosomal ftsN gene. This PCR product was then used to replace the native ftsN gene in DY329/pWM1152, using lambda-mediated recombineering (Yu et al., 2000), to make WM1932. A linked argE::Tn10 marker was introduced from CAG12185 (Singer et al., 1989) to permit selection for TcR as well as CmR. To construct the FtsN depletion strain, the ftsN gene in pBAD30-FtsN (Geissler and Margolin, 2005) was cloned as a SacI–HindIII fragment into the thermosensitive ApR plasmid pTSA29 (Phillips, 1999). A correct plasmid clone was then selected in Top10 cells, and plasmid DNA was transformed into WT W3110 to make WM2245. This strain in turn was transduced with a P1 lysate from the strain with ΔftsN::cat linked to argE::Tn10, selecting ApR TcR CmR transductants. Several transductants were picked, and did not form colonies when streaked on LB tet cam plates and incubated at 42°C, suggesting that they were FtsN depletion strains. One strain was chosen and called WM2355. Depletion of FtsN in WM2355 was confirmed by the formation of long filamentous cells after shifting growth from 30°C to 42°C and loss of viability.
Table 1.
Strains used in this study.
| Strains | Description | Source |
|---|---|---|
| CAG12185 | argE::Tn10 linked to ftsN | E. coli stock centre |
| WM640 | TOE44 (ftsK44) | Begg et al. (1995) |
| TX3772 | MG1655 lacU169 | Laboratory collection |
| W3110 | Wild-type strain | Laboratory collection |
| DY329 | W3110 ΔlacU169 nadA::Tn10 gal490 λcI857 Δ(cro-bioA) | Yu et al. (2000) |
| EC448 | MC4100 (λ ΔattL-lom)::bla lacI q Ptrc-ftsZ-gfp | Weiss et al. (1999) |
| WM1115 | TX3772 leu::Tn10 ftsA12(Ts) | Geissler et al. (2003) |
| WM1281 | CH2 (recA::Tn10, ftsA0)/pDB280 (repA tsftsA+) | Hale and de Boer (1999) |
| WM1657 | TX3772 ftsA* (R286W) ΔzipA::kan | Geissler et al. (2003) |
| WM1932 | ΔftsN::cat in DY329/pWM1152 | This study |
| WM2109 | DY329+pWM1747 (GFP-FtsK1-859) ΔftsK:kan | This study |
| WM2177 | W3110 leu::Tn10 ftsA* (R286W) | This study |
| WM2227 | W3110 + pZAQ | This study |
| WM2245 | W3110 + pWM2245 | This study |
| WM2354 | WM2177 ΔzipA::kan | This study |
| WM2294 | W3110 + pWM1747 (ftsK) ΔftsK::kan | Geissler and Margolin (2005) |
| WM2355 | W3110 + pWM2245, argE::Tn10, ΔftsN::cat | This study |
| WM2391 | WM2355 leu::Tn5 | This study |
| WM2417 | WM2391 ftsA(E124A) | This study |
| WM2600 | EC448/pBAD18kan-ftsA sup, ΔftsN::cat | This study |
| WM2701 | W3110/pRR48-ftsA* | This study |
| WM2702 | W3110/pRR48-ftsA (E124A) | This study |
| WM2706 | W3110/pRR48-ftsA sup | This study |
| WM2886 | W3110 leu::Tn5 ftsA(E124A) | This study |
| WM2887 | W3110 leu::Tn5 | This study |
| WM2888 | WM2701 ΔftsK::kan (P1 from WM2294) | This study |
| WM2889 | WM2706 ΔftsK::kan (P1 from WM2294) | This study |
| WM2890 | WM2702 ΔftsK::kan (P1 from WM2294) | This study |
| WM2893 | WM2701 ΔzipA::kan (P1 from WM1657) | This study |
| WM2891 | Ptrc-ftsZ-gfp in WM2886 | This study |
| WM2892 | Ptrc-ftsZ-gfp in WM2887 | This study |
| WM2894 | WM2706 ΔzipA::kan (P1 from WM1657) | This study |
