

Summary
The crystallizable fragment (Fc) of the immunoglobulin class G (IgG) is a very attractive scaffold for the design of novel therapeutics due to its quality of uniting all essential antibody functions. This article reviews the functionalization of this homodimeric glycoprotein by diversification of structural loops of CH3 domains for the design of Fcabs, i.e. antigen‐binding Fc proteins. It reports the design of libraries for the selection of nanomolar binders with wildtype‐like in vivo half‐life and correlation of Fc receptor binding and ADCC. The in vitro and preclinical biological activity of selected Fcabs is compared with that of clinically approved antibodies. Recently, the great potential of the scaffold for the development of therapeutics for clinical use has been shown when the HER2‐binding Fcab FS102 entered clinical phase I. Furthermore, methods for the engineering of biophysical properties of Fcabs applicable to proteins in general are presented as well as the different approaches in the design of heterodimeric Fc‐based scaffolds used in the generation of bispecific monoclonal antibodies. Finally, this work critically analyzes and compares the various efforts in the design of highly diverse and functional libraries that have been made in the engineering of IgG1‐Fc and structurally similar scaffolds.
Keywords: IgG1‐Fc, Antibody engineering, Library design, Fcab
This article is part of a series of reviews covering Immunoglobulins: from genes to therapies appearing in Volume 270 of Immunological Reviews.
Introduction
Monoclonal antibodies (mAbs) undoubtedly belong to the most prominent therapeutics of the last two decades. Since 1986, more than 40 mAbs have been approved by the Food and Drug Administration (FDA) for the use in various diseases, including cancer, immune disorders, and infectious diseases, and many more are in clinical trials, with full‐size IgG still representing the majority of these approved mAbs. However, based on the success of these drugs, several different antigen‐binding formats such as antibody fragments or domains (i.e. antibody‐based scaffolds) and non‐antibody protein scaffolds have been investigated for their potential as binding molecules. The postulated advantage of such molecules is the reduced size that potentially leads to enhanced tissue penetration and facilitated production.
For example, three full‐size antigen‐binding fragments (Fab) of IgG have been approved by the FDA for clinical use so far [certolizumab pegol 1, ranibizumab 2, and abciximab 3]. At a considerable smaller size, single‐chain variable fragments (scFv) comprise the variable domains of heavy and light chain connected by a polypeptide linker and therefore contain the complete binding sites of antibodies. It has been reported that scFvs can be readily expressed in various hosts like bacteria, yeasts, and plants 4, 5, 6. An advancement of this technology was the construction of bispecific T‐cell engagers [BiTEs 7], comprising two scFvs with specificities for CD3 and a target antigen expressed as one polypeptide chain. Dual‐affinity re‐targeting molecules (DARTs) follow a similar principle: As a further development of the diabody technology 8, a C‐terminal disulfide bond covalently links two scFvs that are expressed as separate polypeptides to generate a more stable bispecific molecule 9. Xiao et al. reported the selection of molecules based on the isolated CH2 domain of IgG1 binding to the HIV‐1 envelope glycoprotein 10. Another concept uses nanobodies, i.e. the single‐domain antigen‐binding fragments of camelid heavy chain antibodies, which, for example, have the potential for application in oral immunotherapy due to the increased resistance to extreme pH and proteolytic digest as compared to conventional antibodies 11, 12. Examples for non‐antibody‐binding scaffolds comprise, e.g. designed ankyrin repeat proteins (DARPins). DARPins are small, single‐domain proteins derived from natural repeat proteins that can be engineered to bind diverse antigens 13. And finally, the immunoglobulin‐like structure of the tenth type III unit of human fibronectin (10Fn3) has served as the basis for the engineering of one further novel binding scaffold 14.
The potential therapeutic value of these formats is, however, largely reduced due to the absence of the crystallizable fragment (Fc) and the resulting inability to trigger effector functions such as antibody‐dependent cellular cytotoxicity (ADCC) or complement‐dependent cytotoxicity (CDC). Accordingly, the in vivo half‐life is limited due to the absence of the Fc‐located binding site of the neonatal Fc receptor FcRn, and a lot of effort is necessary to overcome these drawbacks, which to some degree outweighs the advantage of the smaller size.
In 2009, Rüker and Wozniak‐Knopp 15 reported the engineering of the structural loops of immunoglobulin constant domains to generate novel binding sites (Modular Antibody Engineering). Based on the observation that immunoglobulin‐like domains are structurally conserved in the β sandwich core regions while at the same time exhibiting high variability of the loops 16, the three C‐terminal loops of the CH3 domain of IgG1‐Fc were engineered to bind diverse antigens in initial studies, yielding antigen‐binding Fc fragments termed ‘Fcabs’. In this review, the therapeutic potential of such Fcabs and efforts in functional engineering as well as the engineering of biophysical properties will be discussed. There will be no focus on the engineering of individual antibody domains like monomeric CH2 or CH3 domains as scaffolds for the design of novel binders, as this topic has been reviewed recently 17.
Structure of IgG‐Fc
The structure and function of human IgG‐Fc has been described extensively. However, it is necessary to provide the background for discussion in the present review. Immunoglobulins of isotype G are the predominant antibody class in circulation and comprise two identical light (L) and heavy (H) chains forming a ‘Y‐shaped’ structure 18. An IgG molecule can be dissected into two distinct fragments (Fab, Fc) that are responsible for the in vivo properties (Fig. 1): An antigen‐binding fragment (Fab) is a heterodimeric protein composed of one light chain and the N‐terminal half of one heavy chain. It encompasses two variable (V) domains (VH, VL) whose complementarity‐determining regions (CDRs) form the antigen‐binding site, and two constant (C) domains (CL, CH1) (Fig. 1). By contrast, the crystallizable fragment (Fc) is a homodimeric glycoprotein with one monomer consisting of two constant immunoglobulin domains (CH2, CH3) from the C‐terminal half of one heavy chain (Fig. 1). Through interaction with its ligands [Fcγ receptors (FcγR), C1q and FcRn] it mediates various immune effector functions and increases the half‐life of the antibody molecule 19. The Fc and the two Fab fragments meet at a central flexible segment, the hinge region, which plays an important role in the mediation between antigen recognition and effector functions and forms interchain disulfide bonds between the two heavy chains. X‐ray crystallography of IgG1‐Fc comprising amino acids 216–446 20 (Fig. 1) normally reveals structure for residues 238–443, underlining that the hinge region is highly flexible 21, 22, 23, 24. In addition, the conformation of the hinge proximal region of the CH2 domain is shown to be ‘soft’, i.e. to be relatively mobile in comparison to the more defined structure observed at the CH2–CH3 interface.
Figure 1.
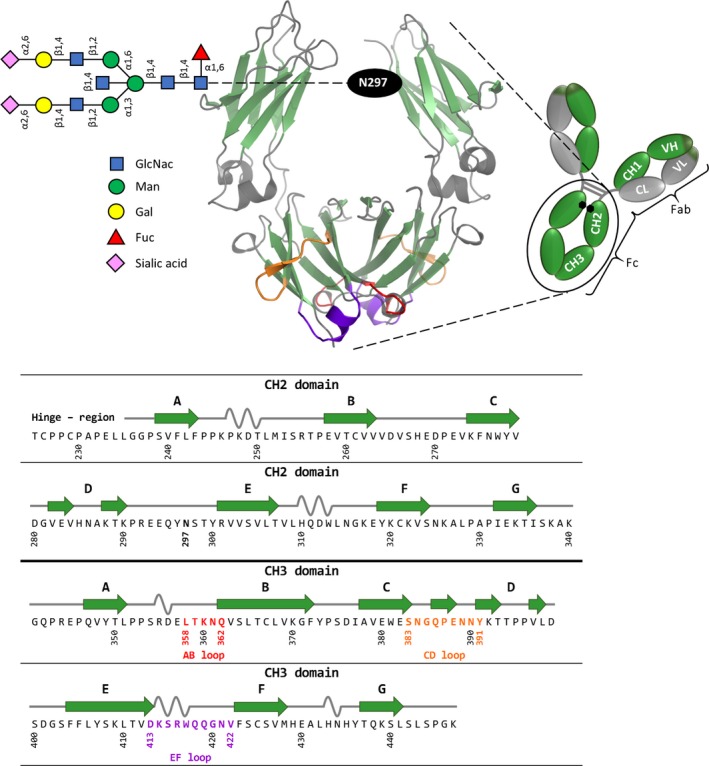
Schematic representation of homodimeric human IgG1‐Fc (PDB‐ID 1OQO), generated using PyMOL, with the corresponding amino acid sequence. IgG1 is composed of two heavy and two light chains, with the Fab region carrying the antigen‐binding sites and the Fc part mediating various effector functions. The homodimeric Fc part comprises the CH2 and CH3 domains, where β strands are depicted in green, and α helices and random coils are shown in gray. The C‐terminal loops of the CH3 domain are colored in red (AB loop), orange (CD loop), and purple (EF loop). The asparagine at position 297 carries the glycan (NaNaFbi), graphically represented according to Anthony et al. 44. The amino acid sequence of the CH2 and CH3 domain of wildtype IgG1‐Fc is shown and numbered according to Eu numbering system 20. On top of the sequence, the secondary structure elements of CH2 and CH3 according to the crystal structure (PDB‐ID 1OQO) are shown.
Four distinct IgG subclasses differing in their heavy chains (and consequently in their Fc), exist in humans (hIgG1‐4), and in mice (mIgG1, 2a, 2b, 3). This review will mainly focus on the structure–function relationships of the Fc of human IgG1 as a basis for engineering strategies for the design of novel therapeutic proteins. In the following, IgG‐Fc and IgG1‐Fc will always mean the basic homodimeric scaffold including two monomers, each being composed of the N‐terminal hinge region followed by CH2 and CH3 domains (i.e. Thr225 to Lys446; for amino acid numbering and assignment of secondary structures, see Fig. 1).
A single N‐linked glycan is attached to asparagine 297 (CH2 domain) of each heavy chain. The glycan has a complex biantennary structure (Fig. 1) and can vary by the addition of sugar residues to specific parts of the core structure. The latter is composed of N‐acetylglucosamine and mannose and can be modified by the addition of fucose, bisecting N‐acetylglucosamine, and two arms defined by α1,3 and α1,6 mannose linkages (Fig. 1). The arms can be extended by the addition of galactose and sialic acid. There is a tremendous heterogeneity in the IgG‐Fc glycan, with over 30 distinct glycans detected on circulating IgG in healthy individuals 25. The two Fc glycans are essential for the structural integrity of Fc (and IgG) by contributing mainly to the interface of the CH2 domains, where they face the center of the Fc with the α1,3 arm protruding into the cavity between the heavy chains and the α1,6 arm extending along the heavy chain backbone 26, 27. Crystal structures of IgG‐Fc reveal a distinct conformation for the oligosaccharide resulting from multiple non‐covalent interactions with the protein 23. This was confirmed by molecular dynamics (MD) simulations demonstrating that the glycans form more hydrogen bonds with the individual protein chains at the cost of glycan–glycan interactions 28.
The thermodynamic parameters (obtained by differential scanning calorimetry) describing the unfolding of IgG1‐Fc reflect these interactions, with the thermal unfolding of the CH2 domains showing a progressive reduction in stability with loss of sugar interactions while the unfolding of the CH3 domain is unaffected 29. Typically, IgG1‐Fc exhibits two thermal transitions (best modeled using two sequential two‐state transitions) with the lower temperature transition (Tm1 approximately 71°C) reflecting unfolding of the CH2 domain and the second transition (T m2 approximately 82°C) unfolding of the CH3 domain at pH 7.4 30. The latter underlines the importance of the extensive hydrophobic CH3–CH3 interface for the stabilization of IgG1‐Fc. During unfolding, the CH2 domains behave as a single cooperative unit, and unfolding is reversible as long as the protein sample is taken to approximately 75°C at maximum, whereas unfolding of the CH3 domains is irreversible. Typically, the thermal stability of the Fc fragment decreases with decreasing pH as demonstrated experimentally 31 and by computational means 28.
Based on the importance of glycosylation for the conformational and thermal stability of IgG1‐Fc, yeast surface display has been the method of choice to screen IgG1‐Fc libraries for variants with desired properties 32. The advantage of yeast display over phage, bacterial, and ribosome display is the existence of a eukaryotic protein production machinery that is necessary for post‐translational modifications (including glycosylation) and quality control of expressed proteins in the endoplasmic reticulum (ER) and the Golgi complex of eukaryotes 33. Also, in contrast to phage and ribosome display, yeast display selections usually include one or more flow cytometric sorting steps, facilitating quantitative analysis during selection. In addition, normalization for protein expression is possible by including expression tags 34.
Natural binding partners of IgG1‐Fc
The most obvious advantage of IgG‐Fc over other antibody‐derived or non‐antibody binding scaffolds is its quality of combining all essential antibody functions except for antigen recognition, including binding to FcγRs. These may be expressed constitutively on hematopoietic cells (e.g. macrophages, eosinophils, neutrophils, natural killer cells, lymphocytes) and other tissues and may be induced or upregulated, differentially, on each cell type when exposed to cytokines or other activating agents 21. Stimulation of cells through FcγRs may result in the activation or deactivation of one or more of a variety of effector functions, including ADCC, CDC, phagocytosis, oxidative burst, release of inflammatory mediators, etc. 35. Due to the high sequence homology between the FcγR types and between the subclasses of IgG, spatial homology for interaction sites is found. In general, binding sites are located at the lower hinge and adjacent regions in the CH2 domain 36, 37 (Fig. 2).
Figure 2.
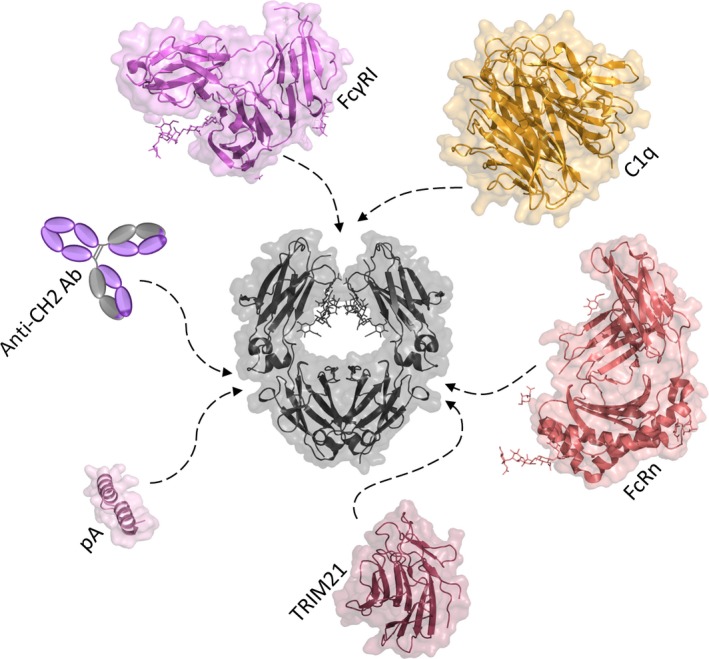
Representation of secondary structural elements with transparent molecular surface of IgG1‐Fc and its binding ligands. IgG1‐Fc (PDB‐ID 1OQO) is colored in gray. Glycan structures are depicted as lines. Dashed arrows mark the binding site of the neonatal Fc receptor [PDB‐ID 1I1A 50]; Fc gamma receptor I [PDB‐ID 4W4O 119]; the mini‐Z domain of protein A (PDB‐ID 1OQO); a globular head of the complement system protein C1q [PDB‐ID 1PK6 47]; the C‐terminal PRYSPRY domain of TRIM21 [PDB‐ID 2IWG 120]; and the anti‐CH2 antibody (clone MK 1 A6, AbD Serotec). The epitope of C1q was obtained by docking studies combined with MD simulations by Schneider et al. 121. Traxlmayr et al. 86 defined the binding site of the anti‐CH2 antibody to be located at the C‐terminal part of the CH2 domain. The proportions regarding size of all shown crystal structures are true to scale.
A dynamic model of Fc–FcγR recognition was proposed in which oligosaccharide/protein interactions within the Fc protein generate an equilibrium population of conformers, with distinct structures that recognize and bind individual ligands 21. Besides contribution to the conformational and thermal stability, the two glycan chains of Fc are an absolute requirement for binding of Fc (and IgG) to FcγRs, as this interaction is lost after deglycosylation 27. The Fc glycan apparently maintains an open conformation of the Fc heavy chains required for interaction with FcγRs as supported by the structure of aglycosylated Fc 38. In the latter, the two heavy chains arrange in a closed conformation and therefore lack the FcγR‐binding pocket. However, it was demonstrated that mutations in the Fc backbone can alter the overall structure of aglycosylated Fc and restore binding to FcγRs to some extent, suggesting that the Fc glycan chains primarily affect protein–protein interactions by altering the Fc backbone conformation 39. The interplay between the primary and secondary N‐acetylglucosamine residues with the protein structure seems to be particularly critical for FcγR recognition 40.
The human FcγRI (CD64) is expressed on monocytes and macrophages and a number of myeloid cell lines and binds to IgG1 and IgG3 with similar affinity at the lower hinge region around residues 234–238 and residues in the hinge proximal region of the CH2 domain 41, 42. In humans, two forms of FcγRII (CD32) are found, namely FcγRIIa and FcγRIIb. Both are widely expressed on multiple cell types, constitutively and/or following induction or upregulation 43 and bind to IgG1 and IgG3 with similar affinity. While their external domains are highly homologous they transduce opposite signals (activating versus inhibitory) via their intracytoplasmatic domains. The binding site at the Fc is similar to that of FcγRI 21. Finally, FcγRIII (CD16) is a low‐affinity receptor either expressed as an intrinsic (FcγRIIIa) or glycosphingolipid‐linked protein (FcγRIIIb). Binding of natural killer (NK) cell‐expressed FcγRIIIa to the lower hinge region activates ADCC 37.
As mentioned above there is some variation in composition of IgG‐Fc glycans in vivo and this may directly contribute to modulation of interaction (and affinity) with individual FcγRs classes, thereby mediating activating, inhibitory, or anti‐inflammatory processes. Specific glycan forms have been associated with distinct immunological milieus 44. For example, increase in fucosylation and decrease in sialylation and galactosylation on the Fc glycan were observed during inflammatory conditions 45. There is an ongoing discussion whether these modifications are related to the expression of glycan‐modifying enzymes like glycosyltransferases or whether other regulatory mechanisms are involved. Further studies are necessary to dissect the regulation of antibodies in vivo and this knowledge will also lead to the design and production of more efficient (glycoengineered) therapeutics, i.e. full‐size mAbs or Fcabs, respectively.
Fig. 2 illustrates that C1q, the recognition subunit of C1 (i.e. the complex triggering activation of the classical pathway of complement) also binds at the N‐termini of the CH2 domains. Similar to FcγRs, binding depends on the presence and mode of glycosylation at Asn297. It includes acidic and basic residues at CH2 but might also involve in vivo interaction with the CL domains of the Fab arms 46, 47. There are six heads on C1q, connected by collagen‐like stems to a central stalk, and the isolated heads bind to the Fc rather weakly. Recently, it has been shown that antigen‐binding on cell surfaces can facilitate the formation of IgG‐hexamers and that these IgG‐hexamers engage the headgroups of C1q 48. The resulting avidity effect increases the apparent affinity for C1q and triggers complement lysis. The IgG‐hexamers are formed by non‐covalent Fc–Fc interactions involving residues I253, H433, and N434. Mutation of these residues strongly reduces complement activation. Moreover, the authors also defined mutations in the IgG‐Fc molecule that increased the formation of hexamers and thus resulted in improved activation of CDC. Thus, this study not only defined the molecular mechanism that triggers the classical pathway of complement but it also enabled the construction of Fc‐mutants that activate the complement system more potently.
As mentioned above, one of the advantages of IgG‐Fc‐based therapeutic antibody fragments is the presence of a natural binding site for the neonatal Fc receptor FcRn (Fig. 2). FcRn mediates the transport of maternal IgG across the placenta in humans, thereby conferring humoral immunity to the fetus against antigens encountered by the mother 49. In addition, FcRn binds IgG with nanomolar affinity at acidic pH (≤6.5) in intracellular vesicles and releases it upon encountering the basic pH of the bloodstream (7.4). This recycling process prevents lysosomal degradation, ultimately resulting in prolonged serum half‐life. The FcRn/Fc‐binding interface spans a large surface area at the CH2–CH3 interdomain region. The center of the FcRn/Fc interface includes a hydrophobic core with surrounding salt bridges. At the Fc, the interface encompasses residues in the AB loop and the EF loop of the CH2 domain, as well as the G‐strand of the CH3 domain 50.
Another highly specific receptor binding to the IgG‐Fc region via its C‐terminal PRYSPRY domain (Fig. 2) is the tripartite motif‐containing 21 (TRIM21) protein. This cytosolic receptor recognizes antibody‐opsonized pathogens and targets them to the proteasome through auto‐ubiquitylation of E3 ubiquitin ligase. The neutralization mechanism of TRIM21 is thought to link the adaptive immune system with intracellular defense 51, 52.
Numerous other ligands bind to IgG‐Fc, including proteins produced by microorganisms. The most prominent example is Staphylococcus aureus Protein A that binds with high affinity at the CH2–CH3 interface of IgG1 and IgG2 and is used for purification of mAb formats containing Fc (e.g. Fcabs).
Generation of novel and improvement of existing functions in IgG1‐Fc
As outlined above, binding of ligands to IgG‐Fc involves the N‐termini of the CH2 domains as well as the CH2‐CH3 interface (Fig. 2), whereas the C‐termini of the CH3 domains are not affected. Each CH3 domain provides three C‐terminal (structural) loops that can be diversified for the generation of novel antigen‐binding sites: residues 358 to 362 (AB loop), residues 383 to 391 (CD loop) and residues 413 to 422 (EF loop) (Fig. 1). These loops correspond to regions with the most pronounced flexibility within IgG1‐Fc 28. Upon engineering the loops, Fcabs, homodimeric Fc proteins with two potential antigen‐binding sites in close vicinity, can be generated.
Wozniak‐Knopp et al. 53 were the first to report the incorporation of antigen‐binding properties in human IgG1‐Fc. Five residues in each the AB loop (358–362) and the EF loop (413–415, 418–419) of the CH3 domain (compare with Fig. 1) were randomized using NNB degenerate codons and five additional random residues were inserted at position 415 to enlarge the potential binding surface. A 7.4 × 107 yeast surface display library was constructed and probed for binding to Protein A and FcγRI for comparison with surface‐displayed wildtype IgG1‐Fc. It was shown that a considerable amount of clones retained the binding to these proteins, suggesting structural integrity of the displayed molecules. Fc fragments binding to the extracellular domain of HER2 (an oncoprotein of the ERBB receptor family) were selected by fluorescence‐activated cell sorting (FACS) using decreasing concentrations of the antigen. Next, the Fcab exhibiting the highest binding affinity was matured, yielding the final clone H10‐03‐6, for which specific binding to HER2 at a K D value of 8.6 nM could be determined by surface plasmon resonance (SPR) spectroscopy. Moreover, H10‐03‐6 was shown to elicit ADCC in an experiment involving a HER2‐expressing cell line and primary human NK cells to an extent that was approximately 20‐fold lower than for trastuzumab, a monoclonal antibody binding to HER2 which is applied in the treatment of certain breast cancers 54. Importantly, no adverse effect of loop engineering on the in vivo half‐life of the molecule could be determined after injection of H10‐03‐6 or wildtype IgG1‐Fc in BALB/c mice.
The correlation of FcγRIIIa‐binding affinity and ADCC in Fcabs was further investigated by Kainer et al., who performed mutational studies to modulate ADCC potency. In a similar experiment as described above, HER2 overexpressing cells and NK cells were mixed and treated with variants of a HER2‐binding Fc fragment carrying mutations that were previously reported to affect the affinity for FcγRIIIa. The authors concluded that known effects of the affinity‐potency correlation can be assumed for the Fcab format 55.
A recently published study describes a more elaborate investigation of the in vivo and in vitro activity of H10‐03‐6 with a special emphasis on the comparison with the clinically approved anti‐HER2 antibody trastuzumab 56. Complementing the work described above, the authors demonstrated the simultaneous binding of FcγRIIIa and HER2 to H10‐03‐6 in an SPR‐spectroscopic experiment as well as binding of FcγRIIIa to H10‐03‐6 on the surface of HER2‐expressing SKBr3 cells. It was demonstrated that the epitopes that are recognized by trastuzumab and H10‐03‐6 overlap. Interestingly, a human tumor cell proliferation assay revealed that, in contrast to trastuzumab, H10‐03‐6 is not able to inhibit proliferation in a dose‐dependent manner. However, when a preclinical in vivo tumor xenotransplant model using human HER2 expressing BT‐474 cells was used to confirm tumor‐killing via ADCC, a significant retardation of tumor growth could be determined which provided proof of the biological activity of the Fcab despite its obvious differences in functionality compared to trastuzumab.
A complementary study on the ADCC potency of H10‐03‐6 was published by Jez et al. 57, who expressed the Fcab in human cells as well as wildtype and glycoengineered plants to generate four different glycoforms of H10‐03‐6. The authors describe the crucial importance of the glycosylation pattern on ADCC activity, thereby confirming the applicability of concepts reported for full‐size IgG to the Fcab format.
Recently, the discovery and preclinical activity of a novel HER2‐targeting Fcab, FS102, was reported 58. Residues 358–362 and 413–419 in the AB and EF loops, respectively, were randomized to construct a yeast surface display library, which was screened for binding to the extracellular domain of HER2. The structural integrity of specific clones was confirmed by their uncompromised binding to anti‐CH2, FcγRI, and Protein A. One resulting Fcab, FS102, contained a total of nine amino acid substitutions in both loops (compared to wildtype IgG1‐Fc) but exhibited similar biophysical properties, meaning no major structural deviation between the two molecules. FS102 binds HER2 with high affinity comparable to that of trastuzumab and pertuzumab, but does not compete with either mAb in binding to the receptor, suggesting that it is targeting a different epitope. Interestingly, FS102 induces profound HER2 internalization and degradation and, finally, tumor cell apoptosis, which is an important mode of action for antibody therapeutics. The antitumor effect of FS102 in patient‐derived xenografts correlated strongly with the HER2 amplification status of the tumors. At gene copy numbers of >10 HER2 per cell, superior activity of FS102 over trastuzumab or the combination of trastuzumab and pertuzumab was observed both in vitro and in vivo, and FS102 induced complete and sustained tumor regression in a significant portion of HER2‐high patient‐derived xenograft tumor models. The mode of action of FS102 is still under discussion but might be related to the structure of the Fcab and its two potential HER2‐binding sites that are close together (20–40 Å) and relatively inflexible compared to the two typical IgG antigen‐binding sites (120–170 Å). It is possible that the Fcab favors a more ordered and tightly packed interaction with the antigen and thereby initiates pronounced aggregation and internalization of the Fcab/antigen complexes. Binding of the Fcab might also favor a conformation of HER2 that is more susceptible to degradation.
Aside from the successful introduction of novel binding sites into IgG1‐Fc, it was demonstrated that the C‐terminal loops in the CH3 domains can also be engineered to generate pH‐dependent binding between the Fcab and the respective antigen. As serum half‐life controlled by FcRn is a major parameter to be considered for the applicability of therapeutic antibodies, engineering pH‐sensitivity into the interaction of antibodies with their targets may also increase the clinical potential of these molecules. Increasing the affinity to the antigen in the plasma (pH 7.4) while simultaneously decreasing the interactions at acidic pH potentially reduces antigen‐mediated clearance in the lysosome and therefore allows for administration of therapeutic antibodies at lower frequencies and doses. It was demonstrated that prolonged half‐life of tocilizumab, a humanized antibody against IL‐6 receptor, and antibodies binding to proprotein convertase subtilisin kexin type 9 can be engineered by subjecting several residues within the CDR loops to histidine scanning. The authors suggested that the antibody–antigen complex dissociates at endosomal pH and the antibody is salvaged from degradation by binding to FcRn which recycles it back to the cell surface 59, 60.
To apply this concept to IgG1‐Fc, the HER2‐binding variant H10‐03‐6 was used as model Fcab and a library was constructed by applying parsimonious mutagenesis to the regions coding for the binding loops of H10‐03‐6 in order to generate pH‐dependent binding sites. The resulting yeast surface display library was subjected to alternating selections for binding at pH 7.4 and non‐binding at pH 6.0 61. Fcab variants could be selected whose interaction with HER2 was pH‐dependent not only in the yeast display format but also when HER2‐positive SKBr3 cells were titrated with soluble Fcabs. Importantly, this effect was not caused by conformational changes, as shown by DSC and MD simulations. Only one variant contained a single His‐substitution, but all selected mutations were in the close proximity of existing histidines. The application of a surface display method for selections of pH‐dependent binders in order to circumvent the labor‐intensive histidine scanning approach was also demonstrated in a very recent study by Bonvin et al. 62. Here, scFv variants binding to the chemokine CXCL10 and enriched in histidine residues in the CDR‐H3 were selected de novo from a phage library. Based on the selected lead clone, optimized libraries were constructed. When reformatted into human IgG1, the isolated antibody 1A4 inhibited CXCL10‐induced chemotaxis with an IC50 of 1.6 nM. In addition, the strong pH‐dependency was evidenced by performing a dose–response ELISA, where 1A4 exhibited a 167‐fold lower affinity to CXCL10 at pH 6.0 than at pH 7.4.
Of course, not only can IgG1‐Fc be engineered to obtain novel functionalities but also to improve existing functions. One example is the enhancement of the intrinsic property of ADCC elicitation. For instance, Lazar et al. 63 combined algorithms for computational prediction and high throughput screening methods to selectively optimize the affinity and specificity for FcγRs. Designed Fc variants of the HER2‐binding monoclonal antibody trastuzumab exhibited ADCC enhancements over wildtype with the enhancement levels being proportional to the increase in affinity for FcγIIIa. Importantly, ADCC was observed for the engineered variants even with a cell line whose low surface levels of HER2 did not allow for ADCC detection after using wildtype trastuzumab. The authors concluded that especially for antibodies that fail in inhibiting proliferation, enhanced engagement of the immune system by Fc engineering and thus mediated killing, i.e. by ADCC and CDC, could prove to be important.
Another very promising and constantly growing field in which the engineering of IgG1‐Fc plays an important role is the generation of bispecific antibodies (bsAbs), which are able to simultaneously target two different antigens while at the same time maintaining important functions of a mAb. A number of different bispecific formats are currently in clinical trials or already approved for cancer therapy, with catumaxomab (Fresenius Biotech), a mouse IgG2a and rat IgG2b hybrid antibody combining binding to CD3 and EpCAM that was approved in 2009, being a prominent example 64. Two other bsAbs include RG7221 (Roche, Basel, Switzerland), a clinical phase II bsAb designed to bind Angiopoietin 2 and VEGF‐A, and LY3164530 bsAb (Eli Lilly, Indianapolis, IN, USA), currently being evaluated in clinical phase I and comprising binding sites for HER1 and c‐Met 65, 66, 67, 68.
The technologies proposed for the construction of the latter two molecules use different strategies to solve the light chain association problem. The CrossMAbCH1‐CL technology from Roche ensures correct pairing of the light chains by rearranging the CH1 domain of one heavy chain with the CL domains of the corresponding light chain 69, while bispecificity of the LY3164530 antibody is partly achieved by creating an orthogonal interface through mutations in V and L domains for the correct assembly of the different Fab domains.
The second prerequisite for the generation of bispecific antibodies is the enforcement of the correct heavy chain heterodimerization over wrong homodimerization, which requires modification of the Fc fragment. The knob‐into‐holes (KiH) approach mimics a key‐locks system by introducing a bulky residue in one CH3 domain that favors binding to a small residue in the other CH3 domain 70. Atwell et al. 71 extended the KiH technology by construction of a phage library of CH3 ‘hole’ mutants which were tested for binding to the T366W knob mutant and were able to select a heterodimer variant with improved thermal stability. To further stabilize and increase the purity of the heterodimeric Fc, an artificial disulfide bond was introduced by Merchant et al. 72. The latter development was exploited for the production of the above‐mentioned bsAb RG7221.
Another approach represents the SEEDbody technology, which is based on the fact that the CH3 domains of human IgG and IgA do not dimerize 73. By using molecular modeling, the authors investigated interdigitating β‐strand segments of IgG and IgA CH3 domains for the generation of complementary heterodimer contact surfaces.
One further technology to enforce correct dimerization includes mutations at the interface of the CH3 domains altering charge polarity of the heavy chain monomers. Two negatively charged residues in chain A pair with two positively charged residues that were introduced in chain B (DD‐KK variant) resulting in a high degree of heterodimer purity, but also a decrease in thermal stability of the CH3 domain 66, 74. Similarly, Strop et al. 75 identified mutations in the hinge region of human IgG1 and IgG2 that produce stabilizing ionic interactions. By combining these changes of the amino acid sequence with an additional mutation at the canonical position 409 in the CH3 domain, bispecificity was further enhanced. However, in this case both homodimers were first produced separately and then purified by affinity chromatography using protein A. Formation of heterodimers was induced by mixing both samples at equimolar ratios and incubating them under reducing conditions at 37°C for 24 h, resulting in the most stably paired variant EEE‐RRR for hIgG1. Moore et al. 76 identified residues in the CH3 interface promoting Fc heterodimerization by combining structural considerations with sequence information. Several variants were screened for the highest degree of heterodimerization, and an HA‐TF variant was identified, where His and Thr are stabilized through a hydrogen bond and Ala and Phe act as key and lock.
Von Kreudenstein et al. described the heterodimer format ZW1 providing high thermal stability and 95% purity, with the residual 5% being present as monomeric, but not homodimeric CH2–CH3 subunits. By combination of in silico design and experimental screening, variants with high degrees of heterodimerization but low thermal stabilities were selected. Based on these initial results, modifications of the selected heterodimeric variants were designed toward increasing purity and stability. The leading format ZW1 carries four mutations on each CH3 domain located in the CH3–CH3 interface and exhibits wildtype‐like Tm values 77.
In a very recent study, Choi et al. 78 combined electrostatic with asymmetric hydrophobic interactions and generated the EW‐RVT Fc heterodimer. By introducing a disulfide bond between the CH3 domains they could further improve purity and thermal stability 79.
All of the above‐mentioned approaches to the heterodimerization of heavy chains and the correct pairing of light chains aim at the production of full‐size bsAbs providing one distinct specificity at each of the two Fab fragments. By contrast, application of the modular antibody technology would allow for the combination of these Fab‐located binding sites with novel, artificial antigen‐binding sites in the structural loops of constant domains. For example, the Fc of an existing mAb could be replaced with an Fcab to generate a so‐called mAb2 that bivalently binds to one antigen via its Fab fragments and mono‐ or bivalently to a second antigen via the Fcab portion (Fig. 3).
Figure 3.
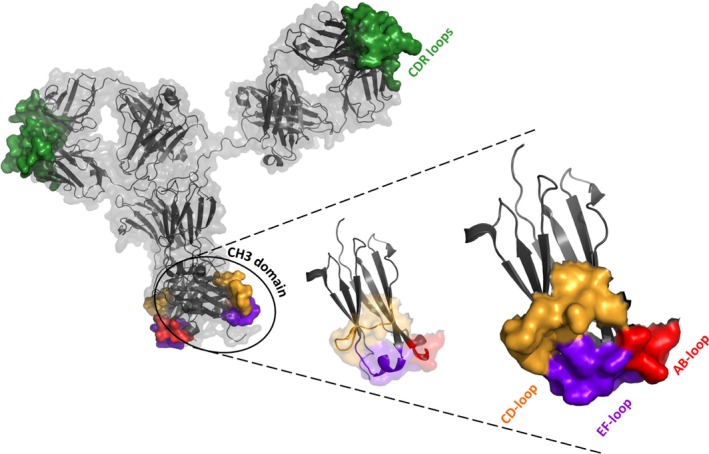
Schematic representation of a hypothetical mAb2 [based on PDB‐ID 1HZH ( 122 )] and one isolated CH3 domain thereof. Secondary structure elements of the antibody are colored in gray with the molecular surface of the CDR loops shown in green. Loop areas in the CH3 domains are displayed in red (AB loop), orange (CD loop), and purple (EF loop). The surface approximations of the three loops indicate the putative region for the generation of binding sites.
Engineering of the biophysical properties of IgG1‐Fc
The correctness of the overall fold of IgG1‐Fc variants was confirmed in many cases. Under some circumstances it was necessary to improve the biophysical properties of Fcabs, either in advance by including stabilizing measures in the construction of libraries to make use of the full versatility of the scaffold or by ‘repairing’ functionalized, yet impaired, Fc fragments, which were mainly selected from early generation combinatorial libraries. Also, even though the Fc fragment is an intrinsically stable protein, the engineering of its properties could further accentuate the advantages over other non‐antibody‐based protein therapeutics.
A detailed systematic study on the effect that the engineering of the C‐terminal structural loops of the CH3 domain of IgG1‐Fc has on the biophysical properties of the Fc fragment was published by Traxlmayr et al. 80, who inserted the αvβ3 integrin‐binding RGD motif in the respective loops. This motif, in the context of the heptapeptide GCRGDCL, forms a cyclic and rather rigid structure as a consequence of disulfide bond formation by the flanking cysteines, resulting in increased interaction with integrins. Single‐, double‐, and triple‐insertion variants (i.e. grafting of GCRGDCL into AB loop, CD loop, EF loop, AB+CD, AB+EF, or CD+EF) were expressed in Pichia pastoris and evaluated for the binding to soluble or cell surface‐expressed αvβ3 integrin as well as for the effect of the engineering efforts on the overall fold and the structural integrity of the protein. Expression levels determined for the variants did not differ significantly from those determined for the wildtype protein, except for the heavily mutated triple‐variant, where grafting of the rigid circular motif into each of the C‐terminal loops apparently resulted in severe misfolding and degradation. Aside from wildtype‐like expression levels, all other constructs exhibited high purity and homogeneity and eluted as single peaks and at defined elution volumes in size‐exclusion chromatography, suggesting the correct folding of the proteins and the absence of aggregates. Also, using electronic circular dichroism spectroscopy, the overall secondary structure content was determined for each variant. No significant changes were observed for AB‐ and CD loop variants, whereas engineering of the EF loop apparently affected the helical content of the protein. This effect was also reflected in the strongly decreased stability of EF loop variants as determined by DSC. In contrast to that, manipulation of AB and CD loop destabilized the protein to a lesser degree. As expected, simultaneous insertion in two loops had a more pronounced effect on protein stability than single insertions. Furthermore, the interaction with natural ligands was analyzed by SPR spectroscopy and biolayer interferometry: FcRn‐, Protein A‐, and FcγRIIIa‐binding revealed to be wildtype‐like, which again confirmed the structural integrity of the recombinant proteins.
The importance of biophysical properties of antibody fragments for large‐scale production and/or therapeutic efficacy has been demonstrated in various studies. Willuda et al. 81 described the correction of an anti‐EGP‐2 scFv fragment which, despite its high affinity, did not enrich at tumor xenografts. By grafting the binding residues of this scFv onto the framework of a more stable fragment and identification and introduction of stabilizing mutations, a functionally improved variant could be generated for which tumor localization was observed.
In a different study, antibodies directed against the chemokine CCL17 were selected from a Fab phage library designed by Shi et al. 82, many of them raised from the VH1‐69 germline gene family abundant in the human immune repertoire 83. Sub‐nanomolar binders could be isolated, which, however, exhibited strong non‐specific protein–protein interaction and low solubility. The authors suggested that the high hydrophobicity of the germline CDR‐H2 leads to these unfavorable properties. By generating a library with randomized surface‐exposed residues within CDR‐H1 and CDR‐H2 to a group of biochemically distinct amino acids, a panel of novel clones could be isolated comprising both high affinity and reduced non‐specific interactions. Surprisingly, the overall hydrophobicity of the selected clones was not significantly reduced, but three residues turned out to be critical in terms of undesired protein–protein interaction. Clones exhibiting a mutation at position I51 to polar or charged residues as well as at position F54 to small polar residues demonstrated higher solubility. Furthermore, mutation of P52 suggested a structural change to the loop associated with higher stability. This study reveals the challenge in selecting for high affinity and good biophysical properties and demonstrates the importance of combinatorial library design, which we will discuss later in this review.
One further study describes a monoclonal antibody that neutralizes binding of angiopoietin 2 to its receptor in vitro and inhibits tumor growth in vivo 84. Even though the pharmacological activity could be demonstrated, production of the antibody coincided with heterogeneity of the preparations, rapid aggregation, and poor expression yields. Exchange of a susceptible surface‐exposed cysteine by all other 19 amino acids yielded a threonine‐variant that exhibited reduced proneness to aggregation, improved homogeneity, largely increased expression levels, retained activity and, interestingly, the midpoint of denaturation shifted by 11°C, corresponding to a strongly improved thermal stability. The authors stated that the engineering process resulted in the fulfillment of requirements necessary for large‐scale production in order to provide amounts of material sufficient for clinical trials.
Considering the conclusions from the above‐mentioned work, comparable efforts proved to be beneficial for the Fc scaffold. One successful attempt to contribute to the development of a ‘tool box’ that would help the correction of impaired Fc fragments was published recently 85. In this work, IgG1‐Fc scaffolds with increased thermal stabilities were constructed by following a directed evolution approach. The authors implemented a novel method that allows screening for stabilizing mutations in proteins that exhibit an already high thermal stability and T m values of up to 85°C. Two libraries of IgG1‐Fc variants differing in their mutation rates were constructed by error‐prone PCR targeting the entire gene. These libraries were then expressed in the yeast surface display format and incubated at 79°C in order to denature those variants displayed on yeast that were not stabilized by mutation. Variants that were still conformationally intact were stained with fluorescently labeled structure‐specific markers, either FcγRI or anti‐CH2 antibody, and thereby tagged for sorting of the displaying yeast cell by FACS. As a consequence of heat denaturation of the yeast cells, plasmid DNA coding for the stabilized variants had to be isolated, followed by re‐transformation of S. cerevisiae and construction of novel libraries, now being enriched in variants carrying favorable mutations. A total of four sorting rounds were performed to select for the most stabilized variants in the libraries which were eventually identified by sequencing of the isolated plasmid DNA. Biophysical characterization of 17 single, double, and triple mutants expressed in P. pastoris revealed that all of the variants exhibited increased thermal stabilities and wildtype‐like binding to relevant ligands, i.e. FcRn, FcγRIIIa, and Protein A. It was concluded that possible adverse effects of the artificial amino acid composition in the engineered loops might be counteracted by the introduction of the identified stabilizing mutations without negatively affecting the intrinsic functionality of the Fc fragment.
In addition, this original IgG1‐Fc library pool generated by error‐prone PCR and the selected libraries after one round of FACS were analyzed by high‐throughput sequencing 86. For each amino acid position, the change in the mutation rate during selection was determined, indicating the tolerance to mutation at the respective position. As expected, selection for binding either to FcγRI or the anti‐CH2 antibody resulted in reduced mutation rates. This was more pronounced in the CH3 domain reflecting the thermal denaturation pathway of IgG1‐Fc with the reversibility of unfolding of the CH2 domain, as long as the CH3 domain remains natively folded. These data indicate a lower selection pressure for the CH2 domain, as only mutations either located in the binding site of the structure‐specific markers or impeding the correct folding of the CH2 domain were eliminated. As both ligands that were used in this study bind to the CH2 domain, the changes in the mutation rates at positions in the CH3 domain were solely dependent on the impact of the respective side chain on foldability and/or stability, enabling the generation of a stability landscape of the CH3 domain. Importantly, positions that are evolutionarily conserved among different species were significantly less tolerant to mutation in these in vitro selections, validating the quality of the stability landscape. Furthermore, the experimentally derived tolerances to mutation correlated with the changes in the free energy of unfolding determined in silico. The results of this study not only revealed the sequence‐stability relationship of an entire protein at single residue resolution but also proved to be an important tool for protein engineering, because library randomizations can be focused on mutation‐tolerant regions of the protein. In addition, comparison of two selection experiments with different ligands (FcγRI and an anti‐CH2 antibody) enabled the identification of their epitopes on IgG1‐Fc.
In a follow‐up project, this directed evolution protocol was applied to improve the biophysical properties of the HER2‐binding Fcab H10‐03‐6 87, which, even though the interaction with important effector molecules was wildtype‐like, exhibited impaired biophysical properties in an initial characterization. In contrast to the method described above, mutagenesis was directed to the engineered loops only in order to minimize the overall changes the Fc fragment. An initial experiment revealed that the binding of H10‐03‐6 to HER2 was decreased after heat incubation, leading to the assumption that a correct fold is necessary for molecular recognition and that combination of heat shock and selection for ligand binding could be applied to simultaneously screen for improved thermal stability and retained affinity to the antigen. After heat incubation, the yeast displayed‐library was either sorted for (i) binding to the antigen and to structurally specific ligands or (ii) binding to the antigen only. Enriched clones were expressed in P. pastoris and HEK293 cells and their biophysical properties were analyzed by SEC, ECD spectroscopy, and DSC. Stabilized variants resulting from the combined staining strategy (antigen and structurally specific ligand) exhibited higher thermal stabilities, which led to the assumption that this selection strategy should be favored. Importantly, all selected mutants were more stable than their parental clone H10‐03‐6. In addition, some stabilized Fcabs were less prone to aggregation after long term storage, showed more wildtype‐like SEC elution profiles and the solubility of the Fcab exhibiting the highest thermal stability was largely increased when compared to the parental clone H10‐03‐6. The general applicability of this method to improve the biophysical properties of suitable proteins was concluded.
In addition, the differences in thermal stability of IgG1‐Fc and Fcabs between the two expression systems, P. pastoris and HEK293, were analyzed. Generally, the high mannose glycan structures attached to Asn297 of the P. pastoris‐produced Fc variants destabilizes the interface of the CH2 domains and therefore leads to lower T m values. In a study by Schaefer and Pluckthun 88, significant differences between full‐size IgGs produced in both expression systems revealed not only different temperatures of unfolding but also indicated differences in terms of aggregation susceptibility. Antibodies produced in P. pastoris were less prone to aggregate formation due their mannose‐rich glycan structure and the N‐terminal residual EAEA extension, remaining from the α‐factor pre‐pro secretion sequence. Although the thermal stability of P. pastoris‐produced Fc variants was lower, the corresponding SEC elution profiles were highly comparable with the HEK‐produced counterparts 87, as was the stability‐ranking of Fcabs in both expression systems. This clearly underlined that P. pastoris‐produced Fc variants can be used to investigate the biophysical properties and binding characteristics of a panel of selected Fcabs.
A different approach to stabilize IgG1‐Fc in order to provide a stable scaffold for the engineering of antigen‐binding sites or other novel functionalities is based on the insertion of artificial disulfide bonds based on computational prediction 89. Selected cysteine‐variants were expressed in P. pastoris and their biophysical properties were characterized, revealing that the thermal stabilities of two variants bearing novel intradomain disulfide bonds were largely increased while the overall structure remained wildtype‐like. Combination of both novel disulfide bonds resulted in even higher thermal stabilities. Moreover, stabilization of the HER2‐binding Fcab H10‐03‐6 could be achieved by the introduction of only one of the disulfide bonds.
In a second study, the observation that the C‐terminus of the CH3‐dimer closely resembles the C‐terminus of the CH1‐CL‐dimer of the Fab fragment led to the assumption that the three C‐terminal amino acids of the CH3 domains could be replaced by those of the CL domain 90. The resulting variant exhibited a wildtype‐like elution profile in SEC, leading to the assumption that specific disulfide bonding was accomplished and no incorrect cross‐bridging occurred. Thermal denaturation of the protein occurred at significantly higher temperatures compared to wildtype IgG1‐Fc and this effect was observed to be further pronounced in a variant carrying both the stabilizing inter‐ and intradomain disulfides. Interestingly, more detailed analysis by DSC revealed a strong stabilizing cooperative effect that the CH3‐located alterations had on the CH2 domain. The introduction of both the computationally and rationally selected mutants into H10‐03‐6 increased the thermal stability of the protein to be approximately wildtype‐like while antigen‐binding was retained. Those identified mutations could serve as a valuable tool for stabilization of single proteins or entire libraries.
In a similar study, Gong et al. 91 described the stabilization of a monomeric human IgG1‐CH2 domain by the introduction of artificial, rationally designed disulfide bonds. For two variants whose expression levels were comparable to the wildtype domain and which were highly soluble, increased thermal and conformational stabilities were determined in heat and chemical denaturation experiments. However, only for the more stable variant a monomeric state was determined. Structural changes in this variant as a consequence of the cysteine mutations were analyzed by NMR, once again confirming the correct formation of the novel disulfide bond. Nuclear Overhauser effect spectra were recorded and showed that the flexibility of both the wildtype CH2 domain and the stabilized variant were rigid in the framework but highly flexible in the loop regions. Based on these findings, it was suggested that the CH2 domain as well as its stabilized variant can be applied as scaffolds for engineering antigen binders.
In summary, the methods described in this section can be applied in several ways: One possibility is the introduction of thus identified stabilizing mutations in naïve libraries in order to ‘prestabilize’ the scaffold and thereby minimize the risk of selecting an Fcab with impaired biophysical properties. The combination of several beneficial mutations exhibiting additive stabilizing effects could not only compensate for destabilization upon mutation of the structural loops but also further improve the characteristics of the resulting binders. Moreover, starting from a stabilized protein scaffold has been shown to promote evolvability by tolerating a wider range of mutations 92. In other words, a library based on a stabilized protein contains a higher fraction of correctly folded and therefore functional protein mutants. A more elaborate way of exploiting stabilizing effects of single or multiple point mutations would be the ‘repairing’ of existing binders as a reaction to negative effects of loop mutations, as was shown for the HER2 binding Fcab H10‐03‐6.
Optimization of library design
The previous chapters have clearly shown that IgG1‐Fc is a very attractive scaffold for the design of novel binding molecules that possess all antibody functions at only one‐third of the size of a full‐size IgG1 molecule. Like many other therapeutic antibodies, antibody‐based and non‐antibody‐based molecules, Fcabs are selected from combinatorial libraries that became an alternative to conventional methods, i.e. the hybridoma technology. The selection of proteins with the desired binding properties from these libraries is in many cases accomplished by the application of various display technologies such as phage, ribosome or yeast surface display 93. Therapeutic IgG1 molecules are generated by selecting either scFv or Fab from combinatorial phage libraries, or full‐size IgG from yeast or mammalian display libraries. The three main aspects of such libraries that are known to impact library quality are (i) design, (ii) the origin of sequence diversity, and (iii) the method of library generation, while library quality can be assigned to the library size, diversity, and the developability of the selected molecules, i.e. high affinity and good biophysical properties. Maximization of library diversity can be accomplished by increasing the functional size and improving the resulting molecules’ developability, which for example is achieved by restriction to one stable framework or a limited number of consensus frameworks. The issue of immunogenicity is met by choosing framework and CDR compositions as close to the human germline as possible 93.
The introduction of novel binding sites into non‐complementarity‐determing‐region (CDR) loops in immunoglobulin‐like domains of non‐Ig proteins as well as immunoglobulin constant domains as already demonstrated in the context of Fcabs requires more elaborate considerations with respect to library design. While the lack of somatic hypermutation is, as is the case for CDR sequences of scFv or Fab fragments selected from combinatorial libraries, met by different affinity maturation strategies, the design of libraries for naïve selections is more challenging than in scFv or Fab libraries. First of all, there is little or no natural variation in the amino acid sequences of non‐CDR loops and loop lengths are, in contrast to some CDR loops, mostly conserved. Information on typical amino acid compositions that can be drawn from the large pool of existing CDR sequences is not available for alternative scaffolds. And lastly, the knowledge of canonical CDR loop conformations is a design advantage that cannot be exploited in non‐CDR loops as yet. Therefore, these favorable properties that are intrinsic to CDR loops have to be systematically investigated for non‐CDR loops of Ig‐derived and alternative binding scaffolds.
As the term ‘antibody‐based’ is commonly used to describe formats that exploit the original binding site formed by the CDRs of either both or only one of the variable domains, antigen‐binding IgG1‐Fc, albeit derived from IgG1, will be classified as an ‘alternative binding scaffold’. In this respect, the initial strategies that are pursued in order to provide a high degree of diversity, but also functionality, in libraries of different alternative binding scaffolds, are consistent. In this section, we will focus on these strategies and compare the library design efforts that have been made in the engineering of IgG1‐Fc and other formats, especially the fibronectin type III scaffold, which, due to its immunoglobulin‐like fold, is particularly interesting for comparison.
As mentioned above, the generation of diversity in alternative binding scaffolds cannot rely on the natural variation that is present in antibodies through somatic recombination and hypermutation during B‐cell development. While in the course of this maturation process stable frameworks supporting diversity in variable domains are selected for, it is highly probable that diversification of one or several amino acid positions in alternative binding scaffolds impairs the overall protein fold to a certain degree. As mentioned above, in the case of Fcabs, it was shown that the introduction of an integrin‐binding motif into each of the three C‐terminal loops of IgG1‐CH3 is possible while retaining the binding to generic ligands 80. However, overall folds of the proteins as well as thermal stabilities were impaired to some degree. The design of the first combinatorial libraries, i.e. the choice of amino acid positions in the C‐terminal loops to be randomized, was based on the degree of evolutionary conservation and visual evaluation of the crystal structure of IgG1‐Fc according to several criteria (solvent accessibility and structural independence of the respective amino acid side chains; generation of a coherent‐binding surface) 15, and the applicability of this IgG1‐Fc variant library was proved by isolating Fcabs binding to hen eggwhite lysozyme and CD20. Later, using an advanced library, it was shown that randomization of five positions in each the AB loop and the EF loop of IgG1‐CH3 as well as insertion of five additional random amino acids in the EF loop, construction of a yeast surface display library and selection of IgG1‐Fc variants binding to HER2 yielded well‐expressing and biologically functional, yet slightly destabilized proteins 53. To circumvent this minor drawback already on the level of naïve libraries, two approaches have successfully been followed, which resulted in the construction of superior next generation libraries: First, prior to selections, naïve libraries can potentially be cleaned by isolating yeast cells that display IgG1‐Fc still binding to conformationally specific ligands (e.g. Protein A or FcγRI) after diversification. Depending on the applied threshold of residual binding to these ligands, this cleaning step decreases the library size significantly. As an alternative, optimized regions of diversification were identified for the generation of libraries of IgG1‐Fc in order to minimize detrimental effects on the CH3 framework and increase the number of productive clones in the library 94. For this purpose, yeast surface display model libraries were constructed with distinct or overlapping regions randomized in the AB, CD, or EF loop of IgG1‐CH3 and subjected to a protocol that involves the incubation at increasing temperatures of yeast suspensions induced for surface display and subsequent flow cytometric recording of the residual binding to conformationally specific ligands. Library denaturation curves were derived from the resulting mean fluorescent intensities and used for the determination of temperatures of half‐maximal irreversible denaturation (T 1/2) of entire yeast libraries, yielding a clear hierarchy of the distinct loop regions’ tolerance to randomization. IgG1‐Fc libraries were re‐designed based on these findings and are currently evaluated for their potential in the selection of Fcabs.
An approach to library design similar to the basic strategy pursued for Fcabs was published by Koide et al. 14, who identified the tenth type III unit of human fibronectin (10Fn3), an immunoglobulin‐like β‐sandwich protein, as a potent binding scaffold. Similar to the involvement of the N‐terminal loops BC, FG, and C'E of IgG1‐CH2 in the binding of the Fcγ receptors 39, 10Fn3 binds to integrin via the RGD motif in its FG loop. Initially, 10Fn3 library design was based on sequence analyses of several fibronectin type III domains as well as structural evaluation of the loop architectures. Selections using a library with five residues randomized in each the BC and the FG loop yielded a 10Fn3 variant binding to ubiquitin with an IC50 of 5 μM. However, this variant was less soluble at neutral pH than the wildtype 10Fn3 protein and interacted with the column material in size‐exclusion chromatography. In a follow‐up study, it was suggested though that this negative side effect of mutation could be counteracted by the removal of unfavorable electrostatic interactions on the protein surface 95. Xu et al. 96 reported selections of 10Fn3 variants binding to TNF‐α. The ‘master library’ applied in these selections consisted of an equimolar mixture of three sublibraries having either one, two, or all three of the N‐terminal 10Fn3 loops randomized. Interestingly, all but one of the resulting TNF‐α‐binding 10Fn3 variants stemmed from the sublibrary having all three loops randomized, indicating the importance of diversity for the isolation of high‐affinity binders. Again, the stabilities reported for two representative variants were decreased. In a follow‐up study, Parker et al. 97 reported the significant loss of stability and solubility of VEGF‐R2‐binding 10Fn3 variants during affinity maturation, once more suggesting a trade‐off between stability and affinity. The authors counteracted destabilization by structure‐based site‐directed mutagenesis, which also included the systematic reversion of randomized positions to their respective wildtype amino acids. Also, it was suggested to apply alternative designs of the apparently crucial DE loop of 10Fn3. All of these findings were based on the biophysical characterization of a limited set of 10Fn3 variants, which leads to the assumption that it would be possible to further enhance 10Fn3 library design (i.e. at which positions the N‐terminal loops can be randomized) by applying the flow cytometry‐based method developed for libraries of IgG1‐Fc to this scaffold as described above, even though it is focused on stability and does not take into account the effects library design will have on affinity 94. If, on the other hand, results from one further study on the 10Fn3 scaffold by Lipovsek et al. 98 are taken into consideration, it can be expected that a higher number of randomized residues will increase the affinity of selected 10Fn3 variants while, as mentioned above, stability and solubility will be impaired. Consequently, if information on the importance of distinct regions for the overall fold and stability of the scaffold is made available in a systematic manner, as was done for IgG1‐Fc, desired affinities can be approached by manipulating increasing numbers of distinct loop positions while at the same time being aware of the effect this will have on the biophysical properties of the scaffold.
Besides the application of modular antibody engineering to the C‐terminal loops of IgG1‐Fc‐CH3, various other studies have described the introduction of novel binding sites to immunoglobulin constant domains: Xiao et al. 10 identified HIV‐1 inhibitors based on isolated IgG1‐CH2 by randomizing the longest N‐terminal loops in the domain, i.e. BC and FG. An interesting design feature was the addition of a glycine residue at the C‐terminal end of each loop to provide flexibility and favor the accommodation of amino acid alterations necessary for stability and antigen recognition. However, as the stability of native CH2 is relatively low, the solubilities of three HIV‐1‐binding ‘nanoantibodies’ selected from this library were reported to be poor. To overcome these issues, the isolated CH2 domain was stabilized by introducing an additional disulfide bond and removing seven N‐terminal residues 91, 99, 100. Starting from this scaffold, a library was constructed that comprised randomized BC and DE loops and a CDR‐H3 from the HIV‐1 gp120‐binding VH m36 grafted in place of the FG loop 101. While one of two clones selected from this library interacted non‐competitively with an HIV‐1 neutralizing epitope and FcRn, the second was not further characterized due to its aggregation proneness, leading to the authors’ conclusion that further improvements to the library design would be necessary to develop IgG1‐CH2 as a scaffold for the development of novel therapeutics.
Length variation of CDR loops is another important factor in the natural generation of diversity. Among the six CDR loops, H3 is the most diverse, both in terms of sequence and length. In a study on the clustering of antibody CDR loop confirmations, North et al. 102 assigned between two and eight different loop lengths to L1, L2, L3, H1, and H2, and 20 different lengths to H3, ranging from 5 to 26 residues. This length variability is introduced by imprecise joining during the combinatorial rearrangement of VH, DH, and JH genes in the course of B‐cell maturation 103 and is considered to be a main contributor to the recognition of diverse antigens, as H3, given by its location in the center of the antigen‐binding site, potentially controls the relative positions of VH and VL and affects the flexibility and cavity size of the paratope 104.
Consequently, in order to mimic this crucial aspect of the development of antigen specificity by the immune system, loop length variation in the antigen‐binding sites of alternative scaffolds is an important factor in the generation of specific, high‐affinity interactions. In the process of development of the Fcab scaffold, several steps have been made toward a loop elongation strategy that would allow for the selection of variants binding to diverse antigens while at the same time the intrinsic stability can be retained to a satisfactory degree. As mentioned above, the initial design for variant libraries of IgG1‐Fc included the insertion of five additional residues at the N‐terminal part of the EF loop, with this loop elongation aiding in the selection of an Fcab binding to HER2 with a K D value of 8.6 nM 53. However, mutation and insertion negatively affected the biophysical properties of this Fcab 87. Therefore, one further aspect of the detailed analysis of the C‐terminal loops of IgG1‐CH3 was the systematic insertion of five additional random residues at different loop positions according to a sliding window 94. T 1/2 values were determined for the resulting yeast surface display libraries as described above and used for the comparison of the tolerance of different loop positions to insertion. The results from this study, as well as the identification of sites of natural insertion from phylogenetic analysis of IgG1‐CH3 from different species, were also considered for the design of novel variant libraries of IgG1‐Fc. As mentioned above, the evaluation of the effect of these design features is part of current investigations. Furthermore, findings from a study that dealt with the identification of stabilizing point mutations in IgG1‐CH3 by using a directed evolution approach were combined with rational considerations 85, 105. In order to generate stabilizing stem regions that would allow for loop elongation, point mutations were introduced at two intrinsically stable motifs in the EF loop. The central part of this loop is constituted by an arginine residue forming a salt bridge with a glutamic acid in the CD loop, and a tryptophan residue that has been shown to be crucial for protein stability by contributing to the packing of the hydrophobic core. At the C‐terminus of this RW motif, a glutamine residue was replaced by a stabilizing leucin residue. At the N‐terminus of the adjacent F‐strand, a serine between a phenylalanine also contributing to hydrophobic packing and a cysteine forming the intradomain disulfide bond was replaced by a stabilizing threonine. Yeast surface display libraries including or not including these stabilizing stem regions and carrying increasing numbers of additional residues in between them were constructed, and their stabilities were evaluated by using the yeast surface display‐based method described above 94, showing that, indeed, a largely increased tolerance to loop elongation was achieved by this stabilization approach, with the potential of such libraries for the selection of Fcabs binding to diverse antigens being yet to be looked into.
While the work on the design of libraries of IgG1‐Fc to date has focused on the question of how enhanced diversification, both in terms of site‐ or region‐specific randomization and loop elongation, affects the overall fold and the biophysical properties of the scaffold, the trade‐off between this focus and its effect on Fcab affinity is still under investigation. However, this has been addressed for other alternative formats, including the above‐mentioned immunoglobulin‐like 10Fn3 scaffold, which is why the results of the underlying studies should be of interest in this context and a link can be made to ongoing and future projects dealing with IgG1‐Fc. Koide et al. 106 performed basic experiments on the potential for elongation of all the loops in the 10Fn3 domain, thereby providing some first information on the tolerance to insertion of alanines, which, of course, does not reveal the effect of full randomization. This was further investigated by Hackel et al. 107, who diversified all three N‐terminal loops of 10Fn3 both in length and composition. Four different lengths were chosen for each loop based on sequence analysis of 10Fn3 from different species, and NNB degenerate codons were used to incorporate all 20 amino acids. Three FACS sortings of the naïve library against lysozyme were performed. The output of this naïve selection was diversified by loop shuffling and error‐prone PCR and sorted twice, followed by another four rounds of this procedure, yielding 10Fn3 variants of single digit picomolar affinity and high sequence diversity, having accumulated a considerable number of beneficial framework mutations. Interestingly, further mutational studies on the DE loop of one particular high‐affinity 10Fn3 variant revealed that this loop can be either engineered to improve affinity or stability to a considerable degree. These results underline once more that library design for scaffolds based on immunoglobulin and immunoglobulin‐like domains must undergo adjustments based both on the systematic evaluation of naïve libraries and selection outputs in order to efficiently approach an optimum balance of stability and affinity of relevant variant proteins.
The degree of CDR sequence diversification in combinatorial libraries of antibody‐based alternative scaffolds, i.e. the usage of the amino acid repertoire, follows various different strategies. While naïve libraries provide natural diversity by combination of functional V‐gene segments isolated from the lymphoid tissues of non‐immunized donors [e.g. CAT 1.0 and CAT 2.0 libraries of Cambridge Antibody Technology 108, 109], synthetic libraries such as the HuCALs (Morphosys) offer any desired amino acid compositions in the CDRs 110, 111. In general, it was observed that, despite the hypervariability of CDR sequences, there is a bias of functional CDRs to certain amino acid types, most of all tyrosine, glycine, and serine 112. Fellouse et al. showed that while scFv variants binding to some antigens at nanomolar affinities were selected from phage display libraries providing only a binary amino acid repertoire (i.e. tyrosine and serine), selections of binders from the same libraries to other antigens yielded affinities only in the micromolar range 113, 114, 115. Modifications of these libraries included an increased chemical diversity in CDR‐H3 and the consideration of non‐paratope residues potentially important for CDR conformation 116.
As mentioned above, Morphosys’ HuCAL Fab libraries are fully synthetic, and they were developed over the course of several years 110, 111. The latest version, named HuCAL PLATINUM, offers, among other features, length variation in CDR‐H2 and CDR‐H3, and length‐dependent amino acid frequencies as observed in the analysis of rearranged sequences, which was realized by using a large set of different trinucleotide mixtures. In principle, a similar approach is potentially valid for the construction of variant libraries of IgG1‐Fc. Naturally, while the alignment of thousands of VH and VL sequences was used to generate information on the amino acid distributions in CDR loops, it cannot be estimated how, for example, binary codes or natural distributions in CDRs would influence the efficient isolation of well‐developable Fcabs binding to diverse antigens at high affinities. Nevertheless, Hackel et al. investigated the effect of full or restricted diversity in 10Fn3, which, again, can be a valuable source of information for the engineering of IgG1‐Fc due to its immunoglobulin‐like fold 117, 118. In these studies, the sequence‐function landscape of the 10Fn3 scaffold was approached by comparing various amino acid compositions in the N‐terminal loops, leading to important conclusions: First, it was shown that maximal amino acid diversity is more beneficial for the selection of antigen‐binding 10Fn3 variants than a binary tyrosine/serine code as described above. And second, wildtype conservation at positions potentially important for structural integrity, as well as amino acid distribution biases toward the natural occurrence in CDRs made clear that both strategies were valuable for an efficient selection of binding 10Fn3 variants. Consequently, while the identification of positions in IgG1‐Fc‐CH3 at which conservation of wildtype amino acids improves the biophysical properties of selected variants has been accomplished 86, 94, the application of tailored amino acid distributions as described above is a promising approach to be investigated in the future.
Conclusion
Over the past 30 years, therapeutic monoclonal antibodies and antibody‐based alternative formats have been successfully developed, first by using the hybridoma technology and later by selection from combinatorial libraries. In this review, we introduced the potential of IgG1‐Fc as a promising scaffold for the generation of alternative protein therapeutics. By applying the modular antibody engineering technology, novel binding sites for theoretically any desired antigen can be introduced in the C‐terminal loops of the CH3 domains to create Fcabs, antigen‐binding Fcs. In this way, the Fc is engineered to unite all important functions normally conveyed by full‐size IgGs, i.e. (i) the mediation of effector functions through binding to FcγRs; (ii) contribution to a long half‐life in serum through binding to FcRn, and (iii) (novel) binding to an antigen. FS102, a HER2‐binding Fcab that has recently entered the clinic, is believed to accelerate aggregation and internalization of the antigen/Fcab complexes as a consequence of the distinct Fcab structure, with pharmacokinetics not being impaired by the sequence changes in the CH3 domain.
In some cases, as has been described for other alternative scaffolds, the engineering process might negatively affect the biophysical properties of IgG1‐Fc. The current article summarizes a set of methods, mostly based on directed evolution and yeast surface display, which provide a ‘toolbox’ for the correction of impaired biophysical properties of proteins in general. We also reviewed the various approaches to modifying IgG‐Fc for the production of bispecific antibodies and described the highly promising concept of constructing mAb2s, monoclonal antibodies with one specificity in the two antigen‐binding sites of the Fabs and one additional specificity at the C‐terminus of the Fc, which again can be accomplished by modular antibody engineering.
Finally, this review provides comments on potential further developments of libraries of IgG1‐Fc. Loop position‐ and loop region‐specific information has been obtained that will aid in the construction of libraries of high quality, and studies dealing with the modification of the architecture of the putative Fcab binding sites by loop elongation have been performed. Many approaches that have proved successful in the context of other alternative scaffolds, especially those targeting tailored amino acid distributions, can be used as models for the future investigation of libraries of IgG1‐Fc, which will ultimately lead to a further accentuation of the intrinsically favorable properties of Fcabs.
Acknowledgements
This work was supported by the Christian Doppler Research Association (Christian Doppler Laboratory for Antibody Engineering), the company F‐star Biotechnology Ltd., as well as the Austrian Science Foundation (FWF W1224 – Doctoral Program on Biomolecular Technology of Proteins – BioToP). The authors declare no conflict of interest.
References
- 1. Melmed GY, Targan SR, Yasothan U, Hanicq D, Kirkpatrick P. Certolizumab pegol. Nat Rev Drug Discov 2008;7:641–642. [DOI] [PubMed] [Google Scholar]
- 2. Pieramici DJ, Avery RL. Ranibizumab: treatment in patients with neovascular age‐related macular degeneration. Expert Opin Biol Ther 2006;6:1237–1245. [DOI] [PubMed] [Google Scholar]
- 3. Genetta TB, Mauro VF. ABCIXIMAB: a new antiaggregant used in angioplasty. Ann Pharmacother 1996;30:251–257. [DOI] [PubMed] [Google Scholar]
- 4. Bird RE, et al. Single‐chain antigen‐binding proteins. Science 1988;242:423–426. [DOI] [PubMed] [Google Scholar]
- 5. Worn A, Pluckthun A. Stability engineering of antibody single‐chain Fv fragments. J Mol Biol 2001;305:989–1010. [DOI] [PubMed] [Google Scholar]
- 6. Raag R, Whitlow M. Single‐chain Fvs. FASEB J 1995;9:73–80. [DOI] [PubMed] [Google Scholar]
- 7. Baeuerle PA, Reinhardt C. Bispecific T‐cell engaging antibodies for cancer therapy. Cancer Res 2009;69:4941–4944. [DOI] [PubMed] [Google Scholar]
- 8. Holliger P, Prospero T, Winter G. Diabodies – small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA 1993;90:6444–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson S, et al. Effector cell recruitment with novel Fv‐based dual‐affinity re‐targeting protein leads to potent tumor cytolysis and in vivo B‐cell depletion. J Mol Biol 2010;399:436–449. [DOI] [PubMed] [Google Scholar]
- 10. Xiao X, Feng Y, Vu BK, Ishima R, Dimitrov DS. A large library based on a novel (CH2) scaffold: identification of HIV‐1 inhibitors. Biochem Biophys Res Commun 2009;387:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Meyer T, Muyldermans S, Depicker A. Nanobody‐based products as research and diagnostic tools. Trends Biotechnol 2014;32:263–270. [DOI] [PubMed] [Google Scholar]
- 12. Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single‐domain antibody fragments. Appl Microbiol Biotechnol 2007;77:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stumpp MT, Binz HK, Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discovery Today 2008;13:695–701. [DOI] [PubMed] [Google Scholar]
- 14. Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol 1998;284:1141–1151. [DOI] [PubMed] [Google Scholar]
- 15. Rüker F, Wozniak‐Knopp G. Engineering of non‐CDR loops in immunoglobulin domains In: Little M. ed. Recombinant Antibodies for Immunotherapy. New York: Cambridge University Press, 2009:231–242. [Google Scholar]
- 16. Halaby DM, Poupon A, Mornon J. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng 1999;12:563–571. [DOI] [PubMed] [Google Scholar]
- 17. Ying T, Gong R, Ju TW, Prabakaran P, Dimitrov DS. Engineered Fc based antibody domains and fragments as novel scaffolds. Biochim Biophys Acta 2014;1844:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature 1976;264:415–420. [DOI] [PubMed] [Google Scholar]
- 19. Hogarth PM, Pietersz GA. Fc receptor‐targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 2012;11:311–331. [DOI] [PubMed] [Google Scholar]
- 20. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest. 5th edn Bethesda, MD: National Institutes of Health, 1991:688–696. [Google Scholar]
- 21. Jefferis R, Lund J. Interaction sites on human IgG‐Fc for FcgammaR: current models. Immunol Lett 2002;82:57–65. [DOI] [PubMed] [Google Scholar]
- 22. Burton DR, Woof JM. Human antibody effector function. Adv Immunol 1992;51:1–84. [DOI] [PubMed] [Google Scholar]
- 23. Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9‐ and 2.8‐A resolution. Biochemistry 1981;20:2361–2370. [PubMed] [Google Scholar]
- 24. Padlan EA. Anatomy of the antibody molecule. Mol Immunol 1994;31:169–217. [DOI] [PubMed] [Google Scholar]
- 25. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti‐inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006;313:670–673. [DOI] [PubMed] [Google Scholar]
- 26. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 27. Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol 2007;25:21–50. [DOI] [PubMed] [Google Scholar]
- 28. Lai B, Hasenhindl C, Obinger C, Oostenbrink C. Molecular dynamics simulation of the crystallizable fragment of IgG1‐insights for the design of Fcabs. Int J Mol Sci 2014;15:438–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mimura Y, et al. The influence of glycosylation on the thermal stability and effector function expression of human IgG1‐Fc: properties of a series of truncated glycoforms. Mol Immunol 2000;37:697–706. [DOI] [PubMed] [Google Scholar]
- 30. Ghirlando R, Lund J, Goodall M, Jefferis R. Glycosylation of human IgG‐Fc: influences on structure revealed by differential scanning micro‐calorimetry. Immunol Lett 1999;68:47–52. [DOI] [PubMed] [Google Scholar]
- 31. Vermeer AW, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi‐domain protein. Biophys J 2000;78:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Traxlmayr MW, Obinger C. Directed evolution of proteins for increased stability and expression using yeast display. Arch Biochem Biophys 2012;526:174–180. [DOI] [PubMed] [Google Scholar]
- 33. Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T‐cell receptor engineering. Nat Biotechnol 2000;18:754–759. [DOI] [PubMed] [Google Scholar]
- 34. Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 1997;15:553–557. [DOI] [PubMed] [Google Scholar]
- 35. Hulett MD, Hogarth PM. Molecular basis of Fc receptor function. Adv Immunol 1994;57:1–127. [DOI] [PubMed] [Google Scholar]
- 36. Fridman WH. Fc receptors and immunoglobulin binding factors. FASEB J 1991;5:2684–2690. [DOI] [PubMed] [Google Scholar]
- 37. Wines BD, Powell MS, Parren PW, Barnes N, Hogarth PM. The IgG Fc contains distinct Fc receptor (FcR) binding sites: the leukocyte receptors Fc gamma RI and Fc gamma RIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J Immunol 2000;164:5313–5318. [DOI] [PubMed] [Google Scholar]
- 38. Feige MJ, Nath S, Catharino SR, Weinfurtner D, Steinbacher S, Buchner J. Structure of the murine unglycosylated IgG1 Fc fragment. J Mol Biol 2009;391:599–608. [DOI] [PubMed] [Google Scholar]
- 39. Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci USA 2008;105:20167–20172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lund J, Takahashi N, Pound JD, Goodall M, Jefferis R. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J Immunol 1996;157:4963–4969. [PubMed] [Google Scholar]
- 41. Jefferis R, Lund J, Pound J. Molecular definition of interaction sites on human IgG for Fc receptors (huFc gamma R). Mol Immunol 1990;27:1237–1240. [DOI] [PubMed] [Google Scholar]
- 42. Shields RL, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001;276:6591–6604. [DOI] [PubMed] [Google Scholar]
- 43. Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol 2001;19:275–290. [DOI] [PubMed] [Google Scholar]
- 44. Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci 2012;1253:170–180. [DOI] [PubMed] [Google Scholar]
- 45. Gornik I, Maravic G, Dumic J, Flogel M, Lauc G. Fucosylation of IgG heavy chains is increased in rheumatoid arthritis. Clin Biochem 1999;32:605–608. [DOI] [PubMed] [Google Scholar]
- 46. Duncan AR, Winter G. The binding site for C1q on IgG. Nature 1988;332:738–740. [DOI] [PubMed] [Google Scholar]
- 47. Gaboriaud C, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem 2003;278:46974–46982. [DOI] [PubMed] [Google Scholar]
- 48. Diebolder CA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014;343:1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I‐ related receptor FcRn. Annu Rev Immunol 2000;18:739–766. [DOI] [PubMed] [Google Scholar]
- 50. Martin WL, West AP Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH‐dependent binding. Mol Cell 2001;7:867–877. [DOI] [PubMed] [Google Scholar]
- 51. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif‐containing 21 (TRIM21). Proc Natl Acad Sci USA 2010;107:19985–19990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Randow F, MacMicking JD, James LC. Cellular self‐defense: how cell‐autonomous immunity protects against pathogens. Science 2013;340:701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wozniak‐Knopp G, et al. Introducing antigen‐binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu‐binding sites and antibody properties. Protein Eng Des Sel 2010;23:289–297. [DOI] [PubMed] [Google Scholar]
- 54. Chernajovsky Y, et al. Herceptin. Therapeutic Antibodