

Abstract
To understand the precise disease driving mechanisms in acute myeloid leukemia (AML), comparison of patient matched hematopoietic stem cells (HSC) and leukemia stem cells (LSC) is essential. In this analysis, we have examined the value of aldehyde dehydrogenase (ALDH) activity in combination with CD34 expression for the separation of HSC from LSC in 104 patients with de novo AML. The majority of AML patients (80 out of 104) had low percentages of cells with high ALDH activity (ALDH+ cells; <1.9%; ALDH‐rare AML), whereas 24 patients had relatively numerous ALDH+ cells (≥1.9%; ALDH‐numerous AML). In patients with ALDH‐rare AML, normal HSC could be separated by their CD34+ALDH+ phenotype, whereas LSC were exclusively detected among CD34+ALDH− cells. For patients with ALDH‐numerous AML, the CD34+ALDH+ subset consisted mainly of LSC and separation from HSC was not feasible. Functional analyses further showed that ALDH+ cells from ALDH‐numerous AML were quiescent, refractory to ARA‐C treatment and capable of leukemic engraftment in a xenogenic mouse transplantation model. Clinically, resistance to chemotherapy and poor long‐term outcome were also characteristic for patients with ALDH‐numerous AML providing an additional risk‐stratification tool. The difference in spectrum and relevance of ALDH activity in the putative LSC populations demonstrates, in addition to phenotypic and genetic, also functional heterogeneity of leukemic cells and suggests divergent roles for ALDH activity in normal HSC versus LSC. By acknowledging these differences our study provides a new and useful tool for prospective identification of AML cases in which separation of HSC from LSC is possible.
Keywords: acute myeloid leukemia, leukemia stem cell, hematopoietic stem cell, aldehyde dehydrogenase, high risk factor
Short abstract
What's new?
To understand the precise disease‐driving mechanisms in acute myeloid leukemia (AML), comparison of patient‐matched hematopoietic stem cells (HSC) and leukemia stem cells (LSC) is essential. This study demonstrates the relevance of aldehyde dehydrogenase (ALDH) for the prospective identification of AML cases in which separation of functionally normal HSC from LSC is possible. Increased activity of this biomarker also characterizes a subgroup of patients with adverse outcome, which might be helpful in risk stratification prior to therapy. Overall, this study demonstrates functional heterogeneity of leukemia cells and suggests divergent roles for ALDH activity in normal HSC versus leukemia‐initiating cells.
Abbreviations
- ALDH
aldehyde dehydrogenase
- AML
acute myeloid leukemia
- BM
bone marrow
- CFC
colony forming cell
- ARA‐C
cytarabin
- DEAB
diethylaminobenzaldehyde
- FACS
fluorescence‐activated cell sorting
- FLT3
Fms‐like tyrosine kinase 3
- FLT3‐ITD
FLT3‐internal tandem duplication
- HPC
hematopoietic progenitor cell
- HSC
hematopoietic stem cell
- LSC
leukemia stem cell
- LTC‐IC
long‐term colony‐initiating cell
- MSC
mesenchymal stromal cells
- MNC
mononuclear cells
- NSG
non‐obese diabetic/severe combined immune deficiency/interleukin 2 receptor gammanull
- PI
propidium iodide
- TPO
thrombopoietin
- SCF
stem cell factor
- wt
wild type
Although the long‐term outcome of patients with acute myeloid leukemia (AML) has improved significantly in the past 20 years, recurrence of disease has remained a major challenge.1 Present evidence indicates that AML is a stem cell disease, derived from leukemia stem cells (LSC) that might originate from malignant transformation of normal hematopoietic stem cells (HSC), or alternatively, from progenitors in which the acquired mutations have reinstalled a dysregulated self‐renewal program.2 In analogy to normal HSC, LSC have been reported to be quiescent, well protected within the bone marrow niche, resistant to chemotherapy and responsible for recurrent disease.3, 4
Evidence for the concept of LSC was first reported in patients with AML by using xenogenic transplantation models. These studies showed that only a very small leukemia cell population of CD34+CD38− cells was able to initiate leukemic growth.5, 6 The observation that LSC might be enriched within the CD34+CD38− subset has, however, been challenged by more recent studies demonstrating that CD34+CD38+ as well as CD34− cells also harbor LSC candidates.7, 8, 9 These results suggest that LSC in AML are probably much more heterogeneous than previously anticipated.
Other studies have focused on biochemical markers such as the expression of aldehyde dehydrogenase (ALDH) in leukemia cells, which is a cytosolic enzyme involved in the metabolism of retinoic acid and the detoxification of alkylating drugs such as cyclophosphamide. High ALDH activity has been described as a putative stem cell marker in hematopoietic, neural and mammary tissues.10, 11, 12, 13, 14 In AML, results on the tumorigenic potential of cells with high ALDH activity are controversial but studies from others and our group have confirmed that LSC might be enriched among the ALDH+ subsets.10, 15 Increased expression of ALDH in primary samples from patients with AML has also been shown to be associated with adverse clinical outcome.15, 16, 17
Since all the above mentioned markers for LSC identification such as CD34+CD38− and ALDH activity are also characteristic for normal HSC, attempts have been made to identify marker combinations that are suitable to distinguish HSC and LSC from the same individual with AML. For example, CD90 was reported to be exclusively expressed in HSC whereas lacking in LSC.18 Other markers, including CD123, lineage specific aberrant markers, CLL‐1, CD96 and CD47 have been reported to be upregulated in specific AML patients and might potentially represent candidates for the selection and targeting of LSC.19, 20, 21, 22, 23, 24, 25 However, the value of these additional markers for discerning the differences between normal HSC and LSC has remained controversial.
Under consideration of the extreme heterogeneity of LSC and the emergence of potential leukemia driving mutations in HSC derived from normal human subjects, especially upon aging, comparison between LSC and HSC derived from the same individual suffering from leukemia is of utmost importance for appropriate comparisons to identify the molecular mechanisms leading to AML.26, 27, 28 Pearce et al. showed that HSC could be isolated by high ALDH expression in a small subgroup of 6 AML patients with overall low ALDH expression.10 Another study suggested that leukemia cells could be separated from residual HSC by using the marker combination CD34+CD38− in combination with ALDH activity.29 Other studies reported successful separation of residual HSC from selected AML cases based on the absence of TIM3 and/or CD99 surface expression within the Lin−CD34+CD38− fraction.26, 30 In the latter study, Jan et al. have indicated that phenotypically and functionally normal HSC derived from patients with AML already carry leukemia specific mutations and therefore potentially represent “preleukemic HSC” whose role in relapse initiation remains to be determined.26 On the other hand, sequencing studies indicated that leukemia driving mutations could be detected in HSC derived from healthy subjects and that the number of potential leukemia driving mutations in HSC might increase with age.27 Therefore, the ultimate leukemia initiating and driving mechanisms can only be appropriately studied by comparing LSC with functionally normal HSC from the same individual.