| WM2895 | WM2702 ΔzipA::kan (P1 from WM1657) | This study |
| WM2921 | pWM2537 in WM2355 | This study |
| WM2922 | pWM2538 in WM2355 | This study |
| WM2923 | pWM2539 in WM2355 | This study |
| WM2924 | pWM2540 in WM2355 | This study |
| WM2925 | pWM2541 in WM2355 | This study |
| WM2926 | pWM2542 in WM2355 | This study |
| WM2927 | pWM2543 in WM2355 | This study |
| WM2928 | pWM2544 in WM2355 | This study |
| WM2929 | pWM2545 in WM2355 | This study |
| WM2930 | pWM2931 in WM2355 | This study |
| WM2934 | W3110 leu::Tn10 ftsA* (R286W) | This study |
| WM2935 | W3110 leu::Tn10 ftsA (E124A) | This study |
| WM2936 | WM2934 ΔzipA::kan (P1 from WM1657) | This study |
| WM2937 | WM2935 ΔzipA::kan (P1 from WM1657) | This study |
Mutagenesis of cloned ftsA
Plasmid pET28-FtsA, containing the ftsA gene with an N-terminal hexahistidine tag and a T7 tag from pET28 (Geissler et al., 2003), was the template used for PCR mutagenesis and selection. As shown previously, these tags do not affect the ability of FtsA to complement an ftsA mutant. The ftsA gene on pET28-FtsA was subjected to mutagenic PCR, using primers 727 and 728, which anneal to vector sequences just upstream and downstream of the ftsA insert. Amplification was performed with 1 μl (2.6 U) of Fisher Taq DNA polymerase and buffer, 200 μM dNTPs, 5 mM MgSO4 and 1 μM of each primer for 40 cycles. In each cycle, annealing was at 52°C for 30 s, followed by elongation at 72°C for 3 min.
The PCR product was digested with XbaI and EcoRI and ligated to XbaI–EcoRI-cleaved pET28a. Nine of these ligation reactions were transformed into Top10 cells and plated on LB kan at 37°C, yielding an average of 220 colonies per plate for a total of ~2000 colonies. These colonies were pooled and used to make a mutagenized plasmid bank. Two per cent of the bank was then transformed into WM2355 electrocompetent cells after 1 h of outgrowth at 30°C in SOC. Cells were plated on LB kan tet either at 30°C or at 42°C overnight and yielded 1800 and 15 colonies respectively. To determine whether the ability to survive at 42°C was conferred by the mutagenized plasmid, plasmid DNA was isolated from the 15 temperature-resistant strains and re-transformed into WM2355 at 30°C and 42°C, as were pET28 vector alone and pET28-FtsA plasmids as controls.
Plasmid pWM2425 was the initial isolate of the triple ftsAsup mutant. To rule out potential effects of vector sequences, the NdeI–EcoRI fragment containing ftsA from this plasmid was re-cloned into pET28a and a confirmed isolate was saved as pWM2491. Plasmid pWM2491 was then introduced into WM2355. To test for suppression of the loss of FtsN, these cells were incubated for 75 min at 30°C in 1 ml of SOC, and then half the culture was grown at 30°C for 1 h and plated on LB kan tet at 30°C, while the other half was grown at 42°C for 1 h and plated at 42°C. Unlike control cells containing pET28 vector alone, cells with pWM2491 gave rise to colonies at 42°C at the same efficiency as 30°C, indicating that the loss of FtsN was bypassed.
