

Abstract
The success of cancer immunotherapy reveals the power of host immunity on killing cancer cells and the feasibility to unleash restraints of anti-tumor immunity. However, the immunosuppressive tumor microenvironment and low immunogenicity of cancer cells restrict the therapeutic efficacy of cancer immunotherapies in a small fraction of patients. Therefore deciphering the underlying mechanisms promoting the generation of an immunosuppressive tumor microenvironment is direly needed to better harness host anti-tumor immunity. Early works revealed that deregulated metabolic activities in cancer cells support unrestricted proliferation and survival by producing macromolecules. Intriguingly, recent studies uncovered that metabolic switch in immune and endothelial cells modulate cellular activities and contribute to the progression of several diseases, including cancers. Herein, we review the progress on immunometabolic regulations on fine-tuning activities of immune cells and discuss how metabolic communication between cancer and infiltrating immune cells contributes to cancer immune evasion. Moreover, we would like to discuss how we might exploit this knowledge to improve current immunotherapies and the unresolved issues in this field.
Keywords: Immunometabolism, Immune evasion, Immunotherapy, Cancer metabolism, Warburg glycolysis
Background
With more than 8 million cancer related deaths, cancer is clearly a major health burden worldwide. Although radiotherapy and chemotherapy elicit strong response rates in the majority of cancer patients, metastatic diseases are problematic to control via these conventional interventions and cures remain scarce. A burgeoning field in oncology and immunology is the ability to eradicate cancer cells by rejuvenating the tumoricidal functions of tumor-reactive immune cells, predominantly T cells. Cancer immunotherapy provides oncologists with a new weapon among existing cancer treatments, which is demonstrated by the recent developments of checkpoint blockade, adoptive cell transfer (ACT) and cancer vaccines. However, the benefit of cancer immunotherapy is currently compromised by the immunosuppressive tumor microenvironment [1, 2]. While the importance of the immunosuppressive tumor microenvironment on dampening the anti-tumor immunity is appreciated, the mechanism by which cancer cells instruct the development of the immunosuppressive microenvironment remains unclear.
Over the past two decades significant understanding has been gained in cancer cell-immune system interactions. Additionally, how the unrestricted proliferation and survival of cancer cells is sustained via the deregulation of cellular energetic pathways has been expounded upon [3, 4]. However, whether the abnormal metabolic activities of cancer cells influence the nutrient states of the tumor microenvironment or the metabolic fitness of neighboring stromal and immune cells remains elusive. Similar to cancer cells, upon stimuli recognition lymphocytes engage metabolic reprogramming to drive their activation and differentiation [5–7]. Therefore, cancer and immune cells share similarities of utilizing nutrients and engaging metabolic regulation to sustain proliferation and survival. This raises an intriguing possibility that “metabolic competition” within the tumor microenvironment may allow cancer cells to effectively suppress anti-tumor immunity. In this review, we focus on how immunometabolic regulations fine-tune the activation and anti-tumor responses of T cells and myeloid cells. Moreover, we discuss the burgeoning idea that metabolic communication and competition within the tumor microenvironment may support the formation of the immunosuppressive tumor microenvironment. Finally, we summarize unresolved issues in this filed and discuss how these issues impact the development of cancer immunotherapy.