In our systematic study of 104 AML patients, we have shown that due to the heterogeneity of LSC, individual patients with AML need to be stratified to allow prospective identification of cases in which separation of HSC from LSC is possible. In addition, this classification identified ALDH‐numerous AML as a subgroup of patients with increased therapy resistance and adverse clinical outcome.
Methods
Patient samples
Between December 2009 and September 2013 bone marrow (BM) aspirates of 104 patients with de novo AML and 14 healthy donors were collected after written informed consent. Sample collection and data analyses were approved by the Ethics Committee of the Medical Faculty of the University of Heidelberg. Patient characteristics are shown in Supporting Information Table S1. Patients were categorized into high, intermediate and low risk groups according to cytogenetic criteria as reported by Grimwade et al.31
Preparation of primary cells
Mononuclear cells (MNC) were isolated using density gradient centrifugation with Biocoll separation solution (Biochrom, Berlin, Germany) and used freshly or frozen in FCS/12.5% DMSO for subsequent studies.
Mesenchymal stromal cells (MSC) were isolated from MNC of healthy donors as described previously.32 The cells were cultured in MSCGM™ Mesenchymal Stem Cell Growth Medium (Lonza, Basel, Switzerland) at 37 °C and 5% CO2 and served as feeder layer in in vitro assays.
Flow cytometry and sorting of stem cell populations
MNC were labeled with Aldefluor reagent (Stem Cell Technologies, Vancouver, BC, Canada), CD2‐PE, CD7‐PE, CD11b‐PE, CD15‐PE, CD19‐PE, CD38‐PE, CD56‐PE, CD34‐APC, CD45‐APC‐H7 and propidium iodide (PI; BD Bioscience, Heidelberg, Germany) as described previously.33 Cells were analyzed using a FACScan flow cytometry system (BD Bioscience, Heidelberg, Germany) equipped with a Rainbow laser (Cytek Flow Cytometry Products, CA), and sorted with a FACSAria II sorter (BD Bioscience, Heidelberg, Germany).
In vitro colony assays
To evaluate the stem cell potential of AML subpopulations we used the long‐term culture‐initiating cell (LTC‐IC) assay as described previously15 (for detailed information see supplementary methods). Colony forming cell (CFC) assays were performed using HSC‐CFU complete with Epo (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufactureŕs instructions.
NOD/SCID‐IL2Rγnull (NSG) mouse transplantation
Immune deficient NSG mice at the age of 8–12 weeks were sublethally irradiated with 200 cGy, transplanted with AML bulk or cell subpopulations via intra‐bone injection within 24 hr after irradiation and analyzed after 4–5 months. Bones were collected, cells isolated and labeled with monoclonal antibody cocktails against human antigens including CD3‐FITC, CD19‐PE, CD33‐APC (BD Bioscience, Heidelberg, Germany) and CD45‐APC‐eFluor® 780 (eBioscience, Frankfurt, Germany). Human cells were enriched by depletion of mouse cells using mouse CD45 and mouse Ter199 antibodies conjugated with magnetic Microbeads and LD Columns (Mitenyi Biotec, Bergisch Gladbach, Germany). Alternatively, human CD45+ cells were sorted using a FACSAria II sorter. Mutations of enriched human cell fractions were analyzed by interphase FISH or PCR. Animal experiments were performed at the German Cancer Research Center (DKFZ) in compliance with institutional and governmental guidelines.
Fluorescence in situ hybridization
For patients whose chromosomal aberrations were detected by Fluorescence in situ hybridization (FISH) at the time of diagnosis, MNC, FACS‐sorted CD34+ALDH+ and CD34+ALDH− cell populations were expanded in Stemline II medium (Sigma Aldrich, Munich, Germany; for detailed information see supplementary methods). Cells were then analyzed by interphase FISH following the manufactureŕs instructions using probes for detection of the following chromosomal aberrations: translocations t(8;21)(q22;q22) and t(15;17)(q24;q21), inversion inv(16)(p13;q22), MLL(11q23) rearrangement, trisomy 8, trisomy 13, deletion 17p13 and monosomy X (Kreatech, Amsterdam, Netherlands; MetaSystems, Altlussheim, Germany; and Abbott, Wiesbaden, Germany). Interphase nuclei were validated using an automated scanning system SC300‐25A (Applied Spectral Imaging, Edingen/Neckarhausen, Germany) and a DM RXA RF8 epifluorescence microscope (Leica, Wetzlar, Germany; for detailed information see Supporting Information methods). For most samples, at least 100 nuclei were analyzed (see supplementary Supporting Information Table S2). Hybridization efficiency and threshold for positive signals were validated on PB and BM cells of healthy donors according to the internal lab quality assessment protocols. According to the threefold standard deviation thresholds were set to 4% for deletions, gains, translocations and rearrangements.
PCR for analysis of FLT3‐mutation
Total RNA was isolated with Trizol (Invitrogen, Darmstadt, Germany) and reverse‐transcribed with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) according to manufacturer's instruction. FLT3‐internal tandem duplication (ITD) was detected by PCR with the forward primer 5′‐GCAATTTAGGTATGAAAGCCAGCTA‐3′ and reverse primer 5′‐CCTGATCCTAGTACCTTCCCA‐3′ using a DNA engine thermal cycler (Biorad, Munich, Germany). Amplification products were analyzed on 2% agarose gels and stained with ethidium bromide.
Cell cycle analysis
MNC were incubated with 10 µM EdU (Invitrogen, Darmstadt, Germany) for 1 hr at 37 °C, stained with Aldefluor, CD3‐PE, CD19‐PE and CD45‐APC‐H7, and sorted by FACS into ALDH+ and ALDH− blasts. CD3, CD19 and CD45 were used in order to exclude lymphocytes from our sorted populations. Cell cycle was analyzed using the Click‐iT® EdU Alexa Fluor® 488 Cell Proliferation Assay Kit (Invitrogen, Darmstadt, Germany) according to manufacturer's instruction.
Treatment of leukemia cells with ARA‐C
Primary MNC derived from the BM of patients with ALDH‐numerous AML were sorted for ALDH+, ALDH−, CD34+CD38−ALDH+ and CD34+CD38−ALDH− blasts and incubated with 1 µM of ARA‐C in coculture with MSC for 5 days. Viability was subsequently assessed using Annexin V‐PE and PI (BD Bioscience, Heidelberg, Germany) according to manufacturer's instruction. Samples in which <20% of cells survived in untreated conditions were excluded from the analysis to avoid errors related to sample quality.
Statistical analysis
Data were analyzed using the two‐side Student's t test unless otherwise indicated. For the ARA‐C treatment, the nonparametric Mann–Whitney U test was employed due to a small sample size. Kaplan–Meier‐curves were used to visualize the survival properties of AML patient subgroups with the SPSS software. Statistical differences of the survival properties between the subgroups were calculated using log‐rank test. Statistical significance was defined as p < 0.05.
Results
ALDH expression patterns in healthy and AML BM
ALDH+ cells (R1) were defined as a population with high ALDH activity and low SSC compared to ALDH− cells (R2; Fig. 1). The median frequency of ALDH+ cells in BM MNC of healthy subjects was 1.9% with a range from 0.6 to 3.2% (n = 14). This cell population contained a high frequency of CD34+CD38− HSC and CD34+CD38+ HPC [median and range: 12.7% (2.3–27.3%) and 42.6% (24.0–60.1%), respectively, n = 6] (Fig. 1 a). The median value of ALDH+ cells in normal BM was used as cut‐off value for all following experiments to classify AML patients into the categories ALDH‐rare AML (<1.9% of ALDH+ cells) versus ALDH‐numerous AML (≥1.9% ALDH+ cells).
Figure 1.
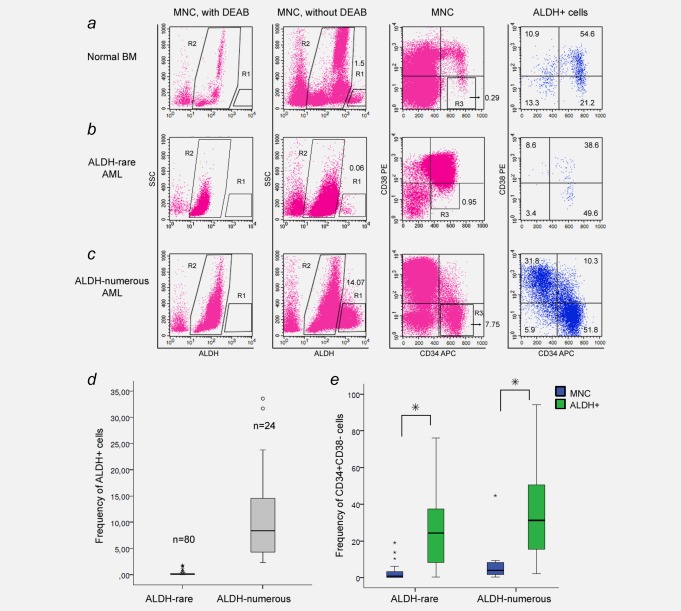
ALDH expression patterns in normal and AML BM. ALDH activity of MNC was determined by flow cytometry using the aldefluor reagent. Diethylaminobenzaldehyde (DEAB), an ALDH inhibitor, was used as a negative control for ALDH staining (first column). In absence of DEAB, ALDH+ cells (R1), which are characterized by high ALDH activity and low SSC, could be detected and distinguished from the ALDH− subset (R2; second column). CD34/CD38 expression of MNC and ALDH+ cells in the R1 gate are shown in the last two columns. R3 marks the CD34+CD38− cell population in MNC. ( a ) ALDH expression in normal BM showed a small population of ALDH+ cells, which was enriched for CD34+CD38− HSC and CD34+CD38+ HPC. Two patterns of ALDH+ populations were observed in patients with AML: ( b ) patients with ALDH‐rare AML were characterized by low frequencies of ALDH+ cells (<1.9%), while ( c ) patients with ALDH‐numerous AML by high numbers (≥ 1.9%). ( d ) The diagram shows the distribution of ALDH+ cell frequencies in BM MNC of patients with ALDH‐rare (median: 0.07%) and ALDH‐numerous AML (median: 8.4%). ( e ) In both groups, ALDH+ cell populations were highly enriched for CD34+CD38‐ cells in comparison to MNC. Percentages of ALDH+ (R1) and CD34+CD38−cells (R3) in MNC and the distribution of ALDH+ cells in the CD34/CD38 plots are depicted.
For patients with AML, the median number of ALDH+ cells was 0.1% with a range from 0.0 to 33.6%. Whereas the majority of patients with AML (80 out of 104 patients) had low percentages of ALDH+ cells, that is, <1.9% of ALDH+ blasts in the bone marrow, 24 patients had relatively numerous ALDH+ cells. The median frequency of ALDH+ cells among patients with ALDH‐rare AML was 0.07% (0.00 to 1.77%; Figs. 1 b and 1 d). The 24 patients with ALDH‐numerous AML had a median of 8.4% ALDH+ cells (2.3 to 33.6%; Figs. 1 c and 1 d). Thus there were two clusters of AML according to ALDH activity. In both groups, ALDH+ cell populations were highly enriched for CD34+CD38− cells in comparison to MNC (Figs. 1 b, 1 c and 1 e) and showed comparable levels of ALDH activity (Supporting Information Fig. S1).
Normal HSC could be enriched in the ALDH+ subset in ALDH‐rare AML
To detect the presence or absence of leukemia clones in our stem cell candidates, we performed cytogenetic (FISH) and molecular genetic (FLT3‐ITD) studies on MNC, CD34+ALDH+ and CD34+ALDH− subsets derived from nine patients with ALDH‐rare AML and nine with ALDH‐numerous AML.
In five out of nine patients with ALDH‐rare AML, cytogenetic evidence of AML could not be found in the CD34+ALDH+ cells whereas MNC and CD34+ALDH−subsets were highly positive. In another sample, deletion 17p13 was observed in 10% of CD34+ALDH+ cells compared to 90% and 96% in MNC and the CD34+ALDH− subset, respectively (Fig. 2 a and Supporting Information Table S2a). In the remaining three patients with ALDH‐rare AML, only FLT3‐ITD but no cytogenetic aberrations were found at diagnosis. FLT3‐wild type was found among the CD34+ALDH+ cells, whereas the FLT3‐ITD mutation was exclusively detected in the MNC and CD34+ALDH− subsets (Fig. 2 b).
Figure 2.