Plasmid constructions
Plasmids are listed in Table 2. Construction of ftsA mutant combinations was performed in pET28a prior to moving the mutant ftsA genes to pBAD18kan or pCSB1. To construct the FtsA K117R single mutant in pET28a (pWM2488), PCR mutagenesis was performed with primer pairs 826 + 863 and 862 + 528 in the first amplification, and primers 826 + 528 in the second amplification (see Table 3 for oligonucleotide sequences). The product was then cloned as a PstI–XhoI fragment into pET28a. To construct the FtsA E124G single mutant (pWM2489), PCR mutagenesis was performed with primer pairs 826 + 861 and 860 + 528 in the first amplification, and primers 826 + 528 in the second amplification. The product was cloned as a SacI–XhoI fragment into pET28a. To construct the FtsA K48R single mutant (pWM2504), PCR mutagenesis was performed with primer pairs 106 + 837 and 836 + 726-1 in the first amplification, and primers 106 + 726-1 in the second amplification. The product was cloned as a NdeI–BamHI fragment into pET28a. To construct the FtsA K117R E124G double mutant (pWM2490), the original pET28-FtsA plasmid was cleaved with BglII, which cuts both the vector portion and between the K48 and K117 residues encoded by ftsA. This fragment, which includes the N-terminal portion of WT ftsA, was then ligated into BglII-cleaved pWM2425 containing the triple mutant ftsA, resulting in restoration of the WT ftsA encoding the K48 residue but leaving the mutant K117R and E124G-encoding residues intact. To construct the K48R K117R double mutant, the BglII fragment of WM2488 (K117R single mutation) was replaced with the BglII fragment from pWM2491 containing the triple mutation. The K48R E124G double mutant (pWM2507) was constructed by PCR mutagenesis of pWM2489 (E124G), using primers 106 + 897 and 896 + 726-1 in the first amplification and 106 + 726-1 in the second amplification, followed by cloning the product as a NdeI–BamHI fragment into pET28a.
Table 2.
Plasmids used in this study.
| Plasmids | Description | Source |
|---|---|---|
| pZAQ | ftsQAZ in pBR322, TcR | Bi and Lutkenhaus (1990) |
| pDSW207 | Ptrc-gfp pBR322 derivative with strong promoter, ApR | Weiss et al. (1999) |
| pDSW209 | Ptrc-gfp pBR322 derivative with weak promoter, ApR | Weiss et al. (1999) |
| pDSW210 | Ptrc-gfp pBR322 derivative with weak promoter, ApR | Weiss et al. (1999) |
| pRR48 | Derivative of pJC30, Ptac expression, ApR | Ames et al. (2002) |
| pBAD18kan | Expression vector with araBAD promoter, KanR | Guzman et al. (1995) |
| pTSA29 | Thermosensitive replication plasmid, ApR | Phillips (1999) |
| pWM2060 | pSDW210 deleted for gfp, ApR | Geissler and Margolin (2005) |
| pCSB1 | pWM2060 with aph (KanR) replacing bla | This study |
| pET28a | Expression vector, KanR | Novagen |
| pWM1152 | pDSW207 expressing gfp-ftsN | Corbin et al. (2004) |
| pWM2022 | pBAD30-ftsN | Geissler and Margolin (2005) |
| pWM2245 | pTSA29-ftsN | This study |
| pWM2425 | pET28-ftsA sup(K48R K117R E124G) | This study |
| pWM2488 | pET28-ftsA (K117R) | This study |
| pWM2489 | pET28-ftsA (E124G) | This study |
| pWM2490 | pET28-ftsA (K117R E124G) | This study |
| pWM2491 | pET28-ftsA sup(K48R K117R E124G) re-cloned | This study |
| pWM2504 | pET28-ftsA (K48R) | This study |
| pWM2506 | pET28-ftsA (K48R K117R) | This study |
| pWM2507 | pET28-ftsA (K48R E124G) | This study |
| pWM2537 | pCSB1-ftsA from pET28-ftsA | This study |
| pWM2538 | pCSB1-ftsA* (R286W) | This study |
| pWM2539 | pCSB1-ftsA (K48R) | This study |
| pWM2540 | pCSB1-ftsA (K117R) | This study |
| pWM2541 | pCSB1-ftsA (E124G) | This study |
| pWM2542 | pCSB1-ftsA (K48R K117R) | This study |
| pWM2543 | pCSB1-ftsA (K48R E124G) | This study |
| pWM2544 | pCSB1-ftsA (K117R E124G) | This study |
| pWM2545 | pCSB1-ftsA sup(K48R K117R E124G) | This study |
| pWM2546 | pBAD18kan-ftsA | This study |
| pWM2547 | pBAD18kan-ftsA* (R286W) | This study |
| pWM2548 | pBAD18kan-ftsA (K48R) | This study |
| pWM2549 | pBAD18kan-ftsA (K48R K117R) | This study |
| pWM2550 | pBAD18kan-ftsA (K48R E124G) | This study |
| pWM2551 | pBAD18kan-ftsA (K117R E124G) | This study |
| pWM2552 | pBAD18kan-ftsA sup(K48R K117R E124G) | This study |
| pWM2658 | pCSB1-ftsA (E124A) | This study |
| pWM2701 | pRR48-ftsA* (R286W) | This study |
| pWM2702 | pRR48-ftsA (E124A) | This study |
| pWM2706 | pRR48-ftsA sup(K48R K117R E124G) | This study |
| pWM2931 | pBAD18kan-ftsA (E124A) | This study |
| pWM2801 | pDSW210 expressing flag–minC | Shiomi and Margolin (2007) |
| pWM2984 | pDSW209 expressing gfp–tolA | This study |
Table 3.
Oligonucleotides used in this study.
| Oligonucleotide No. | Sequence |
|---|---|
| 727 | TAAGATCTCGATCCCGCGAAATT |
| 728 | TAGCTTTGTTAGCAGCCGGATCT |
| 826 | TATATAGAGCTCATGATCAAGGCGACGGACAGA |
| 836 | GGGGAAGTTCTGCCCNNNGGTATGGTCAATATC |
| 837 | GATATTGACCATACCNNNGGGCAGAACTTCCCC |
| 860 | GCGTGTGCGCGATGGCCATCGTGTGCTGCA |
| 861 | TGCAGCACACGATGGCCATCGCGCACACGC |
| 862 | CGTCCATACCGCGCGATCGGTGCGTGTGCGC |
| 863 | GCGCACACGCACCGATCGCGCGGTATGGACG |
| 902 | TATATAGAGCTCAATAATTTTGTTTAACTTTAAGAA |
| 903 | ATATATCTAGATTAAAACTCTTTTCGCAGCCA |
| 528 | TTTCTCGAGTTAAAACTCTTTTCGC |
| 106 | AACATATGATCAAGGCGACGGAC |
| 726-1 | TATATAGGATCCTTAAAACTCTTTTCGCAG |
All cloned ftsA derivatives in pET28a were transferred to pBAD18kan by amplification of the ftsA gene, its ribosome binding site, and sequences encoding the N-terminal histidine tag with primers 902 + 903, then cloning the PCR product as a SacI–XbaI fragment into pBAD18kan. All cloned ftsA derivatives in pET28a were transferred to pCSB1 by amplification of the ftsA gene with primers 826 + 903, and cloning the PCR product as a SacI–XbaI fragment into pCSB1. These constructs fuse ftsA sequences to a ribosome binding site already present in the vector sequences, and as a result only the ftsA open reading frame was transferred. Therefore, unlike the pET28a and pBAD18kan derivatives, none of the pCSB1 derivatives synthesize an N-terminal histidine tag fused to FtsA. No functional differences between FtsA with or without a histidine tag were apparent throughout this study.
The E124A allele was isolated as a spontaneous suppressor in WM2391 after FtsN depletion at 42°C, and called WM2417. The absence of FtsN in this strain was initially implicated by the Aps phenotype of the strain, indicating that the pTSA29-ftsN thermosensitive plasmid was indeed lost. The absence of FtsN was subsequently confirmed by immunoblotting. DNA sequencing of a PCR product from the chromosomal ftsA gene confirmed that the E124A allele resulted from an A to C change at position 371 in the ftsA nucleotide sequence (the E124G mutation of ftsAsup resulted from an A to G change at the same position). This A to C change serendipitously generated a unique FspI site in ftsA that was useful for screening. The E124A allele was introduced into W3110 by co-transduction with the leu::Tn5 marker (~50% linkage) present in WM2391 to make WM2886; WM2887 was another isolate from the same transduction that contained the WT ftsA instead. To enable selection of ΔzipA::kan or ΔftsK::kan transductants, the Tn5 marker was replaced by a TetR Tn10. This was done by transducing WM2886 and WM2887 with a leu::Tn10 marker from the ftsA* strain WM2177, selecting TetR colonies, and screening for the presence of the E124A allele of ftsA by FspI digestion of a PCR product amplified from chromosomal ftsA (WM2934). Isolates containing the ftsA* allele instead of E124A (WM2935) were confirmed by NcoI digestion of the ftsA PCR product (Geissler et al., 2003).
Plasmid pWM2060 was made by removing the gene encoding GFP from pDSW210 (Weiss, 2004); the EcoRI–ScaI fragment of pDSW210 was replaced by the EcoRI–ScaI fragment of pDSW209. Plasmid pCSB1 was then constructed by cloning the kanamycin cassette (aph gene) from pET28a into the ScaI site of pWM2060, replacing the bla gene with aph.
The pRR48 derivatives were made by inserting a PCR-amplified flag-tagged ftsA construct into NdeI–XhoI-cut pRR48, an AmpR plasmid which contains the tac promoter, lac operator and a lacIq gene (kindly supplied by Sandy Parkinson). The N-terminal FLAG tag had no detectable effect on function of any of the ftsA derivatives. Full details of the cloning will be described in a forthcoming paper.
The GFP–TolA fusion was constructed by PCR amplification of chromosomal tolA using the same primers described previously (Gerding et al., 2007) and cloning the product into pDSW209 with BamHI and SalI.
Other strain constructions
WM2600 was made by transforming the FtsZ-GFP expression strain EC448 (Weiss, 2004) with pBAD18kan-ftsAsup (pWM2552), then introducing the ΔftsN::cat allele by P1 transduction. To test complementation of ftsA mutants, pBAD18kan plasmids containing ftsA or ftsAsup were transformed into the ftsA12(ts) strain WM1115 or the FtsA depletion strain WM1281 (Geissler and Margolin, 2005), grown at the permissive temperature of 30°C, then plated at 30°C or 42°C to inactivate or deplete FtsA.
Acknowledgments
We thank Stacie Meaux and Dr Swapna Thanedar for help with the initial construction and verification of the ftsN::cat allele, Jennie Lin for help with immunoblotting, Dr Sandy Parkinson for the pRR48 plasmid and Dr Anne Delcour for her encouragement and support of C.S.B. This work was supported by National Institutes of Health Grant R01-GM61074.
