

Abstract
RIG‐I is a key cytosolic sensor that detects RNA viruses through its C‐terminal region and activates the production of antiviral interferons (IFNs) and proinflammatory cytokines. While posttranslational modification has been demonstrated to regulate RIG‐I signaling activity, its significance for the sensing of viral RNAs remains unclear. Here, we first show that the RIG‐I C‐terminal region undergoes deacetylation to regulate its viral RNA‐sensing activity and that the HDAC6‐mediated deacetylation of RIG‐I is critical for viral RNA detection. HDAC6 transiently bound to RIG‐I and removed the lysine 909 acetylation in the presence of viral RNAs, promoting RIG‐I sensing of viral RNAs. Depletion of HDAC6 expression led to impaired antiviral responses against RNA viruses, but not against DNA viruses. Consequently, HDAC6 knockout mice were highly susceptible to RNA virus infections compared to wild‐type mice. These findings underscore the critical role of HDAC6 in the modulation of the RIG‐I‐mediated antiviral sensing pathway.
Keywords: deacetylation, HDAC6, innate immunity, RIG‐I, virus sensing
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Retinoic‐acid‐inducible gene‐I (RIG‐I)‐like receptors (RLRs), including RIG‐I and melanoma differentiation‐associated protein 5 (MDA5), play key roles in innate immune responses against virus infection (Yoneyama et al, 2004, 2005; Akira et al, 2006; Pichlmair & Reis e Sousa, 2007). Invading RNA viruses are recognized by RLRs in the cytosol. RLRs activate the signaling adaptor protein mitochondrial antiviral signaling protein (MAVS; also known as IPS‐1, VISA, or CARDIF) and initiate downstream signaling pathways to induce antiviral responses, such as the production of type I interferons (IFNs) and proinflammatory cytokines (Akira et al, 2006; Kawai & Akira, 2006; Pichlmair & Reis e Sousa, 2007; Loo & Gale, 2011). RIG‐I has been reported to recognize 5′‐triphosphate‐containing double‐stranded RNA from diverse viruses or short double‐stranded RNA molecules and has been the subject of extensive research interest as a viral sensor (Yoneyama et al, 2004, 2005; Akira et al, 2006; Pichlmair & Reis e Sousa, 2007). RIG‐I comprises two N‐terminal tandem caspase activation and recruitment domains (CARDs), a central DExH‐box‐type RNA helicase composed of two Rec A domains (Hel‐1 and Hel‐2; the SF2 helicase core), and a C‐terminal regulatory domain (CTD) (Wang et al, 2010; Kowalinski et al, 2011). In uninfected cells, RIG‐I exists in an autorepressed conformation in which the CARDs are not available for inducing downstream signal transduction (Takahasi et al, 2008). Upon viral infection, viral RNAs bind both the CTD and the helicase domain, thereby activating the ATPase activity of the helicase domain, which in turn triggers a conformational change to expose the masked CARDs (Kowalinski et al, 2011; Luo et al, 2011; Leung & Amarasinghe, 2012). The CARD of RIG‐I then interacts with its downstream molecule, MAVS, to trigger antiviral responses (Kowalinski et al, 2011; Luo et al, 2011; Leung & Amarasinghe, 2012).
Several positive or negative regulatory mechanisms for the control of RIG‐I‐mediated antiviral signaling have been described, including posttranslational modifications of RIG‐I. The importance of RIG‐I ubiquitination (Gack et al, 2007; Oshiumi et al, 2013), phosphorylation (Gack et al, 2010), and SUMOylation (Mi et al, 2010), which regulate its function, has been demonstrated. However, the role of RIG‐I acetylation on innate immune responses has not been investigated to date. Regarding the mechanisms of RIG‐I‐mediated viral sensing, there is still significant uncertainty as to how RIG‐I binds and recognizes RNAs in virus‐infected cells and how this process is regulated. In addition, the role of RIG‐I acetylation in modulating its binding activity or the recognition of viral RNAs remains to be elucidated.
Histone deacetylase 6 (HDAC6) is a cytoplasmic deacetylase important for the regulation of cytoskeletal and mitochondrial functions (Hubbert et al, 2002; Kovacs et al, 2005; Zhang et al, 2007; Lee et al, 2010a,b, 2014). Recently, HDAC6 has been shown to regulate replication of human immunodeficiency virus (HIV) and influenza A virus (IVA) (Valenzuela‐Fernandez et al, 2005; Mosley et al, 2006; Husain & Cheung, 2014; Lucera et al, 2014), and innate antiviral immunity (Nusinzon & Horvath, 2006; Zhu et al, 2011; Chattopadhyay et al, 2013). However, it remains unknown how HDAC6 is connected to the antiviral signaling network and whether it plays a critical role in antiviral immunity under physiological conditions in animal.
In the present study, we show that HDAC6 knockdown in vitro and HDAC6‐deficient mice show impaired innate immune responses against RNA viruses. Investigation of the underlying mechanism showed that, upon RNA virus infection, HDAC6 transiently binds RIG‐I and deacetylates lysine 909 (K909), which result in activating CTD of RIG‐I to bind and recognize viral RNAs. These findings undercover a novel mechanism of RIG‐I activation mediated by HDAC6‐dependent deacetylation, facilitating the recognition of viral RNA by the CTD domain of RIG‐I to induce innate immune responses against RNA virus infection.
Results
HDAC6 is essential for the protection of mice against VSV‐Indiana infection
To determine whether HDAC6 plays a physiological role in an RNA virus infection in vivo, HDAC6 +/+ and HDAC6 −/− mice were intravenously infected with vesicular stomatitis virus (VSV, Indiana strain). As shown in Fig 1A, HDAC6 −/− mice are more susceptible to VSV‐Indiana infection than HDAC6 +/+ mice and showed significantly decreased survival rate. The virus titer and replication were measured in samples taken from the brain and spleen of mice 5 days after infection with VSV‐Indiana; the results showed that viral titers were significantly higher in HDAC6 −/− mice than in HDAC6 +/+ mice (Fig 1B and C). To determine the effects of HDAC6 deficiency on viral clearance and IFN production in serum, VSV‐GFP, which shows low virulence, was intravenously injected into HDAC6 +/+ and HDAC6 −/− mice and VSV‐GFP titers and IFN‐β and IL‐6 levels in the serum were measured every 6 h. Consistent with previous results, virus titers were significantly higher and IFN‐β and IL‐6 production was markedly lower in HDAC6 −/− mice than in HDAC6 +/+ mice (Fig 1D and E). In addition, we examined the role of HDAC6 in cytokine induction by poly(I:C), which is a synthetic double‐stranded RNA (dsRNA). Intravenous injection of poly(I:C) caused the rapid and robust induction of IFN‐β and IL‐6 in HDAC6 +/+ mice; however, induction of these cytokines was significantly reduced in HDAC6 −/− mice (Fig 1F). These results indicated that HDAC6 plays an important role in the antiviral immune response by producing IFNs and proinflammatory cytokines in responses to foreign RNA viruses.
Figure 1. HDAC6 −/− mice are susceptible to lethal RNA virus infection and show decreased cytokine production.
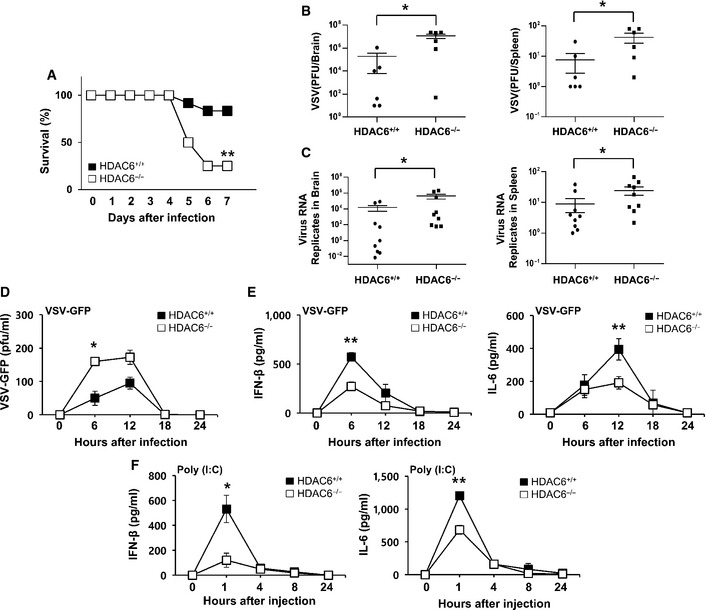
- The survival of age‐ and sex‐matched HDAC6 +/+ mice and HDAC6 −/− mice was monitored for 9 days after intravenous VSV‐Indiana infection (2 × 108 pfu/mouse; n = 12 per group; log‐rank test).
- Determination of the viral load in organs by standard plaque assay. HDAC6 +/+ and HDAC6 −/− mice were intravenously infected with VSV‐Indiana, and the brain and spleen were collected at 5 dpi (2 × 108 pfu/mouse; n = 6 per group; Mann–Whitney test).
- Determination of viral loads in organs by qPCR of VSV viral transcripts. The brain and spleen were collected at 5 dpi after HDAC6 +/+and HDAC6 −/− mice were intravenously infected with VSV‐Indiana (2 × 108 pfu/mouse; n = 9 per group; Mann–Whitney test).
- The viral load in the serum was assessed by standard plaque assay. HDAC6 +/+ and HDAC6 −/− mice were intravenously infected with VSV‐GFP. Serum was collected at the indicated time points and analyzed (4 × 108 pfu/mouse; n = 3 per group; Student's t‐test).
