

Abstract
Background
MicroRNAs (miRNAs) are small endogenous non-coding RNAs that control genes at post-transcriptional level. They are essential for development and tissue differentiation, and such altered miRNA expression patterns are linked to the pathogenesis of inflammation and cancer. There is evidence that miRNA expression is genetically controlled similar to the transcription of protein-coding genes and previous studies identified quantitative trait loci (QTL) for miRNA expression in the liver. So far, little attention has been paid to miRNA expression in the skin. Moreover, epistatic control of miRNA expression remains unknown. In this study, we characterize genetic regulation of cutaneous miRNA and their correlation with skin inflammation using a previously established murine autoimmune-prone advanced intercross line.
Results
We identified in silico 42 eQTL controlling the expression of 38 cutaneous miRNAs and furthermore found two chromosomal hot-spots on chromosomes 2 and 8 that control the expression of multiple miRNAs. Moreover, for 8 miRNAs an interacting effect from pairs of SNPs was observed. Combining the constraints on genes from the statistical interaction of their loci and further using curated protein interaction networks, the number of candidate genes for association of miRNAs was reduced to a set of several genes. A cluster analysis identified miR-379 and miR-223 to be associated with EBA severity/onset, where miR-379 was observed to be associated to loci on chromosome 6.
Conclusion
The murine advanced intercross line allowed us to identify the genetic loci regulating multiple miRNA in skin. The recurrence of trans-eQTL and epistasis suggest that cutaneous miRNAs are regulated by yet an unexplored complex gene networks. Further, using co-expression analysis of miRNA expression levels we showed that multiple miRNA contribute to multiple pathways that might be involved in pathogenesis of autoimmune skin blistering disease. Specifically, we provide evidence that miRNA such as miR-223 and miR-379 may play critical role in disease progression and severity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2455-2) contains supplementary material, which is available to authorized users.
Keywords: MicroRNA, Expression QTL, Epistasis, Autoimmune skin blistering disease, Co-expression analysis
Background
The discovery of microRNAs (miRNAs), a class of small endogenous non-coding molecules ranging from 18–24 nt brought a new level of complexity for understanding the mechanisms that constitute various biological processes [1, 2]. The involvement of miRNAs in the control of gene expression has been thoroughly defined for the cell cycle, metabolism, and immune system and in cancer [3–6]. Binding to the 3′ or 5′ UTR region of genes, miRNAs may yield increased or decreased gene expression levels and have been described to affect various molecular pathways [7].
Approximately half the miRNAs are intergenic with few also located in intronic regions [8, 9]. These are understood to have their own enhancers and promoters and are transcribed by RNA polymerase II [10]. However, it remains unclear whether these are produced as by-products of protein-coding gene transcription or whether their biogenesis has its own machinery [11]. Recent studies in murine and human fibroblasts of liver tissues revealed that miRNA expression can either be regulated by their transcriptional genomic location or by other regions in the genome [12]. Furthermore, mutations in genes involved in miRNA processing, such as AGO1, DGCR8, and DICER, can cause significant changes in the expression of miRNAs, resulting in altered disease susceptibility [13]. Regarding the skin, in vivo studies show that miRNA biogenesis is dependent on both DICER and DGCR8. A lack of these enzymes causes severe phenotypes [14], underscoring the importance of miRNAs in the regulation of morphogenesis and homeostasis of the skin [15]. Differentially expressed miRNAs are associated with different physiological and pathological processes in the skin such as melanoma, Sézary syndrome, psoriasis and atopic dermatitis [16–19]. Advancements have been achieved in understanding various processes regulated by miRNAs. On the other hand, the mechanisms that regulate their own expression have so far remained unknown. One possible approach towards understanding their regulation is constraining on genetic loci whose variations statistically link the variable phenotypic effect. We thus performed an in silico expression QTL-based analysis, which has been widely used for deciphering genetic loci regulating gene expression in various biological processes [20]. Su et al. have further shown that expression QTL-based analysis can provide insights into miRNA regulation [12]. However, no study has yet been performed to determine the impact of genetic variation on miRNAs associated to skin and autoimmune disorders.
This study (i) uses expression QTL-based analysis to define genetic loci that control miRNA expression in the skin and (ii) provides insights into interacting genes to control a single transcript (epistasis). The relevance of these findings is illustrated by employing a mouse model for the autoimmune blistering disease EBA (Epidermolysis Bullosa Acquisita), providing a direct link between miRNA expression and disease phenotype. Taken together, the data provide insights into the complexity of miRNA regulation and possible means to understand various gene interactions altering the expression of miRNAs.
Results
A murine heterogeneous inter-cross line was generated by inter-crossing four parental inbred mouse strains: MRL/MpJ, NZM2410/J and BXD2/TyJ as autoimmunity-prone strains and Cast/EiJ for genetic heterogeneity. As a mouse model for an autoimmune blistering disease, 100 mice from generation 4 of the intercross-line were immunized with recombinant collagen type VII (COL7), an integral component of anchoring fibrils located at the dermal-epidermal junction. This induced a loss of tolerance and production of anti-COL7 autoantibodies in all immunized mice. The incidence of sub-epidermal blisters and the clinical phenotype of epidermolysis bullosa acquisita (EBA) were at 1/3 [21]. All mice were genotyped and miRNA expression profiling was acquired from skin tissue using the Affymetrix’s GeneChip miRNA 2.0 array. To reveal genetic loci that regulate miRNA expression, we then performed an association study between the genotype and the respective expression levels of miRNAs. Further we performed co-expression analysis between expression levels of miRNAs and phenotype. The workflow is presented in a flowchart (Additional file 1: Figure S1).
miRNA expression is genetically controlled
We performed a genome-wide scan to detect genetic loci associated with miRNA expression. Expression levels of miRNA were treated as quantitative trait to yield expression quantitative trait loci (eQTL). Genome-wide significance was determined by a permutation test. Using an E-value cutoff of <0.05, 42 eQTL for 38 miRNAs were mapped to the genome, corresponding to 6.83 % of all murine miRNAs present on the above mentioned Affymetrix GeneChip (Table 1 and Fig. 1). Since the wild derived strain CAST/EiJ was incorporated into the advanced intercross line, we investigated the polymorphic sites located in the transcribed miRNAs that may have an effect of probe hybridization which is derived from C57B6/J [22]. We obtained the SNPs and indels in genome of CAST/EiJ from the database [23]. We found that 4/38 (10.53 %) miRNAs (miR-291a-3p, miR-341, miR-449b and miR-681) exhibits indels in CAST/EiJ strain on chromosome 7 (3.2 Mb), 12 (69 and 109 Mb) and 13 (113 Mb), which may suggest false positive associations for those loci (Table 1). The highest -log P value of 6.57 was observed for miR-298 on chromosome 9 explaining 20.76 % of the variance. The peak SNP (rs3700596) was found within 1 kb of the Ube3D gene, an ubiquitin-conjugating enzyme E2c binding protein. We found only trans – eQTL, except for miR-486 (−log P = 4.10, rs13479880), which was mapped to the same chromosome of its transcriptional site (Chr 8, ~89 Mb). This indicates that other genes rather than their own transcript may contribute to the regulation of miRNA expression which could be tissue specific.