References
- Aarsman ME, Piette A, Fraipont C, Vinkenvleugel TM, Nguyen-Disteche M, den Blaauwen T. Maturation of the Escherichia coli divisome occurs in two steps. Mol Microbiol. 2005;55:1631–1645. doi: 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol. 1997;25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg KJ, Dewar SJ, Donachie WD. A new Escherichia coli cell division gene, ftsk. J Bacteriol. 1995;177:6211–6222. doi: 10.1128/jb.177.21.6211-6222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. FtsZ regulates the frequency of cell division in Escherichia coli. J Bacteriol. 1990;172:2765–2768. doi: 10.1128/jb.172.5.2765-2768.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Beckwith J. FtsQ, FtsL, and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol Microbiol. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- Corbin BD, Geissler B, Sadasivam M, Margolin W. A Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J Bacteriol. 2004;186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Lutkenhaus J. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J Bacteriol. 1992;174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Xu Y, Lutkenhaus J. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts) J Bacteriol. 1993;175:3790–3797. doi: 10.1128/jb.175.12.3790-3797.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Xu Y, Lutkenhaus J. Topological characterization of the essential Escherichia coli cell division protein FtsN. J Bacteriol. 1996;178:1328–1334. doi: 10.1128/jb.178.5.1328-1334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar SJ, Begg KJ, Donachie WD. Inhibition of cell division initiation by an imbalance in the ratio of FtsA to FtsZ. J Bacteriol. 1992;174:6314–6316. doi: 10.1128/jb.174.19.6314-6316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology. 2003;149:3353–3359. doi: 10.1099/mic.0.26580-0. [DOI] [PubMed] [Google Scholar]
- Diez AA, Farewell A, Nannmark U, Nyström T. A mutation in the ftsK gene of Escherichia coli affects cell–cell separation, stationary-phase survival, stress adaptation, and expression of the gene encoding the stress protein UspA. J Bacteriol. 1997;179:5878–5883. doi: 10.1128/jb.179.18.5878-5883.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper GC, McLennan N, Begg K, Masters M, Donachie WD. Only the N-terminal domain of FtsK functions in cell division. J Bacteriol. 1998;180:4621–4627. doi: 10.1128/jb.180.17.4621-4627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Margolin W. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol Microbiol. 2005;58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Elraheb D, Margolin W. A gain of function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci USA. 2003;100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Shiomi D, Margolin W. The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology. 2007;153:814–825. doi: 10.1099/mic.0.2006/001834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. The trans-envelope Tol–Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Beckwith J. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol. 2005;15:514–526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Gueiros-Filho F, Beckwith J. Premature targeting of a cell division protein to midcell allows dissection of divisome assembly in Escherichia coli. Genes Dev. 2005;19:127–137. doi: 10.1101/gad.1253805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Petrovska I, Boyd D, Beckwith J. Mutants, suppressors, and wrinkled colonies: mutant alleles of the cell division gene ftsQ point to functional domains in FtsQ and a role for domain 1C of FtsA in divisome assembly. J Bacteriol. 2007a;189:633–645. doi: 10.1128/JB.00991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Robichon C, Beckwith J. A role for the non-essential N-terminus of FtsN in divisome assembly. J Bacteriol. 2007b;189:646–649. doi: 10.1128/JB.00992-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell. 1997;88:175–185. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ and independent of FtsA. J Bacteriol. 1999;181:167–176. doi: 10.1128/jb.181.1.167-176.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GJ. New cloning vectors with temperature-sensitive replication. Plasmid. 1999;41:78–81. doi: 10.1006/plas.1998.1380. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J. 2002;21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol. 2005;55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- Shiomi D, Margolin W. The C-terminal domain of MinC inhibits assembly of the Z ring in Escherichia coli. J Bacteriol. 2007;189:236–243. doi: 10.1128/JB.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, et al. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev. 1989;53:1–24. doi: 10.1128/mr.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Margolin W. Influence of the nucleoid on placement of FtsZ and MinE rings in Escherichia coli. J Bacteriol. 2001;183:1413–1422. doi: 10.1128/JB.183.4.1413-1422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursinus A, van den Ent F, Brechtel S, de Pedro M, Holtje JV, Löwe J, Vollmer W. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J Bacteriol. 2004;186:6728–6737. doi: 10.1128/JB.186.20.6728-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente M, Rico AI, Martinez-Arteaga R, Mingorance J. Septum enlightenment: assembly of bacterial division proteins. J Bacteriol. 2006;188:19–27. doi: 10.1128/JB.188.1.19-27.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS. Bacterial cell division and the septal ring. Mol Microbiol. 2004;54:588–597. doi: 10.1111/j.1365-2958.2004.04283.x. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol. 1999;181:508–520. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Löwe J. Solution structure and domain architecture of the divisome protein FtsN. Mol Microbiol. 2004;52:651–660. doi: 10.1111/j.1365-2958.2004.03991.x. [DOI] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]