Reviews
The immunosuppressive tumor microenvironment: a challenge for improving cancer immunotherapies
Progress on exploiting the host anti-tumor immunity to combat established and aggressive tumors presents promising effects, including on two of the most deadly forms of cancer: melanomas and pancreatic cancers. However, there are three major hurdles impeding the affect of host anti-tumor immunity and cancer immunotherapy: 1) low number of tumor antigen-specific T cells due to clonal deletion; 2) poor activation of innate immune cells and accumulation of tolerigenic antigen-presenting cells in the tumor microenvironment; 3) formation of an immunosuppressive tumor microenvironment [8]. Cancer vaccines and T cell-based treatment, such as adoptive cell transfer (ACT) and chimeric-antigen receptor (CAR) T-cells, have overcome the first hurdle and produced remarkable results in several tumors. However, the therapeutic efficacy of these treatments remains unsatisfactory due to the incapability to fully cultivate anti-tumor responses in the immunosuppressive tumor microenvironment [9]. Of note, the Speiser’s group demonstrated that tumor-reactive T cells, upon migrating into tumors, lose their effector functions and increase the expression of co-inhibitory receptors compared to circulating cells [10]. Intriguingly, similar findings have also been shown in murine tumor models. Furthermore, short term in vitro culture is able to restore both tumoricidal function and cytokine production in those tumor infiltrating T cells [11]. Taken together, these findings suggest that the tumor microenvironment provides local restraints that abolish the anti-tumor responses of infiltrating T cells. Further investigations uncovered two major underlying mechanisms that disarm anti-tumor immunity in the tumor microenvironment; 1) the accumulation in tumors of immunomodulatory cells, including M2-like macrophages (MΦs), immature dendritic cells, regulatory T cells (Tregs), and myeloid derived suppressor cells (MDSCs), diminishes T cell anti-tumor immune responses through cell-cell contact and cytokine milieu [1, 2]; 2) expression of PD-1 receptor ligands (PD-L1/PD-L2) and reduced expression of tumor antigens and major histocompatibility complex (MHC) in cancer cells [1]. These findings led to the development of anti-CTLA-4 monoclonal antibody treatment, Treg depletion therapy and checkpoint blockade, including PD-1 and PD-L1/L2 inhibition [1, 12, 13].
Metabolic regulation of T cell anti-tumor responses
Metabolic reprogramming guides T cell activation and differentiation
Upon receiving T cell receptor (TCR) and co-stimulatory signals, T cells engage in growth, expansion, and ultimately, differentiation into different cytotoxic, Tregs and helper T cells (Th cells; i.e., Th1, Th2 and Th17) [14, 15]. In addition to these defined signaling cascades, changes in metabolic activity has been shown to intimately support T cell differentiation and effector functions [5]. Naïve T cells rely on oxidative phosphorylation (OXOPHOS) to maintain energy demand; in contrast, activated T cells engage aerobic glycolysis consuming massive amount of glucose [16, 17]. Blocking glycolysis or eliminating glucose uptake during T cell activation period diminishes T cell activation, Th cell differentiation, as well as the generation of effector cytokines, including IFNγ, IL-2, and IL-17a in Th1 cells [5, 18]. Conversly, enhancing glycolytic flux in T cells by overexpressing the glucose transporter 1 (GLUT1) escalates inflammatory responses in mice [17]. In contrast to cytotoxic and effector Th cells, Tregs and memory CD8+ T cells rely on OXOPHOS and fatty acid oxidation (FAO) to support their survival and differentiation. Blocking mitochondrial transport of long-chain fatty acids, a rate limiting step of FAO, with carnitine palmitoyltransferase 1A (CPT1a) inhibitors in Tregs suppresses their activity and survival [19]. Mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) are, mechanistically, the central energy-monitoring system in cells. Disruption of mTOR and AMPK activity is the critical signaling event that integrates metabolic activity with cell activation and differentiation in T cells. Impairment of glycolytic activity in T cells suppresses mTOR, but boosts AMPK activities, by altering the cellular ratio of AMP/ATP. This leads to a failure to switch T cell metabolism from catabolism into anabolism, affecting amino acid uptake and de novo synthesis of amino acids, nucleotides, and fatty acids [5, 18] (Fig. 1). Supporting this, T cell specific mTOR deletion elevates AMPK activity suppressing effector T cell differentiation; but Treg generation from these T cells remains intact. Furthermore, activating AMPK promotes Treg formation in both in vitro and in vitro assays [19, 20]. In addition to aerobic glycolysis, TCR signaling initiates robust amino acid uptake and metabolism in T cells that strengthen mTOR activity and instruct the Th cell differentiation program [21–26]. These findings suggest that mTOR and AMPK antagonize each other to instruct T cell activation and differentiation.