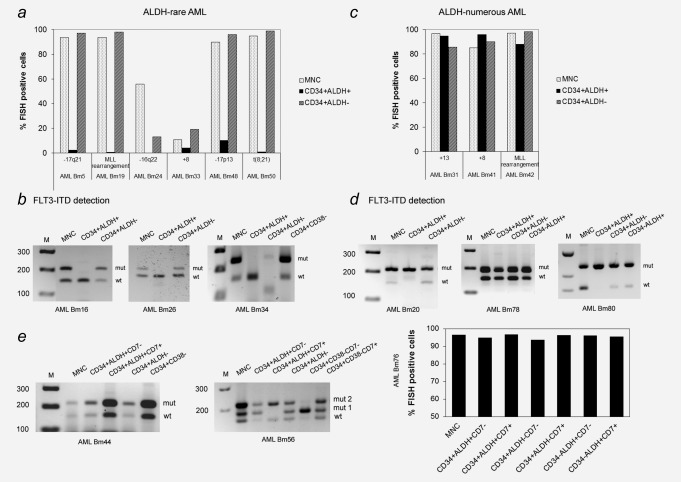
Mutation analysis of CD34+ALDH+ and CD34+ALDH− cells derived from patients with ALDH‐rare and ALDH‐numerous AML. ( a ) FISH analysis indicated that CD34+ALDH+ cells from the BM of patients with ALDH‐rare AML contained normal cells in contrast to MNC and CD34+ALDH−cells. ( b ) FLT3‐ITD PCR for the ALDH‐rare group showed that CD34+ALDH+ cells expressed FLT3‐wild type, while MNC, CD34+ALDH−, and CD34+CD38− cells (when analyzed) contained both wild type and FLT3‐ITD mutation. ( c ) Chromosomal mutations and ( d ) FLT3‐ITD were observed at a high level in all analyzed cell populations derived from patients with ALDH‐numerous AML. ( e ) In patients with ALDH‐numerous AML, coexpression of the aberrant marker CD7 was not able to separate leukemia versus HSC. Clonal markers were found both in CD34+ALDH+CD7− and CD34+ALDH+CD7+ subsets. M: marker, wt: wild type, mut: mutation. *The marker was cut from another part of the gel and put on the appropriate position.
In three patients with ALDH‐numerous AML, we were able to detect comparable numbers of the specific cytogenetic aberrations in all subsets analyzed including MNC, CD34+ALDH+ and CD34+ALDH− (Fig. 2 c and Supporting Information Table S2). For another three patients with FLT3‐ITD positive ALDH‐numerous AML, the mutation was also detectable in all analyzed cell subsets using a PCR approach (Fig. 2 d). In three more ALDH‐numerous AML with aberrant coexpression of CD7, CD34+ALDH+ cells were further subdivided into CD7+ and CD7−, that is, CD34+ALDH+CD7+ and CD34+ALDH+CD7− subsets, followed by clonal marker analysis, that was positive for both subpopulations (Fig. 2 e).
In summary, we showed that cytogenetically and molecularly normal HSC could be isolated using the CD34+ALDH+ phenotype in patients with ALDH‐rare AML, but not in those with ALDH‐numerous AML.
Colony assays confirmed HSC potentials of ALDH+ cells
To verify the hypothesis that in BM of patients with ALDH‐rare AML, normal HSC are enriched within the CD34+ALDH+ subset, we have performed LTC‐IC assays using CD34+ALDH+ and CD34+ALDH− cells from six patients. In five additional patients, we have further separated (a) CD34+ALDH+, (b) CD34−ALDH+, (c) CD34+CD38−ALDH− and (d) all other ALDH− cells (rest of ALDH−cells). LTC‐IC were enriched in CD34+ALDH+ cells (one in 18 cells), followed by CD34−ALDH+ cells (one in 435 cells). Within the ALDH− population, CD34+CD38− cells were more enriched for LTC‐IC (1 in 2,601 cells) than the rest (1 in 13,214 cells; Fig. 3 a). Furthermore, we performed FISH analysis of LTC‐IC colonies derived from single CD34+ALDH+ or ALDH+ cells (in one case since the sample was negative for CD34) and confirmed the absence of primary leukemic aberrations in all colonies tested (Supporting Information Table S3a). In line with this observation, CD34+ALDH+ cells derived from patients with ALDH‐rare AML were able to give rise to all colony subtypes in CFC‐assays confirming their potential of multilineage differentiation (Supporting Information Fig. S2). Hence, our data have shown that leukemia progenitors were not detectable in the CD34+ALDH+ subset.
Figure 3.
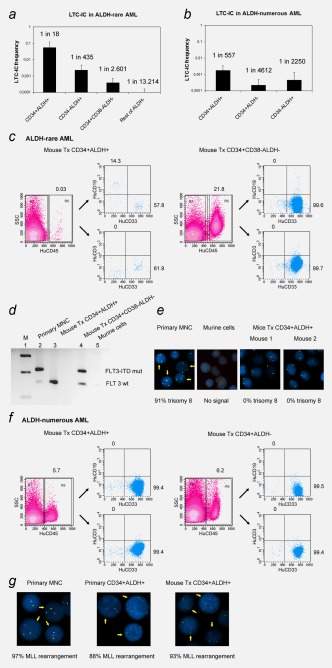
Colony formation potential and engraftment potential of cell subpopulations derived from patients with ALDH‐rare and ALDH‐numerous AML. (a) LTC‐IC frequencies of CD34+ALDH+ cells was highest, followed by CD34−ALDH+, CD34+CD38− ALDH‐ and other ALDH‐ cells (rest of ALDH‐) derived from patients with ALDH‐rare AML. (b) LTC‐IC frequencies of CD34+ALDH+ cells derived from patients with ALDH‐numerous AML were much lower than their counterparts derived from those with ALDH‐rare AML, but significantly higher than other populations including CD34+ALDH− and CD34−ALDH+ cells. (c) CD34+ALDH+ cells derived from patients with ALDH‐rare AML were capable of multilineage engraftment in NSG mice (mouse tx CD34+ALDH+), while CD34+CD38−ALDH‐ gave rise only to CD33+ myeloid cells (mouse tx CD34+CD38−ALDH−). (d) FLT3‐ITD detection revealed that engrafted cells from mice transplanted with CD34+ALDH+ cells expressed FLT3‐wild type. In contrast, FLT3‐ITD was positive in engrafted human cells of mice transplanted with CD34+CD38−ALDH− cells. (e) FISH analysis confirmed the nonleukemic nature of the CD34+ALDH+ progenies with cells being trisomy 8‐negative. (f and g) for patients with ALDH‐numerous AML, AML engraftment was always found upon transplantation of CD34+ALDH+ as well as CD34+ALDH− subsets, demonstrated by FACS and FISH analysis. Percentages of engrafted human cells with the proportions of CD33+, CD19+ and CD3+ cells are depicted in the FACS plots; mutated cells are marked with arrows in the FISH images. M: marker, wt: wild type, mut: mutation.
In analogy, we have performed LTC‐IC assays using (a) CD34+ALDH+, (b) CD34+ALDH−, and (c) CD34−ALDH+ cells derived from five patients with ALDH‐numerous AML. CD34+ALDH+ cells showed the highest LTC‐IC (one in 557 cells) activity compared to the other subsets (CD34+ALDH−: one in 4,612 cells, CD34−ALDH+: one in 2,250 cells; Fig. 3 b). As the frequencies of these LTC‐IC were much rarer compared to the corresponding subsets derived from ALDH‐rare AML, these LTC‐IC were probably derived from residual normal HSC. Clonal marker analysis also revealed that all colonies were non‐leukemic demonstrating that CD34+ALDH+ cells in ALDH‐numerous AML represent a mixture of predominantly leukemic cells and extremely few normal HSC that are phenotypically inseparable (Supporting Information in Table S3 and Fig. S3).
Engraftment and leukemogenesis potential of ALDH+ cells in NSG mice
We studied the engraftment potential of different subpopulations in NSG‐mice using (a) CD34+ALDH+, (b) CD34−ALDH+, (c) CD34+CD38−ALDH− and (d) rest of ALDH− cells derived from two patients with ALDH‐rare AML. In additional three patients, CD34+CD38−ALDH+ and CD34+CD38−ALDH− were sorted and transplanted in NSG‐mice (Table 1a). Both CD34+ALDH+ and CD34+CD38−ALDH+ cells showed multilineage engraftment (CD19+ and CD33+) on transplantation (Table 1a and Fig. 3c) with FISH and FLT3‐ITD PCR confirming the nonleukemic nature of the engrafted human cells (Figs. 3 d and 3 e). In contrast, the CD34+CD38−ALDH− subset initiated human CD33+ engraftment in one of five patients with a larger cell dose required (Table 1a and Fig. 3 c). The leukemic nature of these cells was confirmed by detection of the FLT3‐ITD mutation (Fig. 3 d). The remaining cellular subsets did not engraft, despite transplantation of up to one million cells per animal (Table 1a).
Table 1.
Engraftment of AML cell subpopulations in NSG mice
| a. ALDH‐rare AML | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD34+ALDH+ | CD34‐ALDH+ | CD34+CD38‐ALDH‐ | Rest of ALDH‐ | |||||||
| AML no. | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis | Dose (×103) | Engrafted/total mice (% human CD45+ cells) |
| AML BM67 | 1 | 0/2 | 0.4 | 0/1 | 5–50 | 0/3 | 50–1,000 | 0/4 | ||
| 4.5 | 1/2 (0.03%) | Normal | 150 | 1/1 (21.85%) | AML | |||||
| AML BM72 | 4 | 2/2 (0.07%) | Normal | 5–20 | 0/2 | 5–140 | 0/3 | 50–1,000 | 0/3 | |
| CD34+CD38−ALDH+ | CD34+CD38−ALDH‐ | |||||
|---|---|---|---|---|---|---|
| AML no. | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis |
| AML BM88 | 1 | 1/3 (0.05%) | Normal | 50 | 0/3 | |
| AML BM134 | 1,4 | 1/3 (0.14%) | Normal | 11,4 | 0/4 | |
| AML BM93 | 1,7 | 1/3 (0.02%) | Normal | 145 | 0/3 | |
| b. ALDH‐numerous AML | ||||||
|---|---|---|---|---|---|---|
| CD34+ALDH+ | CD34+ALDH‐ | |||||
| AML no. | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis | Dose (×103) | Engrafted/total mice (% human CD45+ cells) | AML/normal hematopoiesis |
| AML BM42 | 5 | 0/1 | 50 | 0/1 | ||
| 50 | 1/1 (5.86%) | AML | 125 | 0/1 | ||
| 125 | 1/1 (3.39%) | AML | 1,000 | 1/1 (3.55%) | AML | |
| AML BM80 | 50 | 1/1 (67.65%) | AML | NA | ||
| 210 | 2/2 (70.13%) | AML | 210 | 2/2 (63.58%) | AML | |
| 1,000 | 1/1 (68.15%) | AML | NA | |||
| AML BM78 | 350 | 1/2 (0.13%) | AML | 426 | 0/1 | |
| 677 | 1/1 (0.06%) | AML | 750 | 0/1 | ||
| AML BM36 | 5–50 | 0/3 | 5–42 | 0/2 | ||
| AML BM76 | 50–248 | 0/3 | 62 | 0/1 | ||
Percentages or mean percentages of human CD45+ cells in xenografts are indicated. NA: not analyzed.
In analogy, we studied the engraftment potentials of CD34+ALDH+ and CD34+ALDH−cells derived from five individual patients with ALDH‐numerous AML (Table 1b). In two of five samples, leukemia engraftment was observed after transplantation of CD34+ALDH+ and CD34+ALDH− cells with the latter showing inferior engraftment potential. In one case. AML engraftment was only seen after transplantation of CD34+ALDH+ cells (Table 1b and Fig. 3 f). Cytogenetic analyses were performed wherever possible and confirmed the leukemic nature of the engrafted human cells (Fig. 3 g). Two samples failed to engraft.
Thus, we have provided unequivocal evidence that functionally normal HSC can be found within the CD34+ALDH+ subset of patients with ALDH‐rare AML, whereas LSC reside within the ALDH− subset. In contrast, in patients with ALDH‐numerous AML, functional LSC are detectable in both compartments and HSC cannot be separated.
ALDH‐numerous AML samples showed higher rate of AML engraftment
To investigate if ALDH‐numerous cases represent an AML subgroup with increased engraftment potential we performed AML bulk transplantations of 38 unselected diagnostic cases. Of these cases 11 had an ALDH‐numerous and 27 an ALDH‐rare phenotype. 1–2 × 106 unfractionated cells were transplanted in NSG mice and analyzed 12–16 weeks after transplantation. In this analysis ALDH‐numerous, AML showed increased AML engraftment ability with 36.4% positive cases, whereas only 18.5% of ALDH‐rare cases resulted in AML engraftment. Consistent with these data ALDH‐rare AML samples mostly lead to normal engraftment (Fig. 4 a, Supporting Information Table S4).
Figure 4.
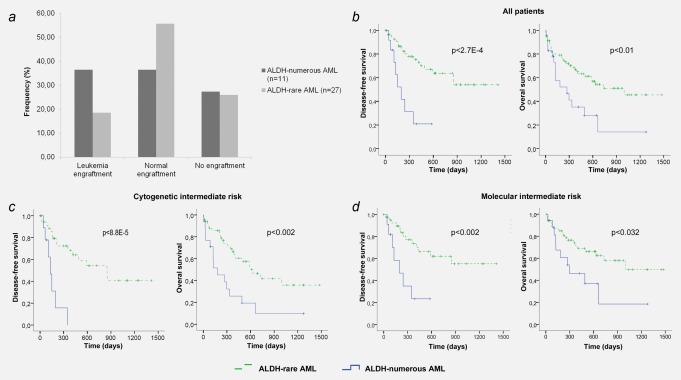
Bulk transplantation results and survival analysis. ( a ) Bulk MNC of AML patients were transplanted into NSG mice and analyzed for leukemia and normal engraftment. Cells derived from ALDH‐numerous AML samples were more likely to induce leukemia in xenografts compared to ALDH‐rare AML. ( b ) Survival analysis revealed significantly shorter DFS and OS for patients with ALDH‐numerous AML compared to those with ALDH‐rare AML. ( c and d ) risk stratification according to the ALDH status in the cytogenetic (c) and molecular intermediate risk group (d) identifies ALDH‐numerous AML as a poor prognosis group within these subsets. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The higher rate of leukemia engraftment suggests a larger pool of LSC in cases with an ALDH‐numerous phenotype. The latter has been shown to be associated with poor survival.34 Thus, our data confirm that the ALDH‐numerous phenotype identifies more aggressive AML cases which might be helpful to further increase the accuracy of risk stratification for subsequent treatment.
Clinical outcome of patients with ALDH‐numerous AML
To investigate the prognostic value of our stratification we have analyzed in parallel to our functional studies the outcome of patients with ALDH‐rare versus ALDH‐numerous AML. This analysis showed significantly shorter disease‐free survival (DFS) as well as overall survival (OS) for the 24 patients with ALDH‐numerous AML compared to the 80 patients with ALDH‐rare AML (p < 2.7E‐4 for DFS, p < 0.01 for OS; Fig. 4 b).
Among patients with ALDH‐numerous AML, only 25% belonged to the cytogenetic high‐risk group with all other cases being intermediate risk (Supporting Information Table S1). Reanalyzing the survival of all 69 cytogenetic intermediate‐risk patients according to our ALDH stratification showed significantly worse DFS (p < 8.8E‐5) and OS (p < 0.002) for patients with ALDH‐numerous disease (Fig. 4 c).
Next, we also stratified AML patients according to their genetic markers distinguishing favorable risk (FLT3‐ITD‐negative/NPM1‐mutated), poor risk (FLT3‐ITD‐positive/NPM1‐wild type) and intermediate risk (all other patients). This stratification again assigned the majority of ALDH‐numerous cases (18 of 24) to the intermediate risk‐group with survival analysis showing adverse outcomes for these patients (DFS: p < 0.002 and OS p < 0.032; Fig. 4 d). Supporting this data, an ALDH‐rare phenotype was highly predictive for better outcome within all analyzed patients independent from their FLT3‐ITD and NPM1 status (Supporting Information Fig. S4).
ALDH+ cells from ALDH‐numerous AML were more ARA‐C resistant and quiescent than ALDH− cells
As patients with ALDH‐numerous AML had a significantly worse outcome we hypothesized that ALDH+ cells might be enriched in AML samples collected after chemotherapy. In four of these patients matched diagnostic (ED) and follow‐up samples after first induction therapy (first) with persistent blasts were available (Fig. 5 a). The ALDH+ cell population was reduced but still detectable after the first induction chemotherapy. In both patients tested, we detected increased numbers of ALDH+ cells that correlated to an expansion of leukemia blasts in the subsequent BM aspirations (second). Despite reduction of ALDH+ cells in the bone marrow upon first induction treatment, ALDH expression persisted and increased within the LSC‐enriched CD34+CD38− subset. Overall, CD34+CD38−ALDH− cells were more effectively eliminated by induction chemotherapy compared to CD34+CD38−ALDH+ cells suggesting that high ALDH activity correlates to increased chemoresistance in vivo (Fig. 5 b).
Figure 5.
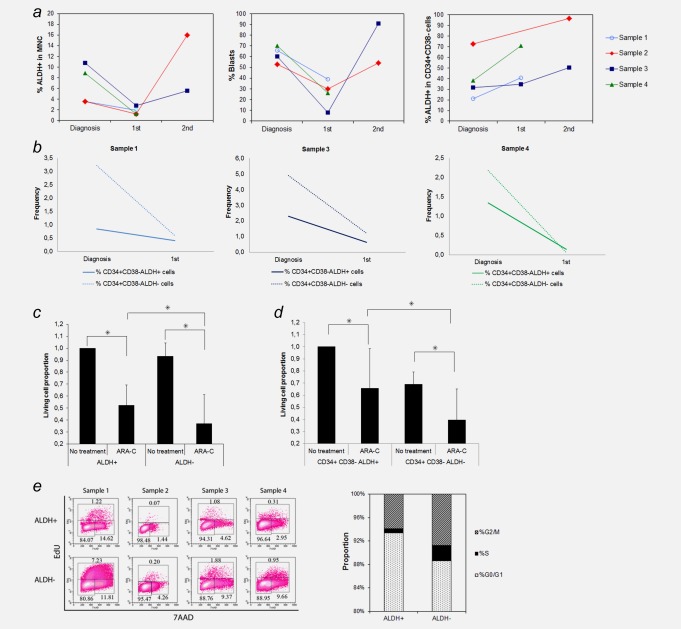
Chemoresistance of ALDH+ and ALDH− blasts derived from patients with ALDH‐numerous AML. ( a ) Follow‐up studies of BM ALDH+ cells derived from 4 patients with ALDH‐numerous AML showed that ALDH+ cells were reduced after the first induction chemotherapy (first time point) and expanded again, parallel to the increase in leukemia blasts (second time point). ALDH activity increased over time within the LSC enriched CD34+CD38− cell population. ( b ) CD34+CD38−ALDH+ cells were more resistant to chemotherapy compared to CD34+CD38−ALDH− cells in all three patients in whom BM aspirates at diagnosis and after first induction chemotherapy were available. ( c ) In vitro treatment with ARA‐C showed that ALDH+ cells were significantly more resistant than ALDH− cells (p < 0.043). ( d ) High ALDH activity of the LSC‐enriched CD34+CD38− population was also associated with increased resistance to ARA‐C in vitro (p < 0.027). ( e ) Cell cycle analysis showed that ALDH+ cells were more quiescent than ALDH−cells (p < 0.02).
We further tested the role of ALDH+ cells in chemoresistance by treating leukemic ALDH+ and ALDH−subsets derived from five ALDH‐numerous AML samples with ARA‐C (1 µM) for 5 days and found that ARA‐C had a more profound effect on ALDH− than on ALDH+ cells (U test, p < 0.043; Fig. 5 c).