Table 1.
eQTL detected for the expression of miRNA
| AffyID | PEAK SNP | Chr | Pos(Mb) | CI | -logP score | α | Transcript Location (Chr:Pos) |
|---|---|---|---|---|---|---|---|
| miR-322 | rs6386920 | 1 | 54.1 | 51.5–58.4 | 4.797697778 | 0.02 | X:50407432-50407526 |
| miR-431 | rs6386920 | 1 | 54.1 | 51.5–59.8 | 5.231417376 | 0.006 | 12:110828657-110828747 |
| miR-26a | rs6265423 | 2 | 47 | 28.1–51.3 | 4.376166299 | 0.021 | 10:126432586–126432669,9:118940914-118941003 |
| miR-291a-3p | CEL.2_50605053 | 2 | 50.5 | 28.1–59.9 | 4.546953451 | 0.018 | 7:3218920-3219001 |
| miR-423-3p | rs6265423 | 2 | 47 | 33.9–51.3 | 4.948513523 | 0.015 | 11:76891566-76891674 |
| miR-671-5p | rs13476472 | 2 | 45.5 | 33.9–50.5 | 5.838570402 | 0.004 | 5:24097932-24098029 |
| miR-23b | rs6250599 | 2 | 48.5 | 44.3–51.3 | 4.335454904 | 0.022 | 13:63401792-63401865 |
| miR-409-5p | rs13476874 | 2 | 159.5 | 136–163 | 4.175101842 | 0.035 | 12:110981368-110981446 |
| miR-409-5p | rs13477083 | 3 | 43.3 | 27.5–45.4 | 4.215972522 | 0.033 | 12:110981368-110981446 |
| miR-546 | rs13477126 | 3 | 56.4 | 52.9–58.3 | 4.493016911 | 0.029 | 10:126435496-126435616 |
| miR-200a-star | rs3671119 | 3 | 126.1 | 117.4–131.3 | 4.688008826 | 0.011 | 4:155429005-155429094 |
| miR-339-5p | rs3660863 | 4 | 7.1 | 3.7–19.5 | 4.878132165 | 0.003 | 5:139845604-139845699 |
| miR-465c-5p | rs13477873 | 4 | 101.1 | 82.8–118 | 4.472478928 | 0.01 | X:64079130-64079210 |
| miR-295 | rs3663950 | 4 | 135.3 | 129.4–141.1 | 4.209508065 | 0.04 | 7:3220774-3220842 |
| miR-878-3p | rs13478002 | 4 | 136.2 | 135.3–141.1 | 5.110152979 | 0.01 | X:64054683-64054760 |
| miR-742 | rs3673049 | 5 | 90.1 | 87.5–96.6 | 4.765858909 | 0.035 | X:64033548-64033612 |
| miR-379 | rs6208251 | 6 | 104.8 | 98.4–116.7 | 4.024954772 | 0.034 | 12:110947270-110947335 |
| miR-154 | rs13479063 | 6 | 136.3 | 133.92–142.4 | 4.309450382 | 0.032 | 12:110976643-110976708 |
| miR-425* | rs3663988 | 7 | 146.5 | 140.2–146.5 | 4.342220512 | 0.035 | 9:108471108-108471192 |
| miR-486 | rs13479880 | 8 | 89.3 | 72.5–95.1 | 4.108040959 | 0.033 | 8:24253027-24253154 |
| miR-487b | rs6257357 | 8 | 88 | 77.7–90.2 | 4.81779528 | 0.019 | 12:110965543-110965624 |
| miR-501-3p | rs13479880 | 8 | 89.3 | 81.7–95 | 4.205948891 | 0.03 | X:6818369-6818477 |
| miR-130b | rs6413270 | 9 | 37.7 | 36.8–44.4 | 4.222559895 | 0.032 | 16:17124154-17124235 |
| miR-298 | rs3700596 | 9 | 86.2 | 85–90.6 | 6.573678024 | 0.001 | 2:174093005-174093086 |
| miR-466c-3p | rs3712394 | 10 | 17.7 | 14.1–24.2 | 4.569565941 | 0.03 | 2:10403161-10403244 |
| miR-466c-3p | rs13480563 | 10 | 27.9 | 24.2–38.7 | 4.591526295 | 0.029 | 2:10403161-10403244 |
| miR-126-5p | rs6374078 | 10 | 60.6 | 30.7–65.9 | 4.177297455 | 0.031 | 2:26446877-26446949 |
| miR-681 | rs13481076 | 11 | 66.5 | 40.1–71.3 | 4.055742697 | 0.043 | 12:70864822-70864931 |
| miR-20a-star | rs3712881 | 11 | 120.9 | 112–121 | 4.160612966 | 0.03 | 14:115443379-115443485 |
| miR-203 | mCV22351241 | 12 | 60 | 55.0–72.6 | 3.838027866 | 0.039 | 12:113369091-113369166 |
| miR-542-3p | CEL.12_84750094 | 12 | 91.4 | 79.7–103.8 | 4.735149963 | 0.023 | X:50402580-50402664 |
| miR-341 | gnf13.079.671 | 13 | 80.5 | 69.5–88.3 | 5.260091403 | 0.008 | 12:110849710-110849805 |
| miR-449b | rs13482231 | 14 | 67.6 | 50.9–72.3 | 4.485232377 | 0.019 | 13:113827627-113827706 |
| miR-7a | CEL.15_4222769 | 15 | 4.4 | 3.2–9.7 | 4.941341377 | 0.044 | 13:58494140–58494247, 7:86033163-86033259 |
| miR-7a | rs13482455 | 15 | 16.7 | 14.8–24.5 | 5.73214998 | 0.022 | 13:58494140–58494247, 7:86033163-86033259 |
| miR-337-3p | rs13482549 | 15 | 45.5 | 38.4–53.9 | 4.298224443 | 0.029 | 12:110823999-110824095 |
| miR-673-5p | rs13482549 | 15 | 45.5 | 38.9–56.6 | 4.592339382 | 0.014 | 12:110810200-110810290 |
| miR-136 | rs13482914 | 17 | 21 | 16.5–27.6 | 4.679083445 | 0.026 | 12:110833537-110833598 |
| miR-466b-5p | rs13483212 | 18 | 12.3 | 0–21 | 4.48515896 | 0.016 | 2:10395846-10395927 |
| miR-493 | rs6211533 | 19 | 57.1 | 50.2–60.2 | 4.98913787 | 0.004 | 12:110818443-110818525 |
| miR-26a | gnfX.023.543 | X | 36.3 | 0–49.3 | 4.718906745 | 0.01 | 10:126432586–126432669, 9:118940914-118941003 |
| miR-466e-5p | rs13483712 | X | 9.1 | 0–33.5 | 4.013512497 | 0.05 | 2:10398257-10398340 |
AffyID Affymentrix ID, Chr Chromosome, Pos (Mb) Peak Position SNP in Mb, CI Confidence Interval (CI) in Mb using 1.5 -log P drop, α Genome wide significance
Fig. 1.
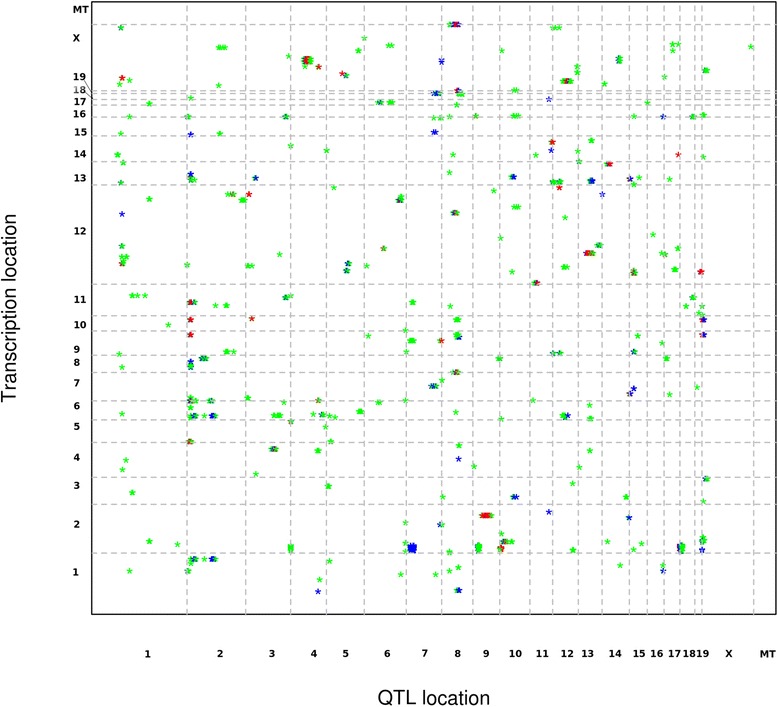
Transcriptome map for eQTL of miRNA . The x-axis in the figure represents the SNP location in the chromosome, and the y axis represents the transcriptional site of miRNAs. In this figure genome wide significant threshold of α < 0.25 is represented in a green. 83 miRNA eQTL were defined as suggestive eQTL for miRNA (genome wide threshold, α < 0.1) are represented in blue. 43 miRNA eQTL with α < 0.05 were observed as significant eQTL are represented in red
Prior knowledge suggests classes of gene families that play a major role in the regulation of miRNA expression i.e. Argonaute proteins and helicases which play a crucial role in its regulation [24]. To examine this assumption, we obtained the coordinates of all helicases and other genes involved in pathways of miRNA biogenesis from ‘The RNA Helicase Database’ and mapped them to the eQTL identified in our study [25]. Helicases like Ddx39, Ddx49, Cd97 and Upf1 were mapped within the confidence interval of miR-486, miR-487b and miR-501 on chromosome 8. Further, four helicases (Ddx50, Ascc3, Ddx21 and Dna2) were mapped within the confidence interval of the eQTL controlling miR-126. Genes that are involved in transcriptional processes, such as Polr3f, Polr2a, Polr3g and Polr2a were also mapped to the eQTL for miR-409, miR-681, miR-34 and miR-449. Other genes which do not belong to these classes, such as lin28a and its homolog lin28b, have been shown to modulate let-7a [26]. These genes were mapped to the eQTL for miR-290 on chromosome 4 and miR-126 on chromosome 10, suggesting that Lin28 might modulate the expression of other miRNAs as well.
Some miRNA eQTL were constrained to a specific location in the genome, thereby suggesting potential eQTL hotspots for the regulation of multiple miRNAs. On chromosome 2, five miRNAs (miR-26a, mir-291a, miR-423, miR-671 and miR-23b) were mapped between 28–51 Mb (Fig. 2). Three nearby SNPs (rs6250599 ~ 48.5 Mb, rs6265423 ~ 47 Mb and rs13476472 ~ 45 Mb) showed significant association (−log P > 4) with all five miRNAs that were mapped to this region. A long non coding RNA 1700019E08Rik was located near SNP rs13476472 (~3 Kb upstream). As for the other two SNPs, the nearest gene to rs6265423 was mapped 6.8 kb apart, coding for the snRNA U7.39-201, while the nearest coding gene for SNP rs6250599 was a pseudogene Gm13489-001 (~13 kb upstream).
Fig. 2.
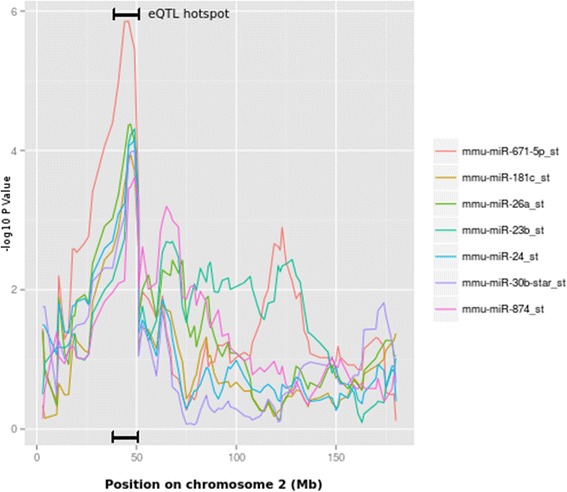
eQTL hot spots on chromosome 2. Position on x axis represents the coordinates in million base pairs on chromosome 2. The y axis is –log P value. The overlapping peaks on y axis represents an eQTL hot spot between 28–51 Mb. miR-181, miR-30b* and miR-874 are additional suggestive eQTL with significant genome-wide (α < 0.1) threshold after permutation
Similarly, on chromosome 8 we identified eQTL for three miRNAs (miR-486, miR487b and miR-501) in confidence interval 72–95 Mb. Three genes Gm1068, Siah1 and Dnaja2 were located near peak SNPs (rs13479880 and rs6257357).
Furthermore, we found several miRNAs that are associated to more than one locus in the genome. For example miR-7a showed significant association with two loci present on chromosome 15 (3–9 Mb, CEL.15_4222769 and 14–24 Mb, rs13482455). Similarly, miR-466-3c was regulated by two nearby loci on chromosome 10 (14–24 Mb, rs3712394 and 24–38 Mb, rs13480563). Additionally, miR-26a was mapped with two loci on chromosomes 2 and X (28–51 Mb, rs6265423 and 0–49 Mb, gnfX.023.543).
Epistatic control of miRNA expression
A defective of single loci may possibly be compensated by another, or only jointly the effect is the strongest. Therefore, for all the miRNAs significant single-locus eQTL, we analyzed the epistatic effect for each SNP pair. As a result, we identified 200 SNP pairs for 8 miRNAs below the significance level (adjusted P value < 0.05) (Fig. 3 and Additional file 1: Table S1). The highest -log P value of 10.38 was found between the SNP pairs rs13480360 (chr 10, ~67 Mb, nearest gene: AK139516) and rs3689658 (chr 2, ~ 85 Mb, nearest gene: Olfr1006) for miR-7a (Table 2). In total, we found 119 SNP pairs for miRNA miR-7a. The hub locus (i.e. SNP with the maximal number of interactions) for miR-7a was observed on chromosome 16 (rs3680665 ~ 84 Mb, nearest gene: AK04263). The same SNP (rs3680665) also showed association with miR-542 additionally with another SNP (rs4200124, nearest gene: Gbe1) present nearby. For miR-742 we observed 47 SNP pairs with SNP rs3657112 (~148 Mb, nearest gene: Snora17) on chromosome 3 showing the highest number of associations (40 SNP pairs). The same SNP i.e. rs3657112 also showed statistical interaction with chromosome 9 loci 67–72 Mb for miR-295. We also found multiple SNPs on chromosome 2 (13–17 Mb) statistical interacting with SNPs on chromosome 1(3–11 Mb) for miR-501. Two miRNAs (miR-136 and miR-337) had only one significant SNP pair: for miR-136 the correspondence was found between SNPs rs37113033 (chr 19, ~5 Mb, nearest gene: Slc29a2) and rs13459176 (chr 15, ~3 Mb, nearest gene: Sepp1), while miR-337 had SNP pair rs3693942 (chr 13, ~55 Mb, nearest gene: Unc5A) and rs3663950 (chr4, ~135 Mb, nearest gene: Il22ra) (Table 2).
Fig. 3.