Fig. 1.
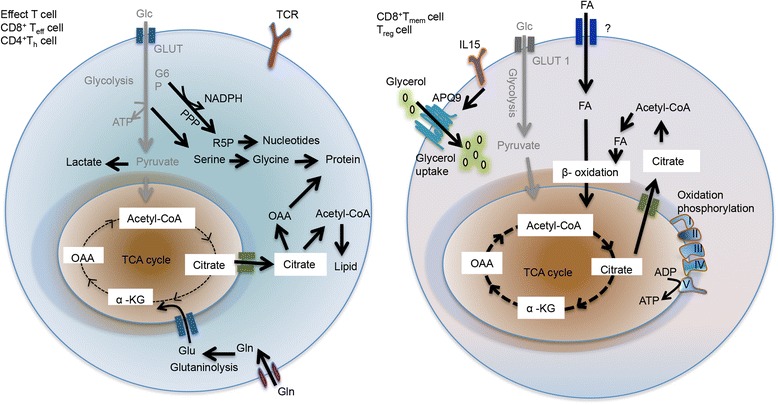
The metabolic profile of activated, effector and memory T cells. CD8+ Tmem cell and Treg cell rely on FAO and OXPHOS for survival and to support function. In addition, Tmem cells acquire glycerol by AQP9 for triglyceride synthesis and then promote Tmem cell proliferation and survival (left panel). In right panel, activated effector T cells (Effect T cells, CD8+Teff cell and CD4+Th cells) use aerobic glycolysis and FAS for proliferation. In activated effector T cell, most pyruvate become lactate and minority of pyruvate enters TCA cycle. However, PPP yields both nucleotide and NADPH for FAS. Glutaminolysis is also important for supplement of TCA cycle intermediates for biosynthesis
Metabolites serve as messengers to govern T cell effector functions
Despite aerobic glycolysis and amino acid uptake being critical for T cell activation, expansion and differentiation, T cells are able to survive in glucose-depleted conditions using mitochondrial OXOPHOS activity to support their energy demand [27]. However, as a trade-off for metabolic adaptation, T cells decrease the production of effector molecules, including as IFNγ, CD40L, and IL-2. Declined mTOR activity contributes to decreased effector functions; however, several new studies have uncovered that weakened generation of glycolytic metabolites also contribute to T cell dysfunction. Production of glyceraldehyde-3-phosphate (G3P) relieves the translational restraint of IFNγ and IL-2 imposed by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [28]. Moreover, accumulation of phosphoenolpyruvate in T cells suppresses ER calcium reuptake sustaining the Ca2+-NFAT pathway, which controls effector molecule production [29]. These studies provide evidence that glycolysis not only provide precursors of biomass and ATP, but also allows activated T cells to sustain effector functions through both transcriptional and translational regulations.
Metabolic pathways govern macrophage polarization
Metabolic preference of M1- and M2-like macrophages
Macrophages (MΦs) are terminally differentiated immune cells that possess high secretory, phagocytic, and antigen-presenting abilities. These activities are tightly controlled by a variety of immune stimuli such as cytokines, ligands of toll-like receptors (TLRs) and other pattern recognition receptors [30]. Resting MΦs encountering microbial stimuli, such as lipopolysaccharide (LPS) or LPS with pro-inflammatory cytokines elevate their phagocytic activity and gain a pro-inflammatory phenotype through the classical “M1-like MΦs” polarization. In contrast, MΦs can engage an alternative activation to become “M2-like”; thereby, sustaining tissue repair, angiogenesis, and Th2 immunity upon IL-4 activation. Importantly, M2-like MΦs produce more anti-inflammatory cytokines and less co-stimulatory proteins to T cells as well as demonstrating a reduced antigen-presenting ability [31]. In addition to defining stimuli and downstream signaling pathways that activate MΦs, recent research has revealed that MΦs must engage the proper metabolic switch when activated with LPS or IL-4. Specifically, TLR-induced signaling promotes MΦs aerobic glycolysis by stabilizing hypoxia-inducible factor 1a (HIF1α) and boosting mTOR activity. This increases glycolytic activity, as well as the pentose phosphate pathway (PPP) and de novo fatty acid synthesis (Fig. 2) [6, 30, 32]. Inhibition of glycolysis or deletion of GLUT1 prevents M1 polarization. This is phenotypically similar to treatment with either a mTOR inhibitor or AMPK activator. Moreover, it is critical that M1-like MΦs reduce mitochondrial OXOPHOS activity by suppressing succinate catabolism in order to maintain their pro-inflammatory properties [33]. In contract to M1-like MΦs, M2-like MΦs display low glucose flux but high rates of FAO and OXOPHOS via a PGC1β–dependent metabolic switch [6, 34, 35]. Of note, impairment of cholesterol efflux in MΦs results in a higher lipid content accompanying a M2-polarized phenotype [36, 37]. However, elevating lipid content with modified low-density lipoprotein (LDL) has also been shown to promote inflammatory activity associated with the M1 phenotype [38]. The underlying mechanisms on this discrepancy remain unclear, but these studies suggest that through modulating nuclear receptor activities, such as PPAR and LXR, the composition of lipid species may influence MΦ polarization [7].
Fig. 2.
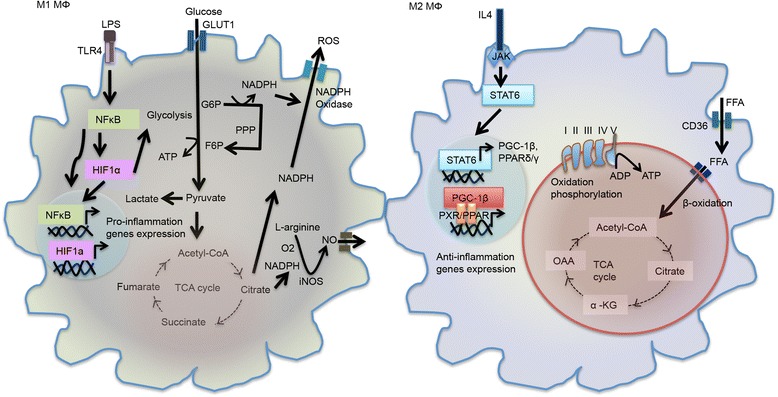
Metabolic reprogramming in M1 and M2 macrophage. LPS-induced M1 macrophages are inflammatory and generate pro-inflammatory cytokine by activation NF-κB and HIFα. M1-like macrophages display increased glycolysis to product lactate and decreased mitochondrial OXPHOS activity. M1-like macrophages also induce PPP to increase generation of NADPH for ROS production and NO synthesis. In contrast, IL4-polarized M2 macrophages activate STAT6 to induce PGC-1β and PPARδ/γ, which promote the expression of anti-inflammatory and metabolic genes. Thus, M2-like macrophages exhibit increased high rates of FAO and mitochondrial OXPHOS activity
Macrophages: double-edge swords in tumor progression and regression
The ability to use immunotherapy to exploit the host immunity to combat malignancy represents a breakthrough for cancer treatment. However, the establishment of the immunosuppressive tumor microenvironment is a major impediment for current immunotherapies [8, 39]. One of the key processes disarming anti-tumor immunity is the accumulation of M2-like tumor-associated MΦs. Tumor outgrowth is facilitated by the accumulation of M2-like MΦ via the prevention of type I immune responses elicited by T cells, the formation of abnormal vasculature and facilitating the dissipation of metastatic cancer cells [2]. However, “re-educating” M2-like MΦs to polarize into M1-like MΦs has been shown to elicit tumor regression. This occurs via restoring an immunosupportive microenvironment marked by elevated phagocytic activity, stronger antigen-presenting ability and secretion of pro-inflammatory cytokines. While the contribution of tumor-associated MΦs to cancer immune evasion and regression is established, many significant questions remain. Two major questions are how cancer cells sustain the pro-tumorigenic phenotypes of MΦs and whether cancer immunotherapy can “re-educate” tumor infiltrating myeloid cells.