To compare the chemoresistance of LSC populations in ALDH‐numerous AML samples, we treated CD34+CD38−ALDH+ and CD34+CD38−ALDH− cells with 1 µM ARA‐C. As expected CD34+CD38−ALDH+ cells showed better survival than CD34+CD38−ALDH− cells in the presence of ARA‐C in six samples analyzed (U test, p < 0.027; Fig. 5 d). Thus, high ALDH activity was also associated with increased chemoresistance in vitro.
Furthermore, cell cycle analysis of ALDH+ and ALDH− cells of four patients with ALDH‐numerous AML using EdU and 7‐AAD showed that the ALDH+ population had less actively cycling cells compared to the ALDH− counterparts (p < 0.02; Fig. 5 e). Increased quiescence of ALDH+ cells from ALDH‐numerous AML might, therefore, explain their relative chemoresistance.
Discussion
The significance of LSC for development, evolution and recurrence of AML has been recognized in numerous studies. A few more recent studies have demonstrated the enormous heterogeneity of LSC, both in patients with ALL as well as with AML.17, 35, 36, 37 Whole genome sequencing studies have also indicated that leukemic clones are heterogeneous at the time of diagnosis.37 In addition, other authors reported that some of the potential leukemia driving mutations could be detected in functionally and phenotypically normal HSC derived from patients with AML or even from normal subjects.26, 27, 28 These cells have been suggested to represent a potential reservoir of “pre‐leukemic” cells that could survive chemotherapy regimens but subsequently transform and become the source of recurrent disease.26, 28 For a proper understanding of the transition from functionally normal hematopoiesis to overt leukemia, appropriate comparative studies of normal HSC and matched LSC are, therefore, essential.
Recent analyses using primary leukemia cells from patients with AML suggested that LSC could be separated from normal HSC based on their differences in ALDH activity.10, 29 In their study of 27 AML patients with focus on those with inversion 16 and translocation (8;21), Gerber et al. reported that the CD34+CD38−ALDH+ subset resembled normal HSC and lacked leukemia‐specific cytogenetic abnormalities in most cases. Functionally, these cells mostly gave rise to normal hematopoietic engraftment in a xenogeneic transplantation model but in some cases separation of HSC from leukemic cells was not feasible.29 An earlier study of 19 AML cases showed that AMLs could be classified in three subgroups based on their ALDH activity (rare‐, numerous‐ and negative‐pattern) with the rare pattern flagging a small subgroup of six patients in which HSC could be separated by their high ALDH‐expression.10
In accordance with these reports, we have demonstrated that, in patients with ALDH‐rare AML (80 of 104 patients), the CD34+ALDH+ subset represents residual and functionally normal HSC with the ability to give rise to multilineage engraftment upon transplantation into immune deficient mice. Cytogenetic and molecular genetic studies have further confirmed their nonleukemic nature before and after transplantation. In contrast, LSC were unequivocally found within the ALDH− subset. Our study was not designed to address the issue of preleukemic stem cells but recent data suggest that CD34+ALDH+ HSC might already harbor leukemia driving mutations.26, 38 In line with these reports, HSC selected by our method were functionally normal with the ability to give rise to multilineage hematopoietic engraftment in xenogeneic transplantation models in vivo. The separation strategy presented in our study will, therefore, provide a valuable tool for future analysis of preleukemic‐HSC in AML.
Albeit we have proven that CD34+ALDH+ cells were associated with features of normal HSC in 77% of our patients, the CD34+ALDH+ subset was unequivocally enriched for LSC in a substantial proportion of our patients, that is, in 24 patients (23%) with ALDH‐numerous AML. The CD34+ALDH+ subset was characterized by the corresponding cytogenetic or molecular genetic aberrations of the original leukemia. Above all, these CD34+ALDH+ cells were able to induce leukemic engraftment in xenogeneic transplantation models. For the group of patients with ALDH‐numerous AML, the leukemia initiating potential was not restricted to the CD34+ALDH+ subset. Indeed, CD34+ALDH− cells from ALDH‐numerous AML were also capable of leukemia engraftment, albeit at a much higher cell dose. Similar to the surface markers CD34 and CD38, ALDH activity alone was, therefore, not sufficient to separate or enrich LSC in this group of patients. This is in line with previous reports that have shown that LSC potential can be detected in different subsets of AML patients.7, 8, 9
By comparing AML engraftment potential of 38 unfractionated AML samples we have shown that ALDH‐numerous AML were twice as likely to give rise to leukemia in vivo compared to ALDH‐rare AML. In this context recent reports have shown that increased engraftment potential is associated with poor prognosis in AML.34 We, therefore, correlated our data to the outcome of all 104 patients and have shown that the ALDH‐numerous pattern identifies a subgroup with overall adverse clinical outcome, as suggested previously.16, 17 Above all, we show that our AML stratification strategy is especially helpful in identifying cases with poor prognosis in the intermediate‐risk subgroup. While Pearce and colleagues have shown that engraftment potential might help to identify cases with poor prognosis in the intermediate subgroup, our FACS approach was able to reliably separate cases with good and poor prognosis tabling it as an additional risk stratification tool.
Furthermore, we have also shown that ALDH+ cells derived from patients with ALDH‐rare versus ALDH‐numerous AML differ from each other in their biology with normal origin in the former group and leukemic origin in the latter. Because ALDH has been reported to be involved in the detoxification of drug‐related aldehydes as well as in the metabolism of retinoic acid, which is known for its ability to block cell cycle,39, 40 its enhanced expression might provide another explanation for the survival advantage of leukemic cells against chemotherapy agents targeting cycling cells. Indeed, our functional studies have demonstrated that the ALDH+ subset derived from patients with ALDH‐numerous AML were more quiescent and resistant to ARA‐C treatment in vitro compared to the ALDH− subset. Furthermore, we have confirmed this ARA‐C resistance of ALDH+ cells compared to ALDH− cells within the LSC‐enriched CD34+CD38− compartment of patients with ALDH‐numerous AML. In accordance with these results, follow‐up studies of the ALDH+ subsets in the marrow of patients with ALDH‐numerous AML showed that ALDH+ cells, especially the CD34+CD38−ALDH+ subset, were able to survive induction chemotherapy with a relative enrichment at the time of relapse. Albeit the leukemic nature of these ALDH+ cells at the follow‐up time‐points was not proven, the blast persistence detected both by conventional cytology and flow cytometry as well as the consistent results from our in vitro analysis strongly suggest the malignant nature of these cells.
Based on the results of our systematic study of 104 AML patients, screening for ALDH activity provides a reliable tool for the prospective identification of ALDH‐rare AML cases suitable for the separation of HSC and LSC. However, for ALDH‐numerous AML, the CD34+ALDH+ subset is unequivocally enriched for LSC. Expression of ALDH in LSC was associated with aggressive disease and adverse clinical outcome. This difference in spectrum and relevance of ALDH activity in the putative LSC population reflects their enormous heterogeneity among AML patients. In conclusion, our study has demonstrated, in addition to phenotypic and genetic, also functional heterogeneity of leukemia cells and suggests divergent roles for ALDH activity in normal HSC versus leukemia initiating cells.
Supporting information
Supporting Information
Acknowledgements
The authors thank Karina Borowski for technical assistance with FACS, Michaela Brough for performance of the FISH analysis, and Katrin Miesala, Anke Diehlmann for isolation and culture of primary cells. We are indebted to Dr. Marieke Essers, Dr. Stephan Wurzer and Dr. Michael Milsom for their help in the transplantation experiments, and Prof. Dr. Meinhard Kieser for his advice in the statistical analysis. We are grateful to clinicians in the Department of Internal Medicine V for their contribution in collection of AML and healthy BM samples and patients who participated in the study.
The copyright line for this article was changed on 28 September 2015 after original online publication.
Authorship Contributions: V.T.H., A.D.H. and C.L. designed the research, V.T.H., E.B., I.H., A.Z.M., S.R., W.W., V.E. and A.J. performed experiments, V.T.H., W.W., A.J., A.D.H. and C.L. analyzed the data, E.B., N.B., P.W., S.R. and C.L. contributed material, A.T. supervised mouse experiments, V.T.H., A.D.H. and C.L. wrote the manuscript with critical input from other authors. All authors reviewed and approved the final version
References
- 1. Maynadie M, De Angelis R, Marcos‐Gragera R, et al. Survival of european patients diagnosed with myeloid malignancies: a HAEMACARE study. Haematologica 2013;98:230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dick JE. Stem cell concepts renew cancer research. Blood 2008;112:4793–807. [DOI] [PubMed] [Google Scholar]
- 3. Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy‐resistant human AML stem cells home to and engraft within the bone‐marrow endosteal region. Nat Biotechnol 2007;25:1315–21. [DOI] [PubMed] [Google Scholar]
- 4. Hoang VT, Zepeda‐Moreno A, Ho AD. Identification of leukemia stem cells in acute myeloid leukemia and their clinical relevance. Biotechnol J 2012;7:779–88. [DOI] [PubMed] [Google Scholar]
- 5. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–7. [DOI] [PubMed] [Google Scholar]
- 6. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–8. [DOI] [PubMed] [Google Scholar]
- 7. Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011;17:1086–93. [DOI] [PubMed] [Google Scholar]
- 8. Taussig DC, Vargaftig J, Miraki‐Moud F, et al. Leukemia‐initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(‐) fraction. Blood 2010;115:1976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac‐deficient mice. J Clin Invest 2011;121:384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pearce DJ, Taussig D, Simpson C, et al. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells 2005;23:752–60. [DOI] [PubMed] [Google Scholar]
- 11. Hess DA, Meyerrose TE, Wirthlin L, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood 2004;104:1648–55. [DOI] [PubMed] [Google Scholar]
- 12. Corti S, Locatelli F, Papadimitriou D, et al. Identification of a primitive brain‐derived neural stem cell population based on aldehyde dehydrogenase activity. Stem Cells 2006;24:975–85. [DOI] [PubMed] [Google Scholar]
- 13. Ginestier C, Hur MH, Charafe‐Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Becker MW, Jordan CT. Leukemia stem cells in 2010: current understanding and future directions. Blood Rev 2011;25:75–81. [DOI] [PubMed] [Google Scholar]
- 15. Ran D, Schubert M, Pietsch L, et al. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Exp Hematol 2009;37:1423–34. [DOI] [PubMed] [Google Scholar]
- 16. Cheung AM, Wan TS, Leung JC, et al. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia 2007;21:1423–30. [DOI] [PubMed] [Google Scholar]
- 17. Ran D, Schubert M, Taubert I, et al. Heterogeneity of leukemia stem cell candidates at diagnosis of acute myeloid leukemia and their clinical significance. Exp Hematol 2012;40:155–65 e1. [DOI] [PubMed] [Google Scholar]
- 18. Blair A, Hogge DE, Ailles LE, et al. Lack of expression of Thy‐1 (CD90) on acute myeloid leukemia cells with long‐term proliferative ability in vitro and in vivo. Blood 1997;89:3104–12. [PubMed] [Google Scholar]
- 19. Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody‐mediated targeting of CD123, IL‐3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009;5:31–42. [DOI] [PubMed] [Google Scholar]
- 20. van Rhenen A, Moshaver B, Kelder A, et al. Aberrant marker expression patterns on the CD34+CD38‐ stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007;21:1700–7. [DOI] [PubMed] [Google Scholar]
- 21. van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL‐1 aids in discrimination between normal and leukemic stem cells. Blood 2007;110:2659–66. [DOI] [PubMed] [Google Scholar]
- 22. Hosen N, Park CY, Tatsumi N, et al. CD96 is a leukemic stem cell‐specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA 2007; 104:11008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009;138:286–99.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kikushige Y, Shima T, Takayanagi S, et al. TIM‐3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010;7:708–17. [DOI] [PubMed] [Google Scholar]
- 25. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009;138:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jan M, Snyder TM, Corces‐Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 2012;4:149ra18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150:264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerber JM, Smith BD, Ngwang B, et al. A clinically relevant population of leukemic CD34(+)CD38(‐) cells in acute myeloid leukemia. Blood 2012;119:3571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA 2011;108:5009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grimwade D, Hills RK, Moorman AV, et al. Research institute adult leukaemia working G. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the united kingdom medical research council trials. Blood 2010;116:354–65. [DOI] [PubMed] [Google Scholar]
- 32. Wagner W, Horn P, Castoldi M, et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS One 2008;3:e2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoang VT, Hoffmann I, Borowski K, et al. Identification and separation of normal hematopoietic stem cells and leukemia stem cells from patients with acute myeloid leukemia. Methods Mol Biol 2013;1035:217–30. [DOI] [PubMed] [Google Scholar]
- 34. Pearce DJ, Taussig D, Zibara K, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood 2006;107:1166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR‐ABL1 lymphoblastic leukaemia‐initiating cells. Nature 2011;469:362–7. [DOI] [PubMed] [Google Scholar]
- 36. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011;469:356–61. [DOI] [PubMed] [Google Scholar]
- 37. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature 2012;481:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506:328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev 2011;7:292–306. [DOI] [PubMed] [Google Scholar]
- 40. Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 2011;6:345–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information