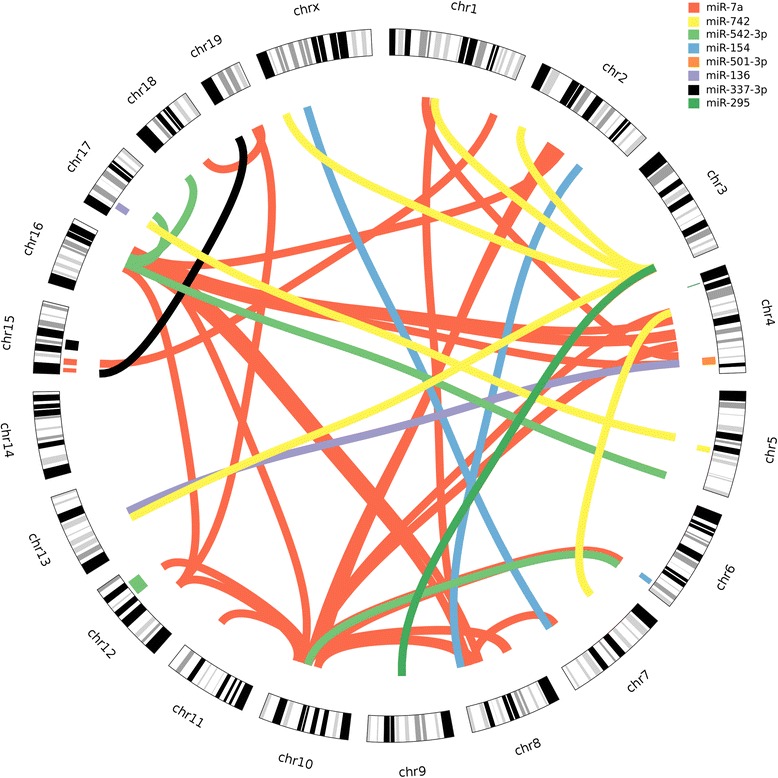
Epistasis in miRNA eQTL. The circular plot shows the chromosomes in their circumference. Each connecting line represents a SNP pair interaction above the significance level (adj. P < 0.05). Each interaction is color coded for different miRNAs. The boxes adjacent to the chromosomal band show eQTL for miRNAs mapped for single locus scans
Table 2.
Epistasis in miRNA. The table includes only the top interacting SNPs for each miRNAs
| miRNA | Top interacting SNP 1 | Top interacting SNP 2 | I.Pval(log) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP ID | chr | Pos(Mb) | Refseq Genes | Pval(log) | SNP ID | chr | pos(Mb) | Refseq Genes | Pval(log) | ||
| miR-7a | rs13480630 | 10 | 67.29 | 0.89 | rs3689658 | 2 | 85.52 | Olfr1006 | 1.12 | 10.4 | |
| miR-136 | rs3713033 | 19 | 5.03 | Slc29a2 | 0.073 | rs13459176 | 15 | 3.23 | Ccdc152, Sepp1 | 1.56 | 7.2 |
| miR-154 | rs3708073 | 8 | 124.28 | Gm20388,Jph3 | 0.3 | rs8246404 | 2 | 136.7 | Mkks | 0.57 | 7.67 |
| miR-295 | rs13480271 | 9 | 72.68 | RP23-461P14 | 0.018 | rs3657112 | 3 | 148.03 | 0.32 | 8.064 | |
| miR-337-3p | rs3693942 | 13 | 55.05 | Unc5a | 0.026 | rs3663950 | 4 | 135.29 | Il22ra1 | 0.2 | 7.17 |
| miR-501-3p | CZECH-2_15618849 | 2 | 15.5 | Gm13364 | 1.33 | rs3716083 | 1 | 9.01 | Sntg1 | 0.72 | 8.46 |
| miR-542-3p | rs4200124 | 16 | 70.7 | 1.12 | rs3718776 | 5 | 150.4 | Wdr95 | 0.14 | 8.5 | |
| miR-742 | rs3657112 | 3 | 148.028 | 0.35 | CEL.1_49993068 | 1 | 49.68 | 0.33 | 9.16 | ||
Chr Chromosome, Pos (Mb) Position in million base pairs, Pval(log) Additive log 10 P value, I.Pval(log) Interacting log 10 P value
To search the candidate interacting gene pairs we investigated the epistatic control of miR-501-3p. Using the Ingenuity pathway analysis (IPA), we searched for all the possible interacting genes present between loci present on chromosome 1 and chromosome 2 [27]. We found three putative interacting gene pairs; namely Commd3 with Cops5, Cacnb2 with Vopip1 and Commd3-Bmi1 with Rb1cc. Interestingly, Cops5 in the Nfkb1 pathway possibly be associated with miR-501 via Tp53 (Additional file 1: Figure S2).
Genetic overlap of QTL for clinical scores and miRNA expression
Any genetic variation with an effect on the clinical phenotype and miRNA levels should map to the same chromosomal region. Previously, genetic loci for the clinical phenotype of EBA, an autoimmune skin blistering disease, were studied using a larger cohort of mice from the same breeding scheme. Previously, we mapped the QTL for the onset of EBA to chromosomes 9 (39.5–46.3 Mb), 12 (83.2–109.9 Mb), 14 (49.1–68.9 Mb) and 19 (46.0-end Mb). Additionally, three QTL were mapped for severity of EBA on chromosomes 1 (3.1–27.3 Mb), 15 (61.0–85.7 Mb) and 19 (43.1-end Mb) [21]. In this study, we found 4 eQTL for miRNAs overlapping with the QTL for EBA (Fig. 4). The eQTL for miR130b (Chr 9: 36–44 Mb, −log P = 4.42), miR-542-3p (Chr 12: 79–103 Mb, −log P = 4.73) and miR-449b (Chr 14: 50–72 Mb, −log P = 4.49) were mapped on QTL for disease onset on chromosome 9, 12 and 14. Additionally, we mapped eQTL for miR-493 (50–60 Mb, −log P = 4.98) to the QTL for both disease severity and onset on chromosome 19. To further evaluate, if the variation in the genome is associated with the miRNA expression levels and also underlies the clinical phenotype, we investigated the genotype association of significant SNPs of miRNA eQTL with clinical phenotype (Fig. 5). We observed that the most significant SNP (rs6211533) mapped for the eQTL of miR-493 on chromosome 19 also shows variation for EBA severity score and onset. In this eQTL minor allele (BB) derived from MRL/MpJ was associated with higher expression of miR-493, late onset of the disease, and higher clinical score (Fig. 5b). Similar observation was also derived for eQTL mapped (peak SNP, rs6413270) for miR-130b where minor allele (BB) derived for MRL/MpJ and NZM/2410J was associated with lower expression of miR-130b, and late onset of disease phenotype (Fig. 5a).
Fig. 4.
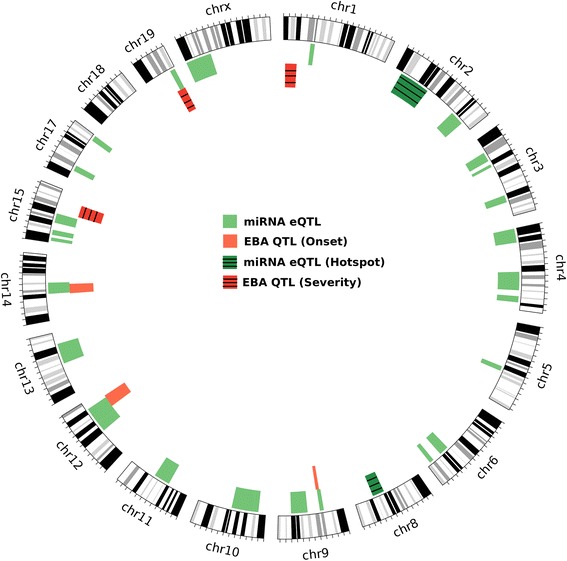
Overlapping QTL for EBA disease and miRNA eQTL. The circular plot shows all the eQTL for miRNAs (green) and QTL for EBA (red). It also presents the eQTL hot spots (dark green) and EBA QTL for onset (red) and severity (dark red). Each circular band represents a chromosome on which QTL and eQTL are mapped. The region within the chromosome which has either red or dark red and green or dark green bands is overlapping eQTL with EBA QTL
Fig. 5.
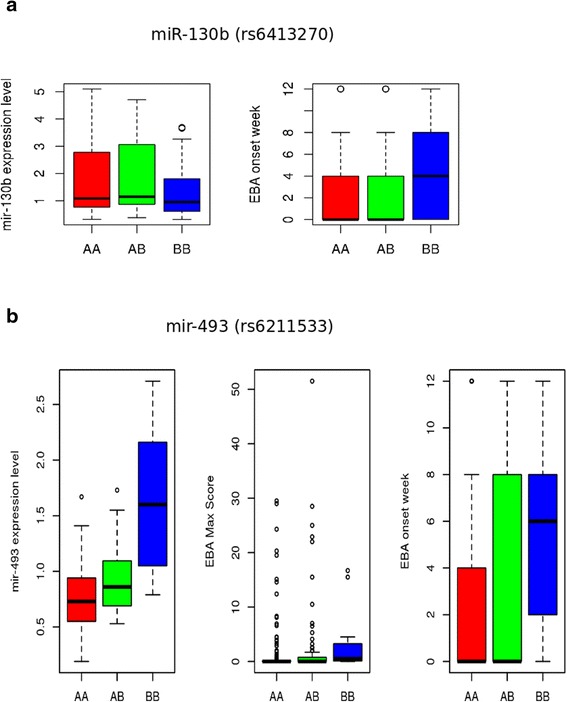
Boxplot for genotype variations for significant SNP in miRNA eQTL for disease QTL. The figure describes the genotype variation for the peak SNP for miRNAs mapped to the previously described EBA QTL. a) In miR-130b eQTL, peak SNP (rs6413270, −logP = 4.22) have genotype AA for BxD2 and CAST/EiJ while BB for NZM/2410J and MRL/MpJ strain. The variation associated with three genotypes AA, AB and BB for expression of miR-130b is presented on left and onset week of EBA on right box. b) In miR-493 eQTL, peak SNP (rs6211533, −logP = 4.99) have genotype AA for BxD2, CAST/EiJ and NZM/2410J while BB for MRL/MpJ strain. The variation associated with three genotypes AA, AB and BB for expression of miR-493 is on left, maximum score of severity of EBA disease in middle and onset week of EBA on right
Expression and co-expression of miRNAs
Immunization of mice with recombinant COL7 leads to development of subepidermal blisters and the clinical phenotype of EBA in 1/3 of the immunized mice, while 2/3 remain clinically healthy. To access differential expression of miRNAs, we divided the 4th generation of our mouse cohort into two separate groups: affected and non-affected mice. Comparing the two groups, only two miRNAs were differentially expressed: miR-379 (adj. P value = 0.044) and miR-223 (adj. P value = 0.044) (Fig. 6 and Additional file 1: Table S2). Both miRNAs were significantly over expressed in mice with disease. Additionally, an eQTL for miR-379 (−log P = 4.7, 98–116 Mb) was also mapped on chromosome 6, with its peak at ~104 Mb (rs6208251, nearest gene: Cntn6).
Fig. 6.
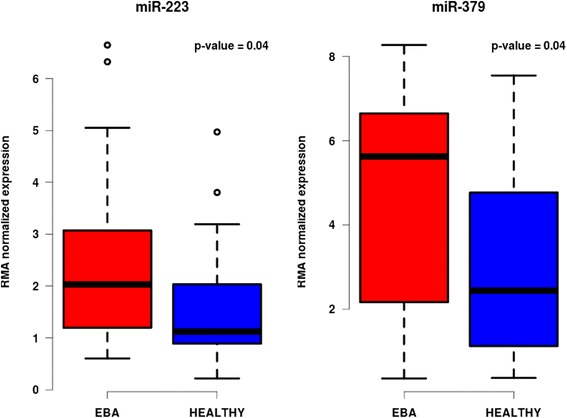
Boxplot showing differentially expressed miRNAs. The box plot shows the most differentially expressed miRNAs miR-223 and miR-379 for the disease phenotype EBA. The plots in blue color show the expression of miRNAs in mice with no clinical phenotype while mice with signs of inflammation are shown in red
The comparison of diseased and non-diseased mice has its drawbacks; the disease severity observed for individual mice might differ depending on the differences in the genome. To investigate miRNA expression affecting disease severity, we performed co-expression analysis among the expression levels of miRNAs (co-expressed miRNAs are in same pathway) in correlation with quantitative scores for severity and onset week of disease. For this purpose, we employed the WGCNA R package, which clusters miRNAs into different modules and further associates them to a phenotypic score, such as EBA severity or disease onset. As a result, we identified 11 clusters (Fig. 7). Using this approach, only the ‘black’ module, consisting of 23 miRNAs, was significantly associated with EBA (ρ = 0.28, P = 0.005) (Additional file 1: Table S3). Additionally, it was significantly correlating to the onset of EBA (ρ = 0.29, P = 0.005). Due to the fact that the ‘black’ module stronger correlates with the onset of the disease than with the maximum score, one can speculate that miRNAs from this module is rather involved in the onset than in the severity of the disease. This would relate to our previous observation, in which miRNA eQTL were overlapping with EBA onset QTL. The pathways associated with the ‘black’ module were predominantly pathways which have been shown to play a crucial role in other autoimmune disorders, such as the MAPK signaling pathway, T-cell receptor and TGF-beta (Table 3).r
Fig. 7.
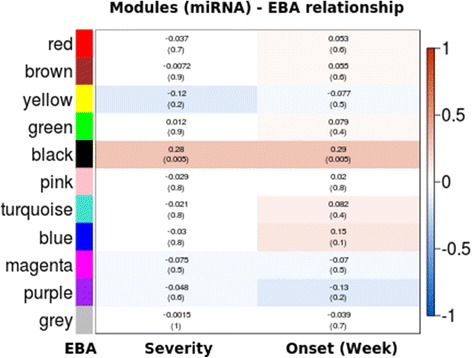
Module-trait relationship between clusters of miRNAs with EBA severity and onset. The graph is a representation of co-expression analyses of miRNAs. The clusters of miRNAs are called modules which are color coded on the y-axis. The disease phenotypes (traits) are EBA onset and EBA severity given on the x-axis. Blocks represent the correlation of module with phenotype using a Pearson correlation coefficient ranging from −1 to 1. The range is color coded with red representing positive correlation and blue representing negative correlation. P values are given in brackets below the correlation coefficients
Table 3.
List of over-represented KEGG pathways for ‘black’ module
| Total Genes of the Term | Union | Targets in the Term | Union miRNAs in the Term | Score |
|---|---|---|---|---|
| AXON_GUIDANCE | 131 | 51 | 17 | 1.88 |
| PATHWAYS_IN_CANCER | 323 | 98 | 18 | 1.788 |
| MAPK_SIGNALING_PATHWAY | 271 | 89 | 17 | 1.69 |
| NEUROTROPHIN_SIGNALING_PATHWAY | 131 | 45 | 16 | 1.557 |
| T_CELL_RECEPTOR_SIGNALING_PATHWAY | 109 | 42 | 16 | 1.424 |
| ENDOCYTOSIS | 219 | 65 | 16 | 1.408 |
| TGF-BETA_SIGNALING_PATHWAY | 85 | 30 | 15 | 1.334 |
| GLIOMA | 65 | 27 | 16 | 1.319 |
| PROSTATE_CANCER | 89 | 32 | 17 | 1.314 |
| UBIQUITIN_MEDIATED_PROTEOLYSIS | 138 | 52 | 16 | 1.277 |
| COLORECTAL_CANCER | 65 | 25 | 16 | 1.271 |
| WNT_SIGNALING_PATHWAY | 153 | 38 | 18 | 1.268 |
| MELANOGENESIS | 100 | 34 | 18 | 1.256 |
| ERBB_SIGNALING_PATHWAY | 87 | 33 | 17 | 1.246 |
| MTOR_SIGNALING_PATHWAY | 53 | 22 | 13 | 1.154 |
| INSULIN_SIGNALING_PATHWAY | 137 | 45 | 16 | 1.151 |
| B_CELL_RECEPTOR_SIGNALING_PATHWAY | 76 | 23 | 16 | 1.137 |
| FOCAL_ADHESION | 197 | 50 | 18 | 1.111 |
| ADHERENS_JUNCTION | 74 | 28 | 17 | 1.097 |
| CHAGAS_DISEASE | 102 | 34 | 16 | 1.094 |
| GNRH_SIGNALING_PATHWAY | 99 | 32 | 15 | 1.042 |
| MELANOMA | 71 | 26 | 16 | 1.037 |
| PANCREATIC_CANCER | 70 | 27 | 15 | 1.028 |
| ENDOMETRIAL_CANCER | 52 | 19 | 16 | 1.022 |
Individual correlation of miRNA expression levels with the disease severity identified 24 miRNAs to be significant (P < 0.05) (Additional file 1: Table S3), with miR-223 showing the strongest association (ρ = 0.4, P = 4.93e-05). To further validate this result we experimentally verified the expression of miR-223 in EBA skin by qRT-PCR. We found that miR-223 is upregulated in EBA skin in comparison to normal skin (Additional file 1: Figure S3). Another miRNA which was highly correlating with EBA was miR-21 (ρ = 0.36, P = 0.00036). Further, investigations of the co-expression module showed that miRNAs were potentially co-regulated i.e. possible overlapping genetic loci among co-expressed miRNAs. We observed that miRNAs in a given locus were either clustered within the same module or showed stronger inter-module membership for a specific module even if they were assigned to different modules. As an example, miR-322 and miR-431, that were mapped on chromosome 1 (51–59 Mb), are clustered in the ‘red’ module. In eQTL hot spots as on chromosome 2 (28–51 Mb), miR-423-3p and miR-23b are clustered in the ‘yellow’ module. Even though other miRNAs in this locus were assigned to a different module, they also show significance for the ‘yellow’ module. Examples are given by miR-671-5p (P = 1.224713e-12), miR-26a (P = 2.654271e-19) and miR-291a-3p (P = 3.530522e-02) (Additional file 1: Table S3). In line with this observation, for the eQTL mapped on chromosome 8, two miRNAs (miR-501-3p and miR-486) were clustered with the ‘brown’ module while miR-487 was assigns to the ‘red’ module but had significant module membership with the brown module as well (P = 0.004). Thus, these genomic loci may be considered as confirmed and strengthen the genetic contribution for the control of miRNAs expression levels.
Discussion
Small non-coding RNAs like miRNAs are known to contribute to the onset and severity of various diseases as well as the defense against them [28]. Thus, miRNAs function as tissue-specific key regulators, affecting some of the major pathways towards an aggravation of disease severity when aberrantly expressed [29]. Accordingly, it is not of surprise that miRNAs have been recently recognized as potential therapeutic targets [30, 31].
However, the underlying mechanisms of such dysregulated miRNA expression patterns are not well characterized. Different studies have shown that gene expression alterations in different tissues are genetically derived [29]. Thus, it is plausible that not only the regulation of gene expression is genetically controlled, but also the expression of miRNAs. In this study we explore the diversity of miRNAs in inflamed skin tissue and genetic loci that control variations in miRNA expression levels across a mouse cohort. We provide evidence that miRNA levels in skin tissue are genetically controlled on transcriptional level by helicases and RNA polymerases that are important for the biogenesis of miRNA. Furthermore, we found that some of the miRNA eQTL are restricted to one particular locus in the genome (eQTL hot spots). Deeper investigation revealed that these miRNAs are under multi-locus and/or epistatic control.
Interestingly, eQTL hot spots were predominantly found in genomic regions coding for non-coding RNA. Hence, it is tempting to speculate miRNA expression might not necessarily be solely controlled by protein-coding RNA, but rather by non-coding RNA which would impose an additional level of post-transcriptional regulation. This in turn leads to the tempting hypothesis that non-coding RNAs do at least in part regulate miRNA expression. Such a scenario is supported by the fact that some non-coding RNAs have been shown to bind to miRNAs at functional level as demonstrated by an interaction of linc-MD1 with miR-133 and miR-135 [32]. Here, linc-MD1 works as a sponge and traps these miRNAs preventing the binding to the canonical targets. Moreover, a recent study even shows an interaction network between lncRNAs and miRNAs [32].
Based on our observed overlap between QTL controlling miRNA expression and EBA, a blistering phenotype in autoimmune skin disease, we conclude that there are interconnected pathways to simultaneously regulate both disease development and miRNA expression. This might explain the findings of earlier studies that show a clear correlation of aberrant miRNA expression and autoimmune diseases [33–36]. Accordingly, initiation and/or progression of the disease do not primarily appear to be caused by aberrant miRNA expression. Quite the contrary seems to be true; aberrant miRNA expression could be a consequence of the disease which in turn would lead to a downward spiral. Hence, miRNAs could provide a large and unexplored reservoir of potential biomarkers for EBA and related cutaneous autoimmune skin blistering diseases and an interesting target for therapeutic intervention.
Conclusion
Taken together, our study provides a complex framework of gene-gene and miRNA-gene-interactions, which eventually leads to disease development and progression. Our data provide evidence that miRNAs are important drivers of cutaneous autoimmune diseases by acting on various pathways. Moreover, the study strongly implies there is yet another, so far largely unexplored level of regulatory network, possibly comprised of by non-coding RNAs which on their part affect miRNA expression. In this sense, aberrant miRNA expression would indeed be one the responsible elements for disease progression, however, the driving force behind might be a different one.
Methods
Generation of a 4-way advanced intercross line
The out bred four-way autoimmune-prone advanced intercross line (AIL) was generated (in our group) from the parental mouse strains BXD2/TyJ, MRL/MpJ, NZM2410/J and CAST/EiJ [21, 37, 38]. All strains were purchased from The Jackson Laboratory (Bar Harbor, ME). The four inbred strains were intercrossed following an equal strain and sex distribution. First generation (G1) offspring mice were then mated based on their parental origin to generate G2 mice in order to maintain an equal distribution of the original strain alleles across the genome. The same procedure was applied for intercrossing G2 and G3 mice. At least 50 breeding pairs were used for each successive generation of mice. Animals were held under pathogen free conditions at a 12-h light/dark cycle with food and water ad libitum. All animal experiments were approved by the Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume des Landes Schleswig Holstein in Kiel, Germany (Refrence number : V 312–72241. 122–5 (12-2/09)).
Induction of experimental EBA
Experimental EBA was induced by immunization with an immune-dominant peptide within the murine NC1 domain of Collagen type VII (GST-mCOL7C) as previously described [39]. In total, 600 mice of the AIL were immunized out of which 100 mice were randomly selected for miRNA expression profiling. Mice were evaluated every 4th week after immunization regarding their development and extent of skin disease, following an established scoring system for a total period of 12 weeks [40]. Murine skin tissues were obtained for analysis at the end of the experiment.
Expression profiling and statistical analysis
Total RNA was extracted from skin tissue and processed as previously described using the Flash Tag Biotin HSR RNA labeling kit and hybridized with the miRNA expression profiling GeneChip miRNA 2.0 Array according to the manufacturer’s protocol [41]. Raw data was pre-processed using R packages and normalized using RMA (Robust Multi-array analysis) to generate expression levels across the samples following instruction of at bioconductors.org [42].
Genotyping and expression QTL analysis
Genomic DNA was isolated from tail clippings and incubated in 500 μl 50 mM NaOH at 95 °C for 2 h. The reaction was neutralized by posterior addition of 50 μl 1 M Tris HCl (pH 8.0). DNA was further processed with DNeasy Blood & Tissue Kit according to manufacturer’s instructions. The extracted DNA was quantified using Nanodrop and normalized to 50 ng/μl in TE buffer (10 mM Tris; 1 mM EDTA; pH = 8). Agarose gel electrophoresis was performed for quality control. A total of 1400 SNPs markers evenly spaced across 19 autosomes and the X chromosome were genotyped on 100 generation 4th mice using Illumina mouse medium density array. 200 SNPs were discarded as they were non informative and 1200 markers were retained. The marker order and position in our map is provided in (Additional file 1). The SNP genotype information for each mouse from generation 4 and four founders is provided in (Additional file 1). These informative SNPs were used for performing eQTL analysis using HAPPY 2.3 on Debian Linux for miRNA expression levels [43, 44]. The software infers the haplotype probabilities for each sample using the progenitor SNP information. The miRNA expression levels of mice (600 probes) were considered as quantitative traits and linkage analysis with haplotype probabilities was done using the additive gaussian model. We considered gender as additive covariate. 1000 permutations were performed across all miRNA probes. A significant threshold (α = 0.05) was defined genome-wide. Correction for family structure was done using a variance component model: A Kinship matrix was obtained by the EMMA R package to account for relatedness among the individual [45] and interpreted as a random effect with sex as fixed effect to perform single marker association with phenotype.
We defined the confidence interval of the given eQTL based on a -log P value drop of 1.5. We considered local regulation if the eQTL for the probe was found on the same chromosome as its genomic location. A trans-regulation was presumed if the QTL controlling the trait was found in a different chromosome than its genomic location. The function “epistasis” from the HAPPY R package was used to find pair wise SNP interactions using F-score for the interaction model for SNPs which were significantly associated with miRNA expression levels in initial genomic scan [43]. All the P-values are corrected for multiple testing using the Bonferroni correction. Circos was used for visualization of eQTL and epistasis [46].
Co-expression analysis
The standard WGCNA procedure was used for module detection [47]. For miRNA we considered 97 samples. 3 samples were excluded as these could not phenotype for EBA score. A weighted adjacency matrix of pair-wise connection strengths (correlation coefficients of gene expression levels) was constructed using the soft-threshold approach with a scale independent topological power β = 7 (miRNA). For each probe, the connectivity was defined as the sum of all connection strengths with all others. Probes were aggregated into modules by hierarchical clustering and refined by the dynamic cut tree algorithm [48]. The Pearson correlation coefficient was determined for each phenotype-module pair. The representative module expression profile, or module eigengene value, is the first principal component of the gene expression profile within a module. The correlation between the module eigengene and the sample trait of interest yields the eigengene significance, as assessed by a correlation test. The modules were assigned by different colors where grey was assigned to traits that could not be clustered in any other module. To determine the enriched pathways involved in a cluster we used ‘miRsystem’ which uses the weighted pathway-ranking method for identifying enriched biological functions [49].
Total RNA preparation, cDNA synthesis and qRT-PCR
Perilesional skin samples from mice injected with NC1 domain of Collagen type VII (GST-mCOL7C) or normal skin from mice injected with GST alone were obtained. Total RNA was extracted using TRIzol® reagent (Invitrogen GmbH, Darmstadt, Germany). RNA concentrations were measured on a Nanodrop 2000c spectrophotometer (Thermo Fischer Scientific GmbH, Bremen, Germany). Total RNA was used for cDNA synthesis as previously described [50]. Briefly, 100 ng of total RNA were poly (A) tailed and reverse transcribed in a single reaction tube containing: 1 μl of 10xpoly(A) polymerase buffer, 0.1 mM of ATP, 0.1 mM of dNTPs, 100 units of MuLV reverse transcriptase (New England Biolabs, Frankfurt am Main, Germany), 1 unit of poly(A) polymerase (New England Biolabs, Frankfurt am Main, Germany), and 1 μM of RT primer (5′-CAGGTCCAGTTTTTTTTTTTTTTTGT-3′) in a final volume of 10 μl. Consequently, the tube was incubated at 42 °C for 1 h followed by 5 min at 95 °C. The cDNA was diluted 1:10 before the qPCR reaction. qPCR analysis was performed on a Mastercycler ep Realplex (Eppendorf AG, Hamburg, Germany) using 8 μl of diluted cDNA, 10 μl of SYBR Select Master Mix (Thermo Fischer Scientific GmbH, Bremen, Germany), and 250nM mmu-mir-223 specific primer set. The cycling conditions were 50 °C for 2 min, 95° for 2 min, followed by 40 cycles of 95 °C 15 s and 60 °C for 1 min. The expression levels of mmu-mir-223 were normalized against β-actin. The sequences of the qPCR primers used in this study are:mmu-mir-223 F: 5′-CGCAGTGTCAGTTTGTCA-3′; mmu-mir-223 R:5′-CCAGTTTTTTTTTTTTTTTGGGGTA-3′; mmu-β-actin F:5-CCCCATTGAACATGGCATTG-3′; mmu-β-actin R: 5′- ACGACCAGAGGCATACAGG-3′.
Availability of the supporting data
Data is accessible at NCBI GEO GSE64276.
Acknowledgement
The project has been funded by Deutsche Forschungsgemeinschaft: the research training programs Modulation of Autoimmunity [GRK1727/1], Genes, Environment and Inflammation [GRK 1743/1] and Excellence Cluster Inflammation at Interfaces [EXC 306/2]. We thank Miriam Freitag, Illona Klamfuss, Susen Möller and Andreia de Castro Marques for help with the AIL line.
Abbreviations
- EBA
epidermolysis bullosa acquisita
- eQTL
Expression quantitative trait loci/locus
- lncRNA
Long non coding RNA
- miRNA
Micro RNA
- QTL
Quantitative trait loci/locus
- SnoRNA
Small nucleolar RNA
- SnRNA
Small nuclear RNA
Additional file
Flowchart describing the workflow of analysis. The flowchart provides an overview of the analysis performed for understanding regulation of miRNAs and their contribution to the disease phenotype. Figure S2 Interaction network accessed via IPA software for epistasis of miR-501. The graph depicts the interacting genes identified from epistasis scan of miRNA miR-501 in chromosome 1 and chromosome 2. The graph shows all known gene interactions between the two loci where genes colored in yellow are from locus on chromosome 2 and green are from locus on chromosome 1. The red line shows the possible pathway for the regulation of miR-501. Figure S3 qRT-PCR validation of miR-223 expression in EBA and normal murine skin. (ZIP 637 kb)
Footnotes
Competing interests
The authors have no competing interest.
Authors’ contributions
GY, MS, WM, BM, AV, NF, BJ and HR performed the experiment and statistical analysis. Ibrahim S, LR, TR, BJ and ZD designed the experiment. TS, MH and CD. S performed the experimental validation. All the authors have read and approved the final manuscript.
Contributor Information
Yask Gupta, Email: Yask.Gupta@uksh.de.
Steffen Möller, Email: Steffen.Moeller@uksh.de.
Mareike Witte, Email: witte_mareike@web.de.
Meriem Belheouane, Email: belheouane@evolbio.mpg.de.
Tanya Sezin, Email: tanya.sez@gmail.com.
Misa Hirose, Email: Misa.Hirose@uksh.de.
Artem Vorobyev, Email: Artem.Vorobyev@uksh.de.
Felix Niesar, Email: niesar@imm.uni-luebeck.de.
Julia Bischof, Email: Julia.Bischof@uksh.de.
Ralf J. Ludwig, Email: Ralf.Ludwig@uksh.de
Detlef Zillikens, Email: Detlef.Zillikens@uksh.de.
Christian D. Sadik, Email: Christian.Sadik@uksh.de
Tobias Restle, Email: restle@imm.uni-luebeck.de.
Robert Häsler, Email: r.haesler@mucosa.de.
John F. Baines, Email: baines@evolbio.mpg.de
Saleh M. Ibrahim, Phone: 0451 500-5250, Email: saleh.ibrahim@uksh.de
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 2.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8(2):93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 3.Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta. 2011;1812(5):592–601. doi: 10.1016/j.bbadis.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13(4):239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136(1):26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 6.Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee I, Ajay SS, Yook JI, Kim HS, Hong SH, Kim NH, et al. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009;19(7):1175–83. [DOI] [PMC free article] [PubMed]
- 8.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A. 2007;104(45):17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin SL, Miller JD, Ying SY. Intronic microRNA (miRNA) J Biomed Biotechnol. 2006;2006(4):26818. doi: 10.1155/JBB/2006/26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22(22):3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghorai A, Ghosh U. miRNA gene counts in chromosomes vary widely in a species and biogenesis of miRNA largely depends on transcription or post-transcriptional processing of coding genes. Front Genet. 2014;5:100. doi: 10.3389/fgene.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su WL, Kleinhanz RR, Schadt EE. Characterizing the role of miRNAs within gene regulatory networks using integrative genomics techniques. Mol Syst Biol. 2011;7:490. doi: 10.1038/msb.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Wang J, Lu X, Song X, Ye Y, Zhou J, et al. Evaluation of six SNPs of MicroRNA machinery genes and risk of schizophrenia. J Mol Neurosci. 2013;49(3):594–599. doi: 10.1007/s12031-012-9887-1. [DOI] [PubMed] [Google Scholar]
- 14.Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, et al. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci U S A. 2009;106(2):498–502. doi: 10.1073/pnas.0810766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider MR. MicroRNAs as novel players in skin development, homeostasis and disease. Br J Dermatol. 2012;166(1):22–28. doi: 10.1111/j.1365-2133.2011.10568.x. [DOI] [PubMed] [Google Scholar]
- 16.Bonazzi VF, Stark MS, Hayward NK. MicroRNA regulation of melanoma progression. Melanoma Res. 2012;22(2):101–113. doi: 10.1097/CMR.0b013e32834f6fbb. [DOI] [PubMed] [Google Scholar]
- 17.Ballabio E, Mitchell T, van Kester MS, Taylor S, Dunlop HM, Chi J, et al. MicroRNA expression in Sezary syndrome: identification, function, and diagnostic potential. Blood. 2010;116(7):1105–1113. doi: 10.1182/blood-2009-12-256719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joyce CE, Zhou X, Xia J, Ryan C, Thrash B, Menter A, et al. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum Mol Genet. 2011;20(20):4025–4040. doi: 10.1093/hmg/ddr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonkoly E, Janson P, Majuri ML, Savinko T, Fyhrquist N, Eidsmo L, et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J Allergy Clin Immunol. 2010;126(3):581–589. doi: 10.1016/j.jaci.2010.05.045. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Stram A, Chen C, Kar S, Gayther S, Pharoah P, et al. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Hum Mol Genet. 2014;23(19):5294–5302. doi: 10.1093/hmg/ddu228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludwig RJ, Muller S, Marques A, Recke A, Schmidt E, Zillikens D, et al. Identification of quantitative trait loci in experimental epidermolysis bullosa acquisita. J Invest Dermatol. 2012;132(5):1409–1415. doi: 10.1038/jid.2011.466. [DOI] [PubMed] [Google Scholar]
- 22.Alberts R, Terpstra P, Li Y, Breitling R, Nap JP, Jansen RC. Sequence polymorphisms cause many false cis eQTLs. PLoS One. 2007;2(7):e622. doi: 10.1371/journal.pone.0000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11(3):228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 25.Jankowsky A, Guenther UP, Jankowsky E. The RNA helicase database. Nucleic Acids Res. 2011;39(Database issue):D338–341. doi: 10.1093/nar/gkq1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Liu H, Lai C, Du X, Su Z, Gao S. The Lin28/let-7a/c-Myc pathway plays a role in non-muscle invasive bladder cancer. Cell Tissue Res. 2013;354(2):533–541. doi: 10.1007/s00441-013-1715-6. [DOI] [PubMed] [Google Scholar]
- 27.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol. 2013;13(9):666–678. doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H, Kohane IS. Tissue and process specific microRNA-mRNA co-expression in mammalian development and malignancy. PLoS One. 2009;4(5):e5436. doi: 10.1371/journal.pone.0005436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt MF. Drug target miRNAs: chances and challenges. Trends Biotechnol. 2014. [DOI] [PubMed]
- 31.De Guire V, Robitaille R, Tetreault N, Guerin R, Menard C, Bambace N, et al. Circulating miRNAs as sensitive and specific biomarkers for the diagnosis and monitoring of human diseases: promises and challenges. Clin Biochem. 2013;46(10–11):846–860. doi: 10.1016/j.clinbiochem.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otaegui D, Baranzini SE, Armananzas R, Calvo B, Munoz-Culla M, Khankhanian P, et al. Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS One. 2009;4(7):e6309. doi: 10.1371/journal.pone.0006309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata K, Furu M, Yoshitomi H, Ishikawa M, Shibuya H, Hashimoto M, et al. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS One. 2013;8(7):e69118. doi: 10.1371/journal.pone.0069118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma X, Zhou J, Zhong Y, Jiang L, Mu P, Li Y, et al. Expression, Regulation and Function of MicroRNAs in Multiple Sclerosis. Int J Med Sci. 2014;11(8):810–818. doi: 10.7150/ijms.8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcet B, Chevalier B, Luxardi G, Coraux C, Zaragosi LE, Cibois M, et al. Control of vertebrate multiciliogenesis by miR-449 through direct repression of the Delta/Notch pathway. Nat Cell Biol. 2011;13(6):693–699. doi: 10.1038/ncb2241. [DOI] [PubMed] [Google Scholar]
- 37.Asghari F, Fitzner B, Holzhuter SA, Nizze H, de Castro MA, Muller S, et al. Identification of quantitative trait loci for murine autoimmune pancreatitis. J Med Genet. 2011;48(8):557–562. doi: 10.1136/jmg.2011.089730. [DOI] [PubMed] [Google Scholar]
- 38.Srinivas G, Moller S, Wang J, Kunzel S, Zillikens D, Baines JF, et al. Genome-wide mapping of gene-microbiota interactions in susceptibility to autoimmune skin blistering. Nat Commun. 2013;4:2462. doi: 10.1038/ncomms3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ludwig RJ, Recke A, Bieber K, Muller S, Marques Ade C, Banczyk D, et al. Generation of antibodies of distinct subclasses and specificity is linked to H2s in an active mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. 2011;131(1):167–176. doi: 10.1038/jid.2010.248. [DOI] [PubMed] [Google Scholar]
- 40.Sitaru C, Chiriac MT, Mihai S, Buning J, Gebert A, Ishiko A, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177(5):3461–3468. doi: 10.4049/jimmunol.177.5.3461. [DOI] [PubMed] [Google Scholar]
- 41.Hasler R, Begun A, Freitag-Wolf S, Kerick M, Mah N, Zvirbliene A, et al. Genetic control of global gene expression levels in the intestinal mucosa: a human twin study. Physiol Genomics. 2009;38(1):73–79. doi: 10.1152/physiolgenomics.00010.2009. [DOI] [PubMed] [Google Scholar]
- 42.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 43.Mott R, Talbot CJ, Turri MG, Collins AC, Flint J. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci U S A. 2000;97(23):12649–12654. doi: 10.1073/pnas.230304397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moller S, Krabbenhoft HN, Tille A, Paleino D, Williams A, Wolstencroft K, et al. Community-driven computational biology with Debian Linux. BMC Bioinformatics. 2010;11(Suppl 12):S5. doi: 10.1186/1471-2105-11-S12-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178(3):1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24(5):719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 49.Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, et al. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One. 2012;7(8):e42390. doi: 10.1371/journal.pone.0042390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busk PK. A tool for design of primers for microRNA-specific quantitative RT-qPCR. BMC Bioinformatics. 2014;15:29. doi: 10.1186/1471-2105-15-29. [DOI] [PMC free article] [PubMed] [Google Scholar]