Cancer cell immune evasion is promoted via the nutrient-specialized tumor microenvironment
Lactic acid and glucose in the tumor microenvironment
In the past decade, it has been demonstrated that increasing aerobic glycolysis and other anabolic metabolism in cancer cells can sustain their unrestricted proliferation by generating macromolecules for biomass synthesis. Oncogenic mutations and dysfunction of tumor suppressor, such as p53, in these cancer cells contribute to their abnormal cellular energetics [3, 39, 40]. These metabolic changes are suspected to alter the nutrient composition in the tumor microenvironment. For instance, the massive generation of lactic acid from cancer cells has been shown to suppress T cell cytotoxic and effector functions; however the mechanism behind this remains unclear. Furthermore, this generation of lactic acid has also been shown to promote M2-polarization in MΦs via monocarboxylate transporter-1(MCT1)-mediated lactic acid uptake [41]. Additionally, Colegio et. al. discovered that lactic acid stabilizes HIF1α to drive M2-polarization [42]. Of note, in contrast to M2-polarization, several other reports have shown that HIF1α stabilization promotes a M1-like macrophage polarization [33, 43, 44]. Although it is unclear how lactic acid acts differentially trigger macrophage polarization via HIF1α stabilization, it has been reported in cancer cells that lactic acid stabilizes HIF1α in cells relying on OXOPHOS but not in cells engaging aerobic glycolysis [45]. Therefore, it is likely that the metabolic states of MΦs can coordinate with lactic acid uptake to determine their polarization fate.
Warburg glycolysis allows cancer cells to consume glucose and increase lactic acid production. Additionally, two recent studies have independently demonstrated that the tumor microenvironment is glucose-depleted contributing to diminished T cell anti-tumor responses [29, 46]. These two studies suggested that cancer cells with higher glycolytic activity have a strong capacity to evade immunosurveillance. Moreover, Ho et. al. discovered that tumor-reactive T cells can be metabolically rewired through phosphoenolpyruvate carboxykinase 1 (PCK1) overexpression. This restores the T cell anti-tumor responses when infiltrating into the glucose-deprived tumor microenvironment through regaining phosphoenolpyruavte production [29]. Moreover, Chang et. al. showed that PD-1 inhibition boosts T cell anti-tumor responses by restoring mTOR activity and glucose flux in tumor-reactive T cells. This has also been observed in the chronic viral infection model and in vitro assays [47–49]. Together, these studies provide proof-of-concept evidence that metabolic competition for nutrients in the tumor microenvironment is involved in establishing and maintaining an immunosuppressive tumor microenvironment. Furthermore, these demonstrate that restoring metabolic fitness in T cells is a promising approach to regain effective anti-tumor responses.
The role of amino acid and fatty acid metabolism in immunosuppressive tumors
In addition to lactic acid production and glucose metabolism, MDSCs, M2-like MΦs and cancer cells, also affect the tumor microenvironment through degradation of extracellular arginine levels via arginase 1 (Arg1) expression [42, 50]. Moreover, tryptophan metabolism in tumors has recently gained attention for how it contributes to immunosuppression. Expression of indoleamine 2,3-dioxygenase (IDO) in antigen presenting cells (APCs) and cancer cells, promotes tumor progression and associates with poor responses to cancer immunotherapies in both clinical reports and murine cancer models [26, 51]. IDO is a metabolic enzyme that degrades tryptophan to kynurenine. Upregulated IDO activity suppresses T cell proliferation by restricting tryptophan to infiltrating T cells. Moreover, the generation of kynurenine may enhance Tregs formation in tumors as it has been shown to disarm gut and neuroinflammation in other disease models [52, 53]. Overall, these findings indicate that the declined amino acid availability in tumors can be harmful for mounting effective T cell anti-tumor immunity.
As well as consuming nutrients, cancer cells also produce massive amounts of fatty acids via de novo fatty acid synthesis, which correlates with more invasive cancer cells [54, 55]. In addition, accumulation of adipocytes and adipocyte-like fibroblasts in tumors can contribute to their metastasis [56]. These findings indicate that the tumor microenvironment may be lipid-enriched. In support of this, tumor infiltrating MDSCs and dendritic cells displayed higher lipid contents that associate with strong immunosuppressive activity and weak antigen presentation, respectively [57–60]. Of note, a fatty acid enriched microenvironment may impinge effector T cell functions and M1-polarization in MΦs while favoring the generation of Tregs and M2-like MΦs [5, 34]. Further investigation is critical to determine whether the fatty acid content in tumors and de novo fatty acid synthesis in cancer cells suppress T cell anti-tumor immunity and macrophage activation.
Unresolved questions and future directions: harnessing metabolic regulation in cancer immunotherapy
Metabolic states of tumor infiltrating lymphocytes and tumor-associated MΦs
Although metabolic pathways and their intermediates control immune responses in T cells and MΦs, much of our understanding has been formed based on in vitro cultures. These cultures generally have a defined nutrient composition and a fixed duration of stimulation. However, the tumor microenvironment in vivo has multiple layers of regulation. For example, the abnormal vasculature and complicated composition of cell types likely all influence the metabolic pathways and nutrients used by tumor infiltrating lymphocytes and tumor-associated MΦs. Therefore, in order to define which metabolic pathways link to effective anti-tumor responses it will be critical to determine the metabolic signature and profile in tumor infiltrating lymphocytes and tumor-associated MΦs, from progressed and regressed tumors. Additionally, metabolic adaption allows T cells to engage OXOPHOS metabolism facilitating their survival. This process requires the electron transport chain and functional mitochondria in T cells. However, it remains unclear if tumor infiltrating lymphocytes are capable of maintaining their mitochondrial fitness in order to adapt to the glucose-depleted tumor microenvironment. Thus, it will be important to examine the mitochondrial health of tumor infiltrating lymphocytes.
Metabolic competition has clearly been shown to contribute to the formation of the immunosuppressive tumor microenvironment. Therefore, it will be vital for cancer immunotherapies to develop new strategies to allow immune cells to acquire sufficient nutrients or metabolites to preserve their anti-tumor responses. Thus, examining how to combine cancer immunotherapies with metabolic inhibitors in order to sustain the metabolic fitness of tumor-reactive T cells and M1-like MΦs will be crucial. In addition, it will also be necessary to elucidate if the therapeutic efficacy of cancer immunotherapies is associated with, as yet undefined, metabolic features in tumors. A better understanding of how immunotherapies respond to different metabolic features in tumors will further improve how we apply cancer immunotherapies in patients.
Why does hypoxia in tumors not improve anti-tumor responses?
Hypoxia is a general feature of established tumors and contributes to elevated aerobic glycolysis by stabilizing HIF1α. Interestingly, overexpressing a degradation-resistant mutant of HIF1α or abolishing the HIF1α-degrading machinery promotes anti-tumor responses in T cells, including cytotoxic activity and production of IFNγ [61]. However, tumor infiltrating T cells fail to maintain anti-tumor responses in tumors where hypoxia is a general environmental stress. This raises an interesting question: why does hypoxia fail to boost T cell anti-tumor responses in tumors. The degree of HIF1α stabilization in cancer cells is associated with the metabolic pathways [45]. Therefore, we postulate that hypoxia fails to stabilize HIF1α in tumor infiltrating T cells due to their higher OXOPHOS metabolism, triggered by PD1 signaling, and the nutrient composition in the tumor microenvironment. If this is true, PD1 inhibition will restore hypoxia-induced HIF1α stabilization, thereby enhancing tumoricidal activity of tumor infiltrating T cells. Another possibility is that hypoxia truly boosts glycolytic rates in tumor-infiltrating T cells by stabilizing HIF1α; however, the available glucose in the tumor microenvironment is insufficient to support T cell anti-tumor responses. More extensive studies are needed to resolve this and will offer an immediate impact on the development of new combined treatments.
How can we improve the metabolic fitness of T cells in ACT therapy?
Reduced expression of tumor antigens and the MHCI complex in cancer cells provides a growth advantage for aggressive tumors by evading T cell mediated immune destruction. The invention of CAR T-cells gives the ability to re-direct the targeting specificity to cancer cells, thereby enhancing the anti-tumor responses in ACT immunotherapy [62]. However, the therapeutic outcome of CAR T-cells on treating solid tumors is disappointing. Interestingly, combining CAR T-cells with PD-1 blockade drastically improves anti-tumor responses, suggesting that restrictions due to the tumor microenvironment limit the CAR T cell induced anti-tumor immunity [63]. Engaging aerobic glycolysis and amino acid metabolism is a prerequisite for proper T cell activation upon TCR stimulation. However, the intracellular signaling pathways and metabolic requirement for sustaining CAR T-cell activation and anti-tumor responses are not fully understood. Given that metabolic competition between cancer cells and infiltrating T cells leads to T cell anergy and dysfunction, it will be important to investigate whether CAR signaling also relies on the metabolic switch to sustain anti-tumor responses. If it is true, it may be possible to improve the metabolic fitness and flexibility of CAR T cells with genetic and chemical approaches.
In addition to CAR T-cells, memory tumor-reactive T cells have been shown to elicit superior anti-tumor responses than effector T cells do [64]. During in vitro assays secondary effector T cells generated from memory T cells, display stronger aerobic glycolysis and IFNγ production than primary effector T cells [65, 66]. Therefore, these secondary effector T cells can plausibly acquire sufficient glucose when in the tumor microenvironment even under “metabolic competition” with cancer cells. Moreover, memory T cells also possess healthier mitochondria, which may help adaptation to a nutrient crisis more effectively [65]. Accordingly, it will be important to gain a detailed understanding of the underlying mechanisms contributing to the stronger metabolic fitness seen in secondary effector T cells. This knowledge will feasibly provide a blueprint for manipulating transferred tumor-reactive T cells. Ultimately, this may allow tumor-reactive T cells to be conferred with enhanced metabolic fitness and effector functions specifically in the nutrient-specialized tumor microenvironment.
Conclusions
Cancer immunotherapy provides an exciting opportunity to introduce a powerful weapon to the arsenal of cancer treatment. However, it is crucial to understand how the intensive communication in the tumor microenvironment efficiently diminishes anti-tumor immunity. Modulation of cellular metabolism has been shown to play a cardinal role in controlling immune responses in a variety of immune cells. Despite this, deregulated cancer metabolism represents a newly appreciated hallmark for cancer cells. Further research in this field will be vital to provide the foundation for new treatments that target cancer metabolism. Here, we hypothesize that changing the immunostimulatory and immunosuppressive nutrients and metabolites within the tumor microenvironment denotes a new immunomodulatory mechanism in tumors (Fig. 3). Altering the nutrient composition allows cancer cells to evade from immunosurveillance and, even coax the formation and accumulation of pro-tumorigenic immune cells. A clear understanding of how the immunometabolic regulation controls immune responses, as well as identifying immunostimulatory and immunosuppressive nutrients, will allow immune responses in tumors to be rebalanced with genetic and/or nutritional approaches.
Fig. 3.
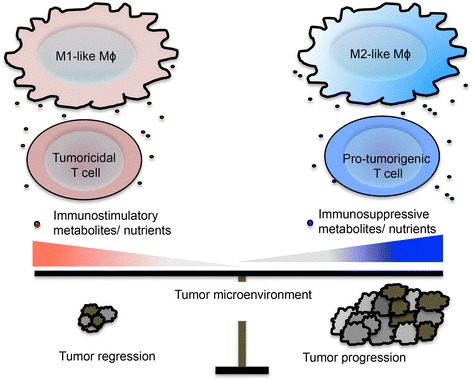
Environmental nutrients control immune cells phenotype. The composition of nutrients within tumor environment affects metabolic fitness of infiltrating immune cells. Thus, new approaches to rewire metabolic fitness in tumor infiltrating immune cells might boost immune response in conjunction with current cancer immunotherapies
Acknowledgements
P.-C. H. is supported by University of Lausanne, Swiss Institute for Experimental Cancer Research (26075483), and Ludwig Center for Cancer Research.
Abbreviations
- ACT
Adoptive cell transfer
- DC
Dendritic cells
- CAR
Chimeric antigen receptor
- APC
Antigen-presenting cell
- OXOPHOS
Oxidative phosphorylation
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contibutions
P.-C. H. and P.-T. L. wrote this manuscript and P.-T. L. composed the figures. Both authors read and approved the manuscript.
Contributor Information
Ping-Chih Ho, Phone: +41-021-692-5947, Email: ping-chih.ho@unil.ch.
Pu-Ste Liu, Email: pu-ste.liu@unil.ch.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–83. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289(11):7884–96. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. 2015;15(1):18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012;33(7):364–72. doi: 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Stauss HJ, Morris EC. Immunotherapy with gene-modified T cells: limiting side effects provides new challenges. Gene Ther. 2013;20(11):1029–32. doi: 10.1038/gt.2013.34. [DOI] [PubMed] [Google Scholar]
- 10.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monu N, Frey AB. Suppression of proximal T cell receptor signaling and lytic function in CD8+ tumor-infiltrating T cells. Cancer Res. 2007;67(23):11447–54. doi: 10.1158/0008-5472.CAN-07-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol. 2012;12(2):136–48. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–82. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–43. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son HJ, Lee J, Lee SY, Kim EK, Park MJ, Kim KW, et al. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm. 2014;2014:973986. doi: 10.1155/2014/973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol. 2013;191(8):4080–5. doi: 10.4049/jimmunol.1300923. [DOI] [PubMed] [Google Scholar]
- 22.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–8. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185(2):1037–44. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–24. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 26.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42(1):41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 28.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–51. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162(6):1217–28. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–84. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–14. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 33.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496(7444):238–42. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–55. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17(4):549–61. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rios FJ, Koga MM, Pecenin M, Ferracini M, Gidlund M, Jancar S. Oxidized LDL induces alternative macrophage phenotype through activation of CD36 and PAFR. Mediators Inflamm. 2013;2013:198193. doi: 10.1155/2013/198193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho PC, Chang KC, Chuang YS, Wei LN. Cholesterol regulation of receptor-interacting protein 140 via microRNA-33 in inflammatory cytokine production. FASEB J. 2011;25(5):1758–66. doi: 10.1096/fj.10-179267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9(10):691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 41.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–9. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 42.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakayama T, Kurobe H, Sugasawa N, Kinoshita H, Higashida M, Matsuoka Y, et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1alpha as a mediator of vascular remodelling. Cardiovasc Res. 2013;99(4):705–15. doi: 10.1093/cvr/cvt146. [DOI] [PubMed] [Google Scholar]
- 44.Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008;10(5):983–95. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- 45.De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0046571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41(5):802–14. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69(4):1553–60. doi: 10.1158/0008-5472.CAN-08-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 52.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 53.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–61. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–6. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao F, Liu C, Guo J, Sun W, Xian L, Bai D, et al. Radiation-driven lipid accumulation and dendritic cell dysfunction in cancer. Sci Rep. 2015;5:9613. doi: 10.1038/srep09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–82. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36(8):494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–46. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 64.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A. 2013;110(35):14336–41. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):1064–72. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]