- ELISA of IFN‐β (left) and IL‐6 (right) in the serum of mice described in (D) (Student's t‐test).
- ELISA of serum IFN‐β (left) and IL‐6 (right) after poly(I:C) injection. HDAC6 +/+ and HDAC6 −/− mice were intravenously injected with poly(I:C). Serum was collected at the indicated time points and analyzed (200 μg/mouse; n = 3 per group; Student's t‐test).
HDAC6 is involved in antiviral response against RNA viruses in macrophages and peripheral blood mononuclear cells
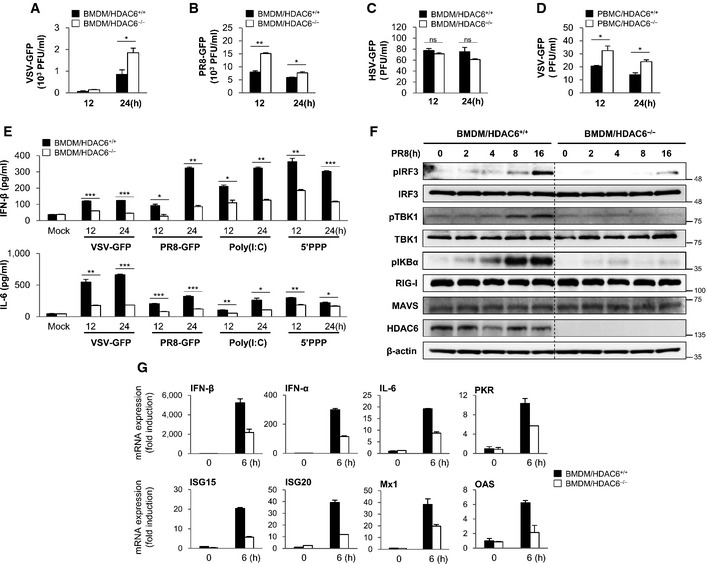
The antiviral role of HDAC6 was evaluated in bone marrow‐derived macrophages (BMDMs) and peripheral blood mononuclear cells (PBMCs) from HDAC6 +/+ and HDAC6 −/− mice. After infection with VSV‐GFP, influenza A virus (PR8‐GFP), and Newcastle disease virus (NDV‐GFP), virus replication was first measured in HDAC6 +/+ and HDAC6 −/− BMDMs. As shown in Fig 2A and B and Appendix Fig S1A and B, virus replication was consistently increased in HDAC6 −/− BMDMs compared with that in HDAC6 +/+ BMDMs. Measurement of IFN‐β and IL‐6 secretion in BMDMs after virus infection or poly(I:C) and 5′‐triphosphate dsRNA (5′ppp‐dsRNA) treatment showed that IFN‐β and IL‐6 production was lower in HDAC6 −/− BMDMs than in HDAC6 +/+ BMDMs (Fig 2E and Appendix Fig S1C). By contrast, there was no significant difference in virus replication or IFN‐β and IL‐6 secretion in BMDMs infected by the DNA virus herpes simplex virus (HSV‐GFP) or in cells treated with poly(dA:dT), a viral DNA‐like molecule (Fig 2C and Appendix Fig S2A). Furthermore, PBMCs isolated from HDAC6 +/+ and HDAC6 −/− mice infected by VSV‐GFP showed a similar pattern to that observed in BMDMs (Fig 2D and Appendix Fig S2B). These results suggested that HDAC6 deficiency suppresses production of type I IFNs and proinflammatory cytokines and selectively enhance the RNA virus infection.
Figure 2. HDAC6 positively regulates the innate antiviral response in bone marrow‐derived macrophages and peripheral blood mononuclear cells.
-
A, BVirus replication at 12 and 24 hpi, in HDAC6 +/+ and HDAC6 −/− BMDMs in response to VSV‐GFP (MOI = 10) infection (A) and PR8‐GFP (MOI = 5) infection (B).
-
CVirus replication in HDAC6 +/+ and HDAC6 −/− BMDMs in response to HSV‐GFP (MOI = 2) infection.
-
DVirus replication in HDAC6 +/+ and HDAC6 −/− PBMCs in response to VSV‐GFP (MOI = 10) infection.
-
EELISA of IFN‐β (upper), IL‐6 (lower) levels in the supernatant of (A) and (B), and in HDAC6 +/+ and HDAC6 −/− BMDMs treated with poly(I:C) (20 μg/ml) or transfected with 5′ppp‐dsRNA (1 μg/ml).
-
FImmunoblot analysis of the phosphorylated and inactive forms of IRF3, IKBα, TBK1, RIG‐I, MAVS, HDAC6, and β‐actin at the indicated times (0, 2, 4, 8, and 16 h) in HDAC6 +/+ and HDAC6 −/− BMDMs. BMDMs were stimulated with PR8‐GFP (MOI = 3).
-
GInduction of mRNA for type I IFN, IL‐6, and other IFN‐related antiviral genes in HDAC6 +/+ and HDAC6 −/− BMDMs in response to a RIG‐I agonist stimulation at 6 h. HDAC6 +/+ and HDAC6 −/− BMDMs were stimulated with 5′ppp‐dsRNA (0.5 μg/ml) for 6 h.
We next examined the effects of HDAC6 deficiency on RNA virus‐mediated activation of downstream signaling pathways in HDAC6 +/+ and HDAC6 −/− BMDMs. HDAC6 +/+ and HDAC6 −/− BMDMs were infected with PR8‐GFP, and phosphorylation of IFN‐related signaling molecules and phosphorylation of IKBα related to NF‐κB activation were confirmed. As shown in Fig 2F and Appendix Fig S3, the phosphorylation levels of IRF3 and TBK1 were markedly lower in HDAC6 −/− BMDMs or siHDAC6‐transfected RAW264.7 cells than in HDAC6 +/+ BMDMs or control RAW264.7 cells, and the levels of phosphorylated IKBα were lower in HDAC6 −/− BMDMs than in their HDAC6 +/+ counterparts. The mRNA levels of IFN‐β, IFN‐α, or antiviral‐related genes were also assessed in BMDMs with 5′ppp‐dsRNA. As shown in Fig 2G, HDAC6 −/− BMDMs showed lower levels of gene expression than HDAC6 +/+ BMDMs. Collectively, these results suggested that HDAC6 positively regulates the type I IFN signaling pathway and antiviral gene expression induced by RNA virus infection in primary immune cells.
HDAC6 positively regulates the innate antiviral response in RAW264.7 cells and fibroblasts
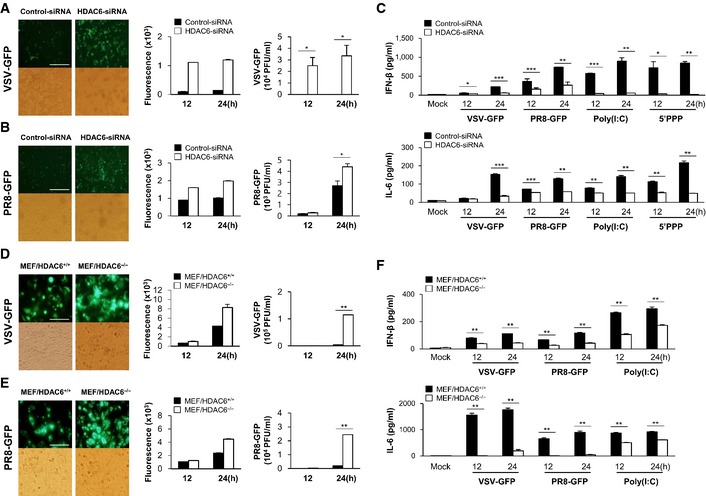
The physiological role of HDAC6 in the response to virus infection was further investigated in RAW264.7 cells and in mouse embryonic fibroblasts (MEFs) isolated from HDAC6 +/+ or HDAC6 −/− mice. We first showed that an HDAC6‐specific small interfering RNA (siRNA) efficiently inhibited the expression of endogenous HDAC6 (Appendix Fig S4A). Similar to HDAC6 −/− BMDMs or PBMCs, HDAC6 knockdown RAW264.7 cells showed higher levels of virus replication than control cells upon infection with VSV‐GFP and PR8‐GFP (Fig 3A and B). Consistent with this, we confirmed that siRNA knockdown of HDAC6 significantly reduced the production of IFN‐β and IL‐6 in RAW264.7 cells infected with virus or in cells treated with poly(I:C) and 5′ppp‐dsRNA (Fig 3C). HDAC6‐dependent virus replication and induction of IFN‐β and IL‐6 were also confirmed in fibroblasts (Fig 3D–F and Appendix Fig S3B). Similar to the results obtained in immune cells, HDAC6 −/− MEFs showed increased replication of VSV‐GFP and PR8‐GFP, and lower levels of IFN‐β and IL‐6 secretion than control cells. Furthermore, when HDAC6 −/− MEFs were reconstituted with wild‐type HDAC6 (Appendix Fig S3C), the replication of VSV‐GFP was reduced (Appendix Fig S3D). Such as the primary immune cell, the mRNA levels of IFN‐β, IFN‐α, or antiviral‐related genes were assessed in HDAC6 knockdown RAW264.7 cells with 5′ppp‐dsRNA (Appendix Fig S5) or in HDAC6 −/− MEFs with PR8‐GFP infection or poly(I:C) transfection (Appendix Fig S6). As shown in figures, HDAC6 knockdown RAW264.7 cells or HDAC6 −/− MEFs showed lower levels of gene expression than control Raw 264.7 cells or wild‐type cells. Taken together, these results strongly suggested that HDAC6 also plays a role in the antiviral response in immune cell lines and fibroblasts.
Figure 3. HDAC6 deficiency reduces the innate immune response on mouse macrophage and mouse embryonic fibroblast.
-
A, BFluorescence microscopy at 24 hpi showing green fluorescence absorbance at 12 and 24 hpi, and virus replication at 12 and 24 hpi, in control and HDAC6 knockdown RAW264.7 cells in response to VSV‐GFP (MOI = 1) infection (A) and PR8‐GFP (MOI = 1) infection (B). Scale bar, 100 μm
-
CELISA of IFN‐β (upper), IL‐6 (lower) levels in the supernatant of cells from (A) and (B), and in control and HDAC6 knockdown RAW264.7 cells treated with poly(I:C) (20 μg/ml) or transfected with 5′ppp‐dsRNA (1 μg/ml).
-
D, EFluorescence microscopy at 24 hpi showing green fluorescence absorbance at 12 and 24 hpi, and virus replication at 12 and 24 hpi, in HDAC6 +/+ and HDAC6 −/− MEFs in response to VSV‐GFP (MOI = 1) infection (D) and PR8‐GFP (MOI = 1) infection (E). Scale bar, 50 μm.
-
FELISA of IFN‐β (upper), IL‐6 (lower) levels in the supernatant of cells from (D) and (E), and in HDAC6 +/+ and HDAC6 −/− MEFs transfected with poly(I:C) (1 μg/ml).
The catalytic activity of HDAC6 is required for the activation of RIG‐I signaling
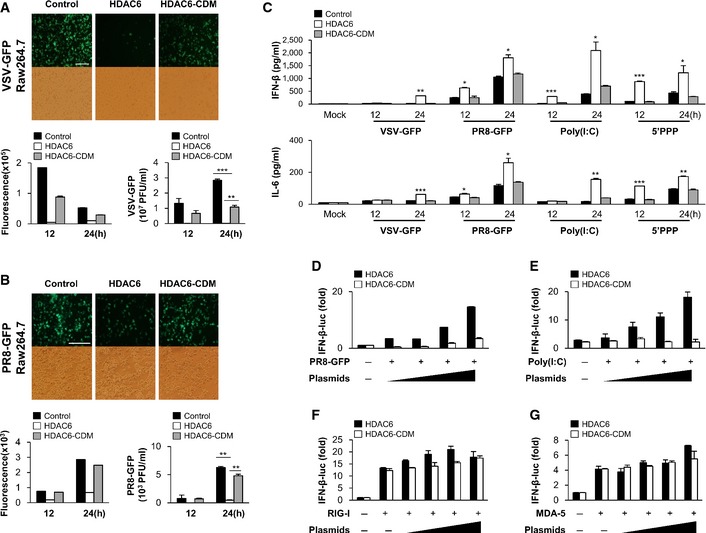
HDAC6 is a well‐known cytoplasmic deacetylase. RAW264.7 cells stably expressing HDAC6 or a catalytically inactive HDAC6 mutant (CD mutant; CDM) were used (Appendix Fig S7) to confirm whether the catalytic activity of HDAC6 is required for the antiviral signaling. HDAC6 or HDAC6‐CDM stable cells were infected with VSV‐GFP and PR8‐GFP. We found that virus replication was significantly inhibited in HDAC6‐overexpressing RAW264.7 cells; however, virus replication in HDAC6‐CDM‐overexpressing RAW264.7 cells was similar to that in control cells (Fig 4A and B). Furthermore, PR8‐GFP infection was performed after transient reconstitution of HDAC6 and HDAC6‐CDM in HDAC6 −/− MEFs. Similarly, we found that virus replication was significantly inhibited in HDAC6‐overexpressing HDAC6 −/− MEFs (Fig EV1). Consistent with this, IFN‐β and IL‐6 production was significantly higher in HDAC6‐overexpressing cells infected with virus or treated with poly(I:C) and 5′ppp‐dsRNA than in HDAC6‐CDM‐overexpressing cells or untreated control cells (Fig 4C), indicating that the catalytic activity of HDAC6 is critical for the antiviral response of HDAC6. Additionally, to confirm the functionality of the catalytic activity of HDAC6, an IFN‐β luciferase reporter assay was performed in cells expressing HDAC6 or HDAC6‐CDM. HDAC6 significantly enhanced the IFN‐β luciferase activity in a dose‐dependent manner in response to PR8‐GFP infection or treatment with poly(I:C). By contrast, HDAC6‐CDM did not enhance PR8‐GFP or poly(I:C)‐mediated IFN‐β luciferase activity (Fig 4D and E). Similar to the virus replication results, these findings indicated that the catalytic activity of HDAC6 is important for the induction of type I IFN signaling.
Figure 4. Deacetylase activity is responsible for the antiviral effect of HDAC6.
-
A, BFluorescence microscopy at 24 hpi showing green fluorescence absorbance at 12 and 24 hpi, and virus replication at 12 and 24 hpi, in vector‐, HDAC6‐, or HDAC6‐CDM‐overexpressing stable RAW264.7 cells in response to VSV‐GFP (MOI = 1) infection (A) and PR8‐GFP (MOI = 1) infection (B). Scale bar, 100 μm.
-
CELISA of IFN‐β (upper) and IL‐6 (lower) levels in the supernatant of cells from (A) and (B), and in vector, HDAC6, or HDAC6‐CDM‐overexpressing stable RAW264.7 cells treated with poly(I:C) (20 μg/ml) or transfected with 5′ppp‐dsRNA (1 μg/ml).
-
D, ELuciferase assay in 293T cells transfected with an IFN‐β luciferase promoter and TK‐Renilla together with HDAC6 or HDAC6‐CDM (100, 200, 400, or 800 ng), followed by PR8‐GFP (MOI = 2) infection (D) or by poly(I:C) (E) (1 μg/ml) transfection for another 12 h.
-
F, GLuciferase assay in 293T cells transfected with RIG‐I (F), MDA5 (G), an IFN‐β luciferase promoter, and TK‐Renilla together with HDAC6 or HDAC6‐CDM (100, 200, 400, or 800 ng). 24 h later, IFN‐β activity was measured by luciferase reporter assay.
Figure EV1. Catalytic activity of HDAC6 is responsible for antiviral effect.
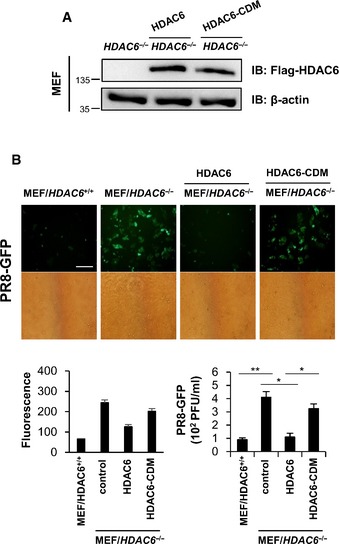
- HDAC6 −/− MEF were transiently transfected with HDAC6‐IRES‐flag and HDAC6‐CDM‐IRES‐flag.
- Fluorescence microscopy, green fluorescence absorbance and virus replication at 24 hpi in WT and RIG‐I −/− MEFs. Cells were transiently transfected with empty vector, HDAC6, HDAC6‐CDM for 36 h, followed by infection with PR8‐GFP (MOI = 1). Data are representative of at three independent experiments. Error bars, mean ± SD. *P < 0.05, **P < 0.01 (Student's t‐test). Scale bar, 100 μm.
Source data are available online for this figure.
We next attempted to determine which signaling molecule in the RLR signaling pathway is targeted by HDAC6. Ligand‐independent IFN‐β induction by RIG‐I, MDA5, MAVS, and 2CARD of RIG‐I expression was not increased by co‐transfection of HDAC6 or HDAC6‐CDM in a dose‐dependent manner (Fig 4F and G and Appendix Fig S8). These results suggested that although HDAC6 functions as a positive regulator of the RLR signaling pathway during viral infections, a different mechanism must be involved in RIG‐I signaling or that upstream of RIG‐I, rather than the downstream signaling of the RLR pathway, which is mediated by RIG‐I and MAVS interactions.
HDAC6 interacts with and deacetylates RIG‐I in response to RNA virus infection
Several lines of evidence indicate that the cytoplasmic deacetylase, HDAC6, plays a role as a positive regulator of the RLR signaling pathway against RNA virus infection, but is not required against DNA virus infection. In addition, HDAC6 affects RIG‐I signaling or signaling upstream of RIG‐I. Since RIG‐I is listed as an acetylated protein in the acetylome database (Choudhary et al, 2009), we explored the possibility that HDAC6 binds and deacetylates RIG‐I upon viral infection. To determine whether HDAC6 interacts with RIG‐I, we performed co‐immunoprecipitation assays with poly(I:C)‐transfected 293T cell lysates using an anti‐HDAC6 antibody. As shown in Fig 5A (lane 1), HDAC6 did not interact with RIG‐I in the absence of poly(I:C). After 4 h of poly(I:C) transfection, HDAC6 interacted with RIG‐I (Fig 5A, lane 2); this interaction was no longer detected after 8 h of transfection (Fig 5A, lane 3). This result suggested that the interaction between HDAC6 and RIG‐I is transiently induced by poly(I:C).
Figure 5. HDAC6 interacts with and deacetylates RIG‐I in response to RNA viral infection.
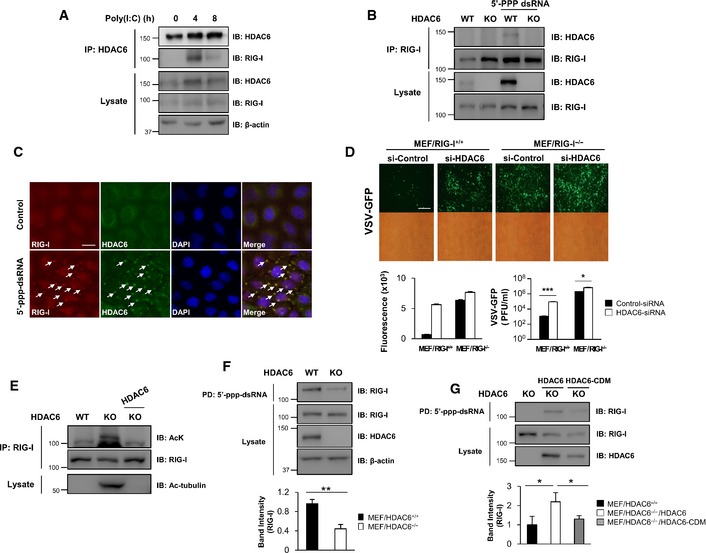
- 293T cells were transfected with poly(I:C) and cell lysates were prepared at the indicated times for co‐immunoprecipitation analysis of endogenous HDAC6 and RIG‐I. Actin was used as the loading control.
- BMDMs were isolated from HDAC6 +/+ and HDAC6 −/− mice and were transfected with 5′ppp‐dsRNA. Whole cell lysates were prepared after 8 h of transfection and co‐immunoprecipitation analysis of RIG‐I and HDAC6 was performed.
- HeLa cells were transfected with 5′ppp‐dsRNA. Cells were stained with anti‐RIG‐I (red) and anti‐HDAC6 H300 (green) antibodies after 8 h of transfection. Nuclei were stained with DAPI (blue) (scale bar, 20 μm). Arrows indicate colocalization of RIG‐I and HDAC6.
- Fluorescence microscopy, green fluorescence absorbance and virus replication at 24 hpi in RIG‐I +/+ and RIG‐I −/− MEFs. Cells were transfected with si‐HDAC6 and si‐control for 36 h, followed by infection with VSV‐GFP (MOI = 1). Scale bar, 100 μm.
- HDAC6 +/+, HDAC6 −/− MEFs, and HDAC6 −/− MEFs reconstituted with HDAC6 wild‐type were subjected to immunoprecipitation using an anti‐RIG‐I antibody and immunoblotted with anti‐acetyl‐lysine and anti‐RIG‐I antibodies. Acetyl‐tubulin was measured in total lysates to determine HDAC6 deacetylase activity.
- Whole cell lysates from HDAC6 +/+ and HDAC6 −/− MEFs were prepared and used in dsRNA pull‐down assays. Pull‐down samples were analyzed by Western blotting with anti‐RIG‐I, anti‐HDAC6, and anti‐β‐actin antibodies. Intensity of the pulled‐down RIG‐I was quantified.
- Whole cell lysates from HDAC6 −/− MEFs and HDAC6 −/− MEFs reconstituted with HDAC6 wild‐type and HDAC6‐CDM were prepared and used in dsRNA pull‐down assays. Samples were analyzed by Western blotting with anti‐RIG‐I and anti‐HDAC6 antibodies. Intensity of the pulled‐down RIG‐I was quantified.
To further confirm the transient interaction between HDAC6 and RIG‐I in response to intracellular double‐stranded RNA, BMDMs from HDAC6 +/+ and HDAC6 −/− mice were transfected with 5′ppp‐dsRNA, a specific ligand for RIG‐I, and analyzed by co‐immunoprecipitation with an anti‐RIG‐I antibody. After 8 h of transfection with 5′ppp‐dsRNA in BMDMs, HDAC6 was co‐immunoprecipitated with RIG‐I (Fig 5B, lane 3). This result indicates that the transient interaction between HDAC6 and RIG‐I in BMDMs is triggered by the intracellular presence of 5′ppp‐dsRNA. And also, we confirmed the co‐localization of HDAC6 and RIG‐I in response to 5′ppp‐dsRNA transfection by immunostaining experiments at 8 h after transfection (Fig 5C). Taken together, these results indicate that HDAC6 interacts with RIG‐I upon RNA virus infection.
Additionally, to investigate the critical correlation between HDAC6 and RIG‐I, knockdown of HDAC6 in RIG‐I +/+ or RIG‐I −/− MEFs was performed using HDAC6‐siRNA (Appendix Fig S9) and infected with VSV‐GFP. As shown in Fig 5D, we found that knockdown of HDAC6 affects for the significant increase of viral replication in RIG‐I +/+ MEFs (about 70‐fold) while RIG‐I −/− MEFs show less changes (about 3‐fold). Our results suggest that antiviral roles of HDAC6 are mainly associated with RIG‐I even though it has still RIG‐I‐independent functions.
Since our results indicate that HDAC6 and RIG‐I transiently interact in response to viral infection (Fig 5A and B) and that the deacetylase activity of HDAC6 is required for inducing innate immune responses against RNA virus infection (Fig 4), we investigated whether RIG‐I is acetylated upon RNA viral infection and whether HDAC6 could deacetylate RIG‐I. As shown in Fig 5E, RIG‐I was acetylated in HDAC6 −/− MEFs and reconstituted HDAC6 successfully deacetylated RIG‐I in HDAC6 −/− MEFs, indicating that HDAC6 is a deacetylase for RIG‐I.
Next, to investigate the functional consequences of HDAC6‐dependent RIG‐I deacetylation, we assessed the effect of hyper‐acetylated RIG‐I on 5′ppp‐dsRNA binding in HDAC6 −/− MEFs. We used 5′ppp‐dsRNA beads to pull down RIG‐I from whole cell lysates of HDAC6 +/+ and HDAC6 −/− MEFs. As shown in Fig 5F, the interaction between RIG‐I and 5′ppp‐dsRNA was significantly reduced in HDAC6 −/− MEFs. Reconstitution of wild‐type HDAC6 in HDAC6 −/− MEFs restored the binding of RIG‐I to 5′ppp‐dsRNA, whereas a catalytically inactive HDAC6 did not (Fig 5G). These results suggested that HDAC6 regulates the binding of RIG‐I to 5′ppp‐dsRNA by deacetylating RIG‐I.
Acetylation of the K909 residue of RIG‐I modulates antiviral innate immune responses
Acetylation of two conserved lysine residue (K858 and K909) in the C‐terminal domain (CTD) of RIG‐I (Appendix Fig S10) was found by mass spectrometry (Choudhary et al, 2009). Interestingly, CTD together with helicase domain of RIG‐I are critical for 5′ppp‐dsRNA binding. We therefore determined whether K858 and K909 acetylation affects the binding of RIG‐I with 5′ppp‐dsRNA. We assessed the binding ability of acetylation‐mimicking (K858Q and/or K909Q) RIG‐I mutants. As shown in Fig 6A, 2KQ (K858Q/K909Q) double‐mutant and K909Q single‐mutant RIG‐I showed complete abolishment of 5′ppp‐dsRNA binding, whereas the K858Q single‐mutant retained 5′ppp‐dsRNA binding ability weakly. By contrast, a K909 acetylation‐resistant RIG‐I mutant (K909R) did not affect the binding to 5′ppp‐dsRNA (Fig 6B). These results suggested that K909 acetylation of RIG‐I regulates the binding of RIG‐I to 5′ppp‐dsRNA. As the supporting result, we have validated acetylation sites in HEK293T cells expressing wild‐type, RIG‐I K858R, RIG‐I K909R or RIG‐I 2KR mutant. After immunoprecipitation and blotting with anti‐acetyl‐lysine antibody, we found that RIG‐I K858R mutant shows similar acetylation level with wild‐type RIG‐I. In contrast, RIG‐I K909R and 2KR mutant showed greater reduction of RIG‐I acetylation, suggesting K909 site could be a major acetylation site of RIG‐I (Appendix Fig S11).
Figure 6. HDAC6‐dependent RIG‐I deacetylation regulates RIG‐I binding to double‐stranded RNA .
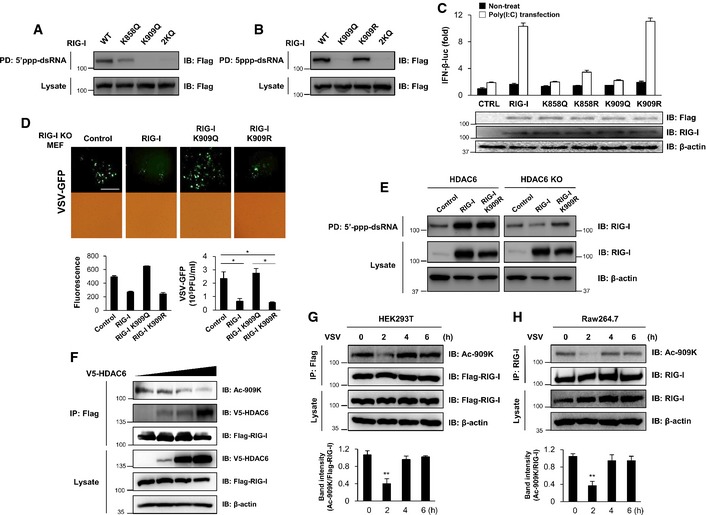
-
A, B293T cells were transfected with the indicated expression plasmids. At 1 day post‐transfection, whole cell lysates were subjected to dsRNA pull‐down assays. Pull‐down samples were analyzed by Western blotting with an anti‐FLAG antibody.
-
CA luciferase assay was performed using 293T cells transfected with the IFN‐β luciferase promoter and TK‐Renilla together with 2 ng of RIG‐I wild‐type, RIG‐I 858Q, RIG‐I 858R, RIG‐I K909Q, or RIG‐I K909R mutant, followed by transfection with poly(I:C) (1 μg/ml) for another 12 h.
-
DFluorescence microscopy, green fluorescence absorbance, and virus replication at 24 hpi in RIG‐I −/− MEFs. Cells were transiently transfected with empty vector, RIG‐I wild‐type, RIG‐I K909Q, or RIG‐I K909R mutant for 24 h, followed by infection with VSV‐GFP (MOI = 1). Scale bar, 100 μm.
-
EHDAC6 −/− MEFs were transfected with the indicated expression plasmids. At 1 day post‐transfection, whole cell lysates were prepared and used in dsRNA pull‐down assays. Pull‐down samples were analyzed by Western blotting with anti‐RIG‐I and anti‐β‐actin antibodies.
-
F293T cells were transfected with 2 μg of FLAG‐tagged RIG‐I and 1, 5, or 10 μg of V5‐tagged HDAC6. At 36 h post‐transfection, whole cell lysates were prepared for co‐immunoprecipitation analysis of V5‐tagged HDAC6 and acetylated K909 (on RIG‐I wild‐type). Samples were analyzed by Western blotting with a K909 acetylation‐specific antibody.
-
G293T cells were transfected with 2 μg of FLAG‐tagged RIG‐I. At 24 h post‐transfection, cells were infected with VSV‐GFP (MOI = 0.1). At 0, 2, 4 and 6 h post‐infection, whole cell lysates were prepared for immunoprecipitation analysis of acetylated K909 (on RIG‐I wild‐type). Samples were analyzed by Western blotting with a K909 acetylation‐specific antibody and intensity of ac‐909K normalized to Flag‐RIG‐I was quantified.
-
HRAW264.7 cells were infected with VSV‐GFP (MOI = 1) for the indicated time and whole cell lysates were prepared for immunoprecipitation analysis of acetylated K909 (on RIG‐I wild‐type). Samples were analyzed by Western blotting with a K909 acetylation‐specific antibody and intensity of ac‐909K normalized to RIG‐I was quantified.
We next investigated whether acetylation‐mimic (K909Q) or acetylation‐resistant (K909R) mutant RIG‐I had an impact on RIG‐I‐dependent antiviral innate immune responses by measuring IFN‐β promoter activation. As shown in Fig 6C, overexpressed RIG‐I wild‐type and acetylation‐resistant mutant (K909R) enhanced activities of the IFN‐β promoter by poly(I:C) stimulation. However, the acetylation‐mimic mutant RIG‐I did not. In addition, we assessed the viral replication of VSV‐GFP after reconstitution of wild‐type RIG‐I, K909Q, or K909R into RIG‐I −/− MEFs. As shown in Fig 6D, wild‐type and acetylation‐resistant (K909R) RIG‐I, which can bind 5′ppp‐dsRNA, significantly inhibited the replication of VSV‐GFP, whereas the acetylation‐mimic K909Q, which cannot bind 5′ppp‐dsRNA, did not inhibit viral replication. These results indicated that deacetylation of the K909 residue of RIG‐I is required for the recognition of 5′ppp‐dsRNA and consequent activation of antiviral innate immune responses against RNA virus infection. To consolidate the hypothesis that HDAC6 deacetylates RIG‐I on K909 residue, which activates CTD of RIG‐I to bind 5′ppp‐dsRNA, we performed 5′ppp‐dsRNA pull‐down assay in HDAC6 +/+ or HDAC6 −/− MEFs expressing wild‐type or acetylation‐resistant RIG‐I mutant (K909R). As expected, wild‐type and RIG‐I K909R mutant were pulled down by 5′ppp‐dsRNA when they are expressed in HDAC6 +/+ MEFs (Fig 6E, left panel). In striking contrast, when they are expressed in HDAC6 −/− MEFs, only acetylation‐resistant mutant RIG‐I K909R partially restores the binding with 5′ppp‐dsRNA (Fig 6E, right panel). These results indicate that the deacetylation of RIG‐I on the lysine 909 residue by HDAC6 stimulates RIG‐I to recognize 5′ppp‐dsRNA.
We also addressed the effect of HDAC6 on RIG‐I‐mediated antiviral signaling by transient transfection of RIG‐I wild‐type and its mutants into HDAC6 +/+ or HDAC6 −/− MEFs. Consistent with Fig 6E, HDAC6 −/− MEFs transfected with RIG‐I K909R mutant showed striking antiviral effect against VSV‐GFP, whereas HDAC6 −/− MEFs transfected with other constructs did not (Fig EV2). Taken together, these results demonstrated that deacetylation of RIG‐I through HDAC6 is essential process in RIG‐I‐mediated antiviral signaling.
Figure EV2. Effect of RIG‐I and RIG‐I mutant in WT and HDAC6 −/− MEFs.
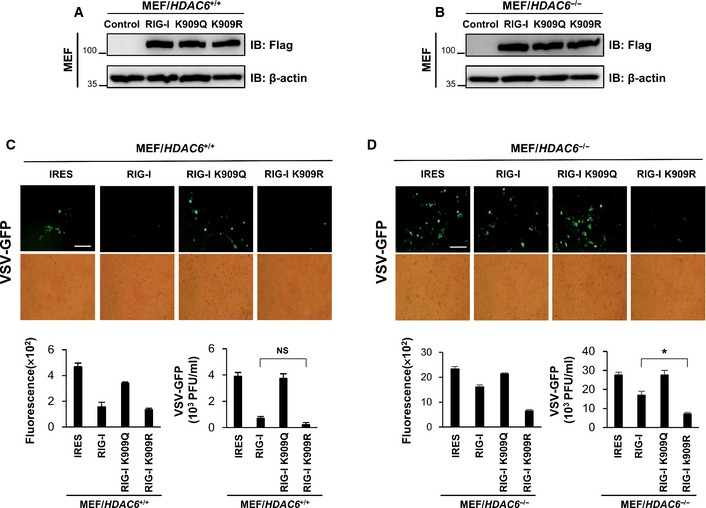
-
A, BHDAC6 +/+ and HDAC6 −/− MEF were transiently transfected with RIG‐I‐IRES‐flag and RIG‐I K909Q, R‐IRES‐flag.
-
C, DFluorescence microscopy, green fluorescence absorbance and virus replication at 24 hpi in WT and HDAC6 −/− MEFs. Cells were transfected with the indicated plasmids for 36 h, followed by infection with VSV‐GFP (MOI = 1). Data are representative of three independent experiments. Error bars, mean ± SD. *P < 0.05 (Student's t‐test). Scale bar, 100 μm.
Source data are available online for this figure.
To further examine acetylation and deacetylation events on RIG‐I K909 mediated by HDAC6 in response to RNA virus infection, we generated the acetyl‐RIG‐I (K909)‐specific antibody. To verify its specificity, FLAG‐tagged constructs of wild‐type, K909R, and K909Q RIG‐I were transfected into 293T cells. Lysates were immunoprecipitated with an anti‐FLAG antibody and detected with the anti‐acetyl‐RIG‐I (K909) antibody. As shown in Fig EV3, anti‐acetyl‐RIG‐I (K909) did not detect the K909R mutant, confirming its specificity. To verify that HDAC6 is a specific deacetylase for RIG‐I on K909, FLAG‐tagged RIG‐I and V5‐tagged HDAC6 were co‐transfected into 293T cells, immunoprecipitated with an anti‐FLAG antibody, and detected with the anti‐acetyl‐RIG‐I (acetylated on K909) antibody. As shown in Fig 6F, HDAC6 deacetylated RIG‐I on K909 in a site‐dependent manner, indicating that HDAC6 is a specific deacetylase for RIG‐I on K909.
Figure EV3. Acetylated K909 lysine‐specific antibody has higher affinity to acetylation‐mimic mutant (K909Q) than acetylation‐resistant mutant (K909R).
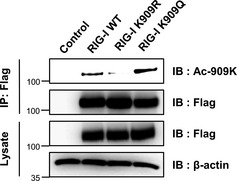
293T cells were transfected with 2 μg of FLAG‐tagged RIG‐I, K909R, K909Q, or empty vector. At 36 h post‐transfection, whole cell lysates were prepared for immunoprecipitation. Samples were analyzed by Western blotting with a K909 acetylation‐specific antibody. Actin was used as the loading control.Source data are available online for this figure.
Next, to investigate RIG‐I acetylation in response to viral infection, we observed acetyl‐RIG‐I on K909 in HEK293T and RAW264.7 after VSV infection. First, after transfection with FLAG‐tagged RIG‐I into 293T cells, VSV‐GFP was infected and immunoprecipitated with anti‐Flag antibody and then detected with the acetyl‐RIG‐I antibody on K909 site. Second, endogenous RIG‐I was immunoprecipitated with anti‐RIG‐I antibody after VSV‐GFP infection and detected with the acetyl‐RIG‐I antibody on K909 site. As shown in Fig 6G and H, the acetylation of RIG‐I on K909 site is decreased at 2 h after infection in HEK293T and RAW264.7 cells. Collectively, these results strongly suggest that acetylation on RIG‐I K909 site is regulated by HDAC6 in response to RNA viral infection.
The structural modeling of acetylation of the K909 residue for RIG‐I/dsRNA complex
Finally, to gain deeper insight of the acetylation of the K909 residue of RIG‐I, 3D modeling program (Discovery Studio 4.0) was used to predict 3D structural changes, which induced by the acetylation of K909. According to the previously published structural data of the RIG‐I CTD domain bound to 5′ppp‐dsRNA (Wang et al, 2010; Luo et al, 2011), the K907 residue forms a hydrogen bond with the phosphate of 5′ppp‐dsRNA, whereas the K909 residue does not directly interact with 5′ppp‐dsRNA (Fig 7A–C). When K909 acetylation is introduced into this 3D structure model, a new hydrogen bond is formed between acetylated K909 and K907, thereby breaking the hydrogen bond between K907 and the phosphate of 5′ppp‐dsRNA (Fig 7B and C). However, deacetylation of K909 by HDAC6 may break the hydrogen bond between K907 and K909 residues, preventing K907 from binding to 5′ppp‐dsRNA. This 3D modeling analysis provides the first structural insight into the changes in RIG‐I induced by K909 deacetylation.
Figure 7. Acetylation of K909 interacts with K907, but K907 does not interact with viral RNA.
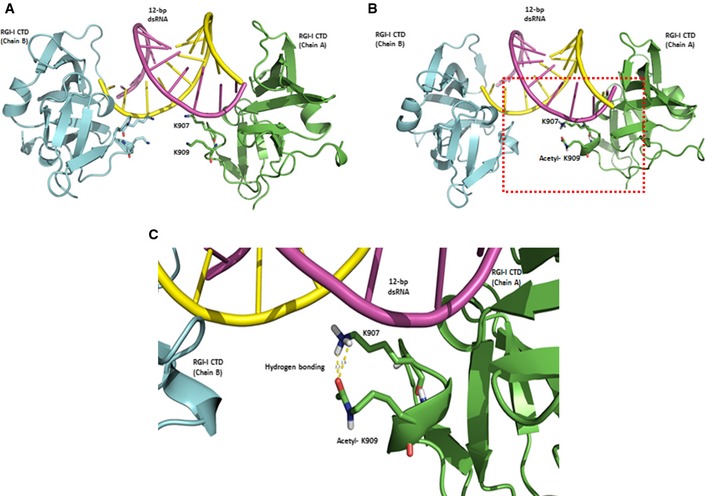
-
AThe structural modeling result of RIG‐I/dsRNA complex and positions of the lysine residue were presented. RIG‐I helicase A and B are shown as green and pale blue ribbons. The dsRNA strands were presented as purple and yellow rods, respectively. Two lysine residues, K907 and K909, on RIG‐I that interact with dsRNA are indicated as violet sticks. K907 forms hydrogen bonding with neither 5′ppp‐dsRNA nor K907.
-
B, CAcetylated K909 and K907 on RIG‐I are indicated as green sticks. There was hydrogen bonding between NH2 of K907 and oxygen of acetyl‐K909.
Discussion
Viral invasion triggers innate immune responses that lead to the activation of IFNs, cytokines, and chemokines to protect the host from viral infection. The ability of the host cells to effectively recognize and detect the viral dsRNA is the most important lines of innate immune defense against virus infection (Kawai & Akira, 2006; Pichlmair & Reis e Sousa, 2007; Kumar et al, 2011). Among the different sensing molecules, RIG‐I is an important pathogen sensor for a variety of RNA viruses in the cytosol. During the antiviral signaling cascade, RIG‐I and its regulatory molecules undergo various posttranslational modifications, including ubiquitination, SUMOylation, and phosphorylation (Gack et al, 2007, 2010; Mi et al, 2010; Oshiumi et al, 2013; Wies et al, 2013), leading to activation of the transcriptional factors, IRF3 and IRF7, as well as NF‐κB, which induce the production of type I IFNs and proinflammatory cytokines (Akira et al, 2006; Kawai & Akira, 2006; Pichlmair & Reis e Sousa, 2007). The regulatory mechanisms involved in RIG‐I signaling, including various posttranslational modifications that control RIG‐I‐mediated antiviral activity, have been well studied (Gack et al, 2010; Mi et al, 2010; Oshiumi et al, 2013; Wies et al, 2013). However, the roles of acetylation and deacetylation of RIG‐I on innate immune responses have not been investigated to date. Especially, the role of RIG‐I acetylation in modulating its binding activity or the recognition of viral RNAs remains to be elucidated.
Here, we provide several lines of evidence suggesting that HDAC6 is an important regulator of RIG‐I‐mediated antiviral innate immune responses. First, HDAC6 −/− mice showed high morbidity after lethal VSV infection due to high rates of viral replication and weak antiviral responses. Second, knockdown or knockout of endogenous HDAC6 in immune cells resulted in reduced RNA virus‐induced production of IFN‐β and proinflammatory cytokines and increased viral replication. Third, overexpression of HDAC6, but not the catalytically inactive mutant, promoted RLR‐mediated inhibition of RNA virus replication. Fourth, HDAC6 interacts with and deacetylates RIG‐I in response to RNA virus infection or RLR stimulation, and deacetylation of RIG‐I by HDAC6 is essential for binding or recognition of 5′ppp‐dsRNA. Finally, HDAC6‐mediated deacetylation of K909 in the CTD of RIG‐I is critical for binding or recognition of 5′ppp‐dsRNA or viral RNAs. Taken together, these findings indicate that HDAC6 is a crucial regulator that induces the binding of RIG‐I to viral RNAs to activate RIG‐I‐mediated antiviral innate immunity.
Recent in vitro and in vivo studies suggest that HDAC6 plays a role in antiviral immune responses (Zhu et al, 2011; Chattopadhyay et al, 2013; Husain & Cheung, 2014). Zhu et al reported that PKCα induces HDAC6‐mediated activation of β‐catenin. Moreover, Chattopadhyay et al showed that HDAC6 −/− mice were highly susceptible to Sendai virus infection. In these studies, it was proposed that β‐catenin is a substrate for HDAC6 important for IRF3‐mediated IFN gene transcription. However, whether other mechanisms are involved in the HDAC6‐mediated regulation of antiviral signaling remains to be determined. To exclude the role of HDAC6 in transcriptional regulation in nuclei, we showed the localization of HDAC6 is mainly in cytoplasm after 5′ppp‐dsRNA transfection instead of nuclear (Fig 5C) and HDAC6 does not affect the level of histone H3 acetylation in HDAC6 KO BMDMs before or after VSV infection (Appendix Fig S12). Additionally, we confirmed β‐catenin‐dependent signaling partially involved in HDAC6‐related antiviral immune response in Figs 5D and EV4. These results strongly suggest that HDAC6 mainly modulates cytoplasmic regulatory machinery to activate antiviral immune responses through RIG‐I and also regulates the antiviral immune responses through β‐catenin partially.
Figure EV4. Effect of knockdown of β‐catenin in control and HDAC6‐overexpressing RAW264.7 cell line against VSV‐GFP infection.
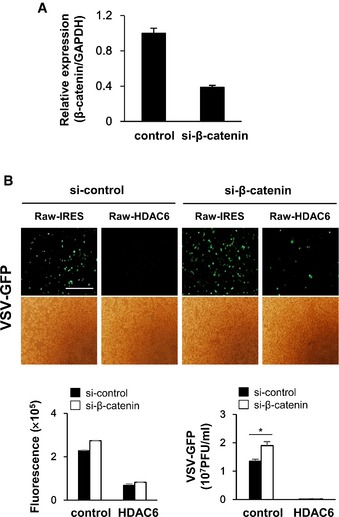
- β‐catenin expression level was decreased in control and HDAC6‐overexpressing Raw264.7 cell line after siRNA transfection.
- Fluorescence microscopy, green fluorescence absorbance and virus replication at 24 hpi in control and HDAC6‐overexpressing RAW 264.7. Cells were transfected with si‐control and si‐β‐catenin for 36 h, followed by infection with VSV‐GFP (MOI = 1). Data are representative of at least three independent experiments. Error bars, mean ± SD. *P < 0.05 (Student's t‐test). Scale bar, 100 μm.
Our in vivo studies showed that HDAC6 −/− mice had higher viral loads and were more vulnerable to killing by VSV‐Indiana than their wild‐type counterparts, suggesting that HDAC6 is critical for antiviral immune defense. The sensitivity of HDAC6 −/− mice to viral killing is likely due to a defect in the production of antiviral molecules such as IFNs and cytokines (Fig 1) (Levy, 2002). When VSV‐GFP or poly(I:C) were injected into HDAC6 +/+ and HDAC6 −/− mice, IFN‐β, and IL‐6 levels in the serum of HDAC6 −/− mice were significantly reduced compared to those in HDAC6 +/+ mice at early time point (Fig 1E and F). Furthermore, IFN‐β luciferase reporter assays in cells expressing HDAC6 showed that HDAC6 significantly enhanced IFN‐β luciferase activity in response to PR8‐GFP infection or treatment of poly(I:C) in a dose‐dependent manner, whereas expression of RIG‐I, 2CARD of RIG‐I, MDA5, and MAVS had no effect (Fig 4D–G and Appendix Fig S8). These results suggest that HDAC6 plays a role in upstream of RIG‐I or RIG‐I molecule during the initial stages of the response against virus infection.
In 2009, Choudhary et al found the acetylation of two conserved lysine residues (K858 and K909) in the CTD of RIG‐I. The CTD of RIG‐I and the helicase domain are recognition sites for viral RNAs containing a 5′‐triphosphate (5′ppp) short double‐stranded structure harboring blunt ends and specific sequence motifs, such as poly‐U/UC (Saito et al, 2007; Takahasi et al, 2008; Kato et al, 2011). Therefore, it is plausible that acetylation and deacetylation of the RIG‐I CTD regulates its binding activity to 5′ppp‐dsRNA to control RIG‐I‐dependent antiviral innate immune responses. Investigation of the mechanisms regulating the acetylation or deacetylation of the RIG‐I CTD resulted in the identification of HDAC6 as a unique cytoplasmic deacetylase with an important role in this process. We showed that the catalytic activity of HDAC6 is indispensable for innate immune responses against RNA viral infection, strongly indicating the existence of HDAC6‐specific substrates (Fig 4). Further assessment of the relationship between HDAC6 and RIG‐I showed that HDAC6 transiently interacts with RIG‐I in response to viral RNA stimulation (Fig 5A–C). Transient nature of the innate immune system molecules including enzymes which can modify the substrates have been well documented in several reviews (Yoneyama et al, 2008; Ma et al, 2013) and RIG‐I‐dependent signaling has been reported as a transient signal (Papon et al, 2009). Moreover, it was predicted that oligomerization of RIG‐I also could be a transient (Louber et al, 2014) and HDAC6 could mediate transient deacetylation of its substrate (Serrador et al, 2004).
In Fig 5E, RIG‐I was hyper‐acetylated in HDAC6 knockout MEFs, and ectopically expressed HDAC6 successfully deacetylated RIG‐I, suggesting that HDAC6 may be a specific deacetylase for RIG‐I. We also showed that deacetylation of RIG‐I by HDAC6 is critical for its ability to bind viral RNA (Fig 5F and G). Taken together, these findings indicate that HDAC6 regulates innate immune responses against RNA virus infection by deacetylating RIG‐I, thereby enabling binding to viral RNA.
To improve our mechanistic understanding, acetylation‐mimic K858Q and K909Q acetylation mutants of RIG‐I was generated and tested their ability to bind viral RNA. As a result, we identified K909 of RIG‐I as a main regulatory acetylation site for viral RNA binding (Fig 6A and B and Appendix Fig S11). Although, our results (Fig 6C) and previous papers (Cui et al, 2008; Papon et al, 2009; Lu et al, 2010; Wang et al, 2010) demonstrated that mutations of K858 inhibits 5′ppp‐dsRNA binding, it remains in vague whether K858 site of RIG‐I is acetylated and what is the exact role of this residue (Appendix Fig S11).
Viral RNA binding and the subsequent innate immune response in RIG‐I −/− MEFs were restored by the deacetylation‐mimic mutant of RIG‐I (K909R), but not by the acetylation‐mimic mutant (K909Q) (Fig 6D). Ectopically expressed wild‐type RIG‐I did not bind 5′ppp‐dsRNA in HDAC6 −/− MEFs, whereas it did bind to 5′ppp‐dsRNA in HDAC6 +/+ MEFs (Fig 6E). Consistently, the acetylation‐resistant mutant (K909R) of RIG‐I partially restored the 5′ppp‐dsRNA binding ability in HDAC6 −/− MEFs (Fig 6E). Although we could not completely exclude the possibility of additional acetylation site of RIG‐I, these results suggested that HDAC6 deacetylates the RIG‐I K909 residue, which is a crucial step for the viral RNA recognition ability of RIG‐I. To further confirm RIG‐I K909 acetylation, we generated an anti‐acetyl K909 RIG‐I‐specific antibody and showed that ectopically expressed HDAC6 deacetylated K909 of RIG‐I (Fig 6F). Taken together, our findings indicate that HDAC6 specifically deacetylates K909 of RIG‐I to activate viral RNA detection.
The crystal structure and NMR spectrum of the RIG‐I CTD in the free state was resolved previously, and a basic RNA binding pocket within the CTD was found to be critical for 5′ppp‐dsRNA recognition (Cui et al, 2008; Takahasi et al, 2008; Lu et al, 2010; Wang et al, 2010). Among the key residues of RIG‐I necessary for binding to 5′ppp‐dsRNA, K907 specifically contributes a hydrogen bond to the phosphate linking nucleosides 2 and 3 or 3 and 4 at the 5′ end of the bound dsRNA. Others have found that mutation of the K907 residue completely abolished 5′ppp‐dsRNA‐induced RIG‐I activation, therefore, phosphate linking via the K907 residue may be critical for the detection of the dsRNA structure (Lu et al, 2010; Wang et al, 2010). Our 3D modeling prediction analysis showed that K907 residue forms a hydrogen bond with the phosphate group of 5′ppp‐dsRNA, whereas the K909 residue did not directly interact with 5′ppp‐dsRNA (Fig 7). However, a new hydrogen bond is formed between acetylated K909 and K907, thereby breaking the hydrogen bond between K907 and the phosphate of 5′ppp‐dsRNA (Fig 7). Based on our analysis, deacetylation of K909 by HDAC6 may break the hydrogen bond between the K907 and K909 residues, preventing K907 from binding to 5′ppp‐dsRNA. However, further studies are needed to investigate the effect of RIG‐I deacetylation on the crystal structure of the complex.
In our present study, the upstream signal molecule which can activate HDAC6 before viral dsRNA recognition by RIG‐I, is still unclear. In the previous study, HDAC6 is known to be phosphorylated by protein kinase C‐alpha (PKCα) so as to regulate the antiviral immune responses upon virus infection (Zhu et al, 2011). PKCα is one of classical calcium activated protein kinases (PKCs) which have been reported to be important for Toll‐like receptor (TLR) signaling (Zhou et al, 2006; Johnson et al, 2007). And recently, it has been reported that the linking virus entry and calcium‐mediated signaling (Collins et al, 2004; Bozym et al, 2010; Cureton et al, 2010; Scherbik & Brinton, 2010). Thus, it is possible that one of PKCs or unknown molecule is activated via calcium‐mediated signaling during virus entry or intracellular trafficking, and can induce the phosphorylation‐mediated HDAC6 activation, which ultimately mediates the deacetylation of RIG‐I for viral RNA detection. However, this plausible hypothesis still awaiting future studies.
In summary, we showed that HDAC6 −/− mice are highly sensitive to RNA virus infection and have a defect in innate immune responses both in vitro and in vivo. Upon RNA virus infection, HDAC6 transiently binds RIG‐I and deacetylates K909, thereby activating the CTD of RIG‐I, enabling it to bind and recognize viral RNAs. These findings suggest a novel mechanism by which HDAC6‐dependent deacetylation positively regulates RIG‐I, thereby facilitating viral RNA recognition by the CTD domain of RIG‐I to induce innate immune responses against RNA virus infection at the early time point of infection.
Materials and Methods
Cell culture and transfection
MEF, HeLa, Vero, RAW264.7 and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; Hyclone), 1% antibiotics/antimycotics (GIBCO) at 37°C with 5% CO2 concentration. U937 cells were cultured in RPMI supplemented with 10% fetal bovine serum (FBS; Hyclone), 1% antibiotics/antimycotics (GIBCO). HEK293T cells were transiently transfected with polyethylenimine (PEI; Polyscience Inc.). Lipofectamine LTX (Life Technologies) and RNAi MAX (Invitrogen) were used for the transfection of poly(I:C), poly(dA:dT), and 5′‐triphosphate double‐stranded RNA (#tlrl‐3pnas, tlrl‐patn, tlrl‐3prna‐100; Invivogen), respectively. MEFs were transfected by NEON transfection system (Life Technologies) according to the manufacturer's instructions. RAW264.7, MEF stable cell lines were generated and maintained using a standard selection protocol with 2 μg/ml of puromycin (GIBCO).
Mice and in vivo experiments
HDAC6−/− mice on a B6.SJL background were kindly given to us by Dr. Yao T.P. (Duke University). All mice were bred in pathogen‐free conditions and genotyped using HelixAmp Direct PCR kit (Nanohelix) before experiments. The conditions for PCR were as follows: 95°C for 5 min, 40 cycles of 95°C for 20 s, 58°C for 30 s, and 72°C for 30 s, followed by 72°C for 5 min (refer to Appendix Table S1 for primer sets).
Sex‐matched (7–8 weeks old) mice were infected with VSV‐Indiana (2 × 108 PFU/mouse) or VSV‐GFP (4 × 108 PFU/mouse), or poly(I:C) (200 μg/mouse) via tail vein injection. After virus infection, mice were monitored daily. Organs and serum were collected at indicated the time point under aseptic conditions to measure the cytokine induction and virus titration. Collected organs were homogenized using TissueLyser instrument (Qiagen) with PBS or RNeasy RNA extraction kit (Qiagen).
Isolation of BMDM and PBMC
BMDMs were isolated from tibia and femur bones. Cells were temporarily incubated with ACK lysing buffer (GIBCO) to lyse red blood cells (RBCs), and cultured in 10‐cm plates with 10% L929 cell conditioned medium. At 4 days after isolation, cells were plated into twelve‐well plates and used in experiments on the following day. PBMCs were isolated from whole peripheral blood using SepMate (Stemcell) with Ficoll‐Paque PREMIUM 1.703 (GE Healthcare) according to the manufacturer's protocol.
Pull‐down assay
The RNA binding assay was carried out as previously described (Oshiumi et al, 2013), with some modifications. To the RNA binding assay, 293T cells were transiently transfected with indicated plasmid DNA using PEI and lysed the following day. The RNA sequences are as follows: (sense strand) AAA CUG AAA GGG AGA AGU GAA AGU G; and (antisense strand) CAC UUU CAC UUC UCC CUU UCA GUU U. Biotin was conjugated at the 3′‐end of the antisense strand. Biotin‐labeled double‐stranded RNA was phosphorylated by T4 polynucleotide kinase (TAKARA). 5′ppp‐dsRNA was incubated for 1 h at room temperature with 14 μg of protein from whole cell lysate. The mixture was added into 10 μl of streptavidin (Cell Signaling), and then rocked at 4°C for 2 h. Beads were washed three times with 100 mM NETN buffer, and analyzed by immunoblotting.
Ethics statement
All animal experiments were managed in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and performed in BSL‐2 and BSL‐3 laboratory facilities with the approval of the Institutional Animal Care and Use Committee of Bioleaders Corporation (Reference number BLS‐ABSL‐14‐010).
Statistical analysis
Determining statistical significance between groups was performed by two‐sided unpaired Student's t‐test and Mann–Whitney test. For mouse survival study, log‐rank test were used to assess statistical significance. Error bars and P‐values are indicated in the figure legends. A P‐value under 0.05 was considered statistically significant.
For more details on the Appendix Supplementary Materials and Methods.
Author contributions
SJC and H‐CL designed and executed biochemical and cell biological experiments, analyzed the data, and wrote the manuscript; SYP, W‐KL, JYM, C‐JK, T‐PY, and JUJ analyzed the data. J‐HK, T‐HK, D‐JJ, J‐EY, Y‐IC, and SK performed the virus infection and HDAC6 mutant experiments. J‐YL and J‐SL designed the overall study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by the Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea (Grant No. 112013032SB010), the Small and Medium Business Administration (S2130867 and S2165234), the Korean Institute of Oriental Medicine by the Ministry of Education, Science and Technology (MEST) (Grant No. K12050), the Korea Healthcare Technology R&D Project funded by the Ministry of Health (Grant No. A103001), National Research Foundation of Korea (Grant No. 2015020957) to J.S. Lee and National Research Foundation of Korea (Grant No. 2013R1A1A2008576 and 2012M3A9C6050087) to J.Y. Lee.
The EMBO Journal (2016) 35: 429–442
References
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783–801 [DOI] [PubMed] [Google Scholar]
- Bozym RA, Morosky SA, Kim KS, Cherry S, Coyne CB (2010) Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog 6: e1001135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Wetzel JL, Sen GC (2013) Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF‐3 are regulated by the acetylation and phosphorylation of its coactivators. MBio 4: e00636–00612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325: 834–840 [DOI] [PubMed] [Google Scholar]
- Collins SE, Noyce RS, Mossman KL (2004) Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 78: 1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, Fujita T, Conzelmann K‐K, Krug A, Hopfner K‐P (2008) The C‐terminal regulatory domain is the RNA 5′‐triphosphate sensor of RIG‐I. Mol Cell 29: 169–179 [DOI] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Whelan SP, Kirchhausen T (2010) The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin‐dependent endocytosis. PLoS Pathog 6: e1001127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU (2007) TRIM25 RING‐finger E3 ubiquitin ligase is essential for RIG‐I‐mediated antiviral activity. Nature 446: 916–920 [DOI] [PubMed] [Google Scholar]
- Gack MU, Nistal‐Villán E, Inn K‐S, García‐Sastre A, Jung JU (2010) Phosphorylation‐mediated negative regulation of RIG‐I antiviral activity. J Virol 84: 3220–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP (2002) HDAC6 is a microtubule‐associated deacetylase. Nature 417: 455–458 [DOI] [PubMed] [Google Scholar]
- Husain M, Cheung CY (2014) Histone deacetylase 6 inhibits influenza a virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J Virol 88: 11229–11239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Albarani V, Nguyen M, Goldman M, Willems F, Aksoy E (2007) Protein kinase Calpha is involved in interferon regulatory factor 3 activation and type I interferon‐beta synthesis. J Biol Chem 282: 15022–15032 [DOI] [PubMed] [Google Scholar]
- Kato H, Takahasi K, Fujita T (2011) RIG‐I‐like receptors: cytoplasmic sensors for non‐self RNA. Immunol Rev 243: 91–98 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol 7: 131–137 [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP (2005) HDAC6 regulates Hsp90 acetylation and chaperone‐dependent activation of glucocorticoid receptor. Mol Cell 18: 601–607 [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S (2011) Structural basis for the activation of innate immune pattern‐recognition receptor RIG‐I by viral RNA. Cell 147: 423–435 [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S (2011) Pathogen recognition by the innate immune system. Int Rev Immunol 30: 16–34 [DOI] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP (2010a) HDAC6 controls autophagosome maturation essential for ubiquitin‐selective quality‐control autophagy. EMBO J 29: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP (2010b) Disease‐causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6‐dependent mitophagy. J Cell Biol 189: 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐Y, Kapur M, Li M, Choi M‐C, Choi S, Kim H‐J, Kim I, Lee E, Taylor JP, Yao T‐P (2014) MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci 127: 4954–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DW, Amarasinghe GK (2012) Structural insights into RNA recognition and activation of RIG‐I‐like receptors. Curr Opin Struct Biol 22: 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE (2002) Whence interferon? Variety in the production of interferon in response to viral infection. J Exp Med 195: F15–F18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M Jr (2011) Immune signaling by RIG‐I‐like receptors. Immunity 34: 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louber J, Kowalinski E, Bloyet L‐M, Brunel J, Cusack S, Gerlier D (2014) RIG‐I self‐oligomerization is either dispensable or very transient for signal transduction. PLoS ONE 9: e108770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Xu H, Ranjith‐Kumar C, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P (2010) The structural basis of 5′ triphosphate double‐stranded RNA recognition by RIG‐I C‐terminal domain. Structure 18: 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucera MB, Tilton CA, Mao H, Dobrowolski C, Tabler CO, Haqqani AA, Karn J, Tilton JC (2014) The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of postentry viral events. J Virol 88: 10803–10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM (2011) Structural insights into RNA recognition by RIG‐I. Cell 147: 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Moore R, Xu X, Barber GN (2013) DDX24 negatively regulates cytosolic RNA‐mediated innate immune signaling. PLoS Pathog 9: e1003721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Z, Fu J, Xiong Y, Tang H (2010) SUMOylation of RIG‐I positively regulates the type I interferon signaling. Protein Cell 1: 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley AJ, Meekings KN, McCarthy C, Shepherd D, Cerundolo V, Mazitschek R, Tanaka Y, Taylor GP, Bangham CR (2006) Histone deacetylase inhibitors increase virus gene expression but decrease CD8 + cell antiviral function in HTLV‐1 infection. Blood 108: 3801–3807 [DOI] [PubMed] [Google Scholar]
- National Research Council (2011) Guide for the Care and Use of Laboratory Animals, 8th edn Washington, DC: The National Academies Press; [Google Scholar]
- Nusinzon I, Horvath CM (2006) Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol 26: 3106–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Matsumoto M, Seya T (2013) A distinct role of Riplet‐mediated K63‐linked polyubiquitination of the RIG‐I repressor domain in human antiviral innate immune responses. PLoS Pathog 9: e1003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, Akira S, Mechti N (2009) The viral RNA recognition sensor RIG‐I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393: 311–318 [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C (2007) Innate recognition of viruses. Immunity 27: 370–383. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo Y‐M, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG‐I and LGP2. Proc Natl Acad Sci 104: 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Cabrero JR, Sancho D, Mittelbrunn Ma, Urzainqui A, Sánchez‐Madrid F (2004) HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity 20: 417–428 [DOI] [PubMed] [Google Scholar]
- Scherbik SV, Brinton MA (2010) Virus‐induced Ca2+ influx extends survival of west nile virus‐infected cells. J Virol 84: 8721–8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M Jr, Inagaki F, Fujita T (2008) Nonself RNA‐sensing mechanism of RIG‐I helicase and activation of antiviral immune responses. Mol Cell 29: 428–440 [DOI] [PubMed] [Google Scholar]
- Valenzuela‐Fernandez A, Alvarez S, Gordon‐Alonso M, Barrero M, Ursa A, Cabrero JR, Fernandez G, Naranjo‐Suarez S, Yanez‐Mo M, Serrador JM, Munoz‐Fernandez MA, Sanchez‐Madrid F (2005) Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell 16: 5445–5454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, Hartmann G, Patel DJ (2010) Structural and functional insights into 5'‐ppp RNA pattern recognition by the innate immune receptor RIG‐I. Nat Struct Mol Biol 17: 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU (2013) Dephosphorylation of the RNA sensors RIG‐I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38: 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T (2005) Shared and unique functions of the DExD/H‐box helicases RIG‐I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175: 2851–2858 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Onomoto K, Fujita T (2008) Cytoplasmic recognition of RNA. Adv Drug Deliv Rev 60: 841–846 [DOI] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas‐Burgos A, Koomen J, Olashaw N, Parsons JT, Yang X‐J, Dent SR (2007) HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell 27: 197–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yang W, Li J (2006) Ca2 + ‐ and protein kinase C‐dependent signaling pathway for nuclear factor‐kappaB activation, inducible nitric‐oxide synthase expression, and tumor necrosis factor‐alpha production in lipopolysaccharide‐stimulated rat peritoneal macrophages. J Biol Chem 281: 31337–31347 [DOI] [PubMed] [Google Scholar]
- Zhu J, Coyne CB, Sarkar SN (2011) PKC alpha regulates Sendai virus‐mediated interferon induction through HDAC6 and β‐catenin. EMBO J 30: 4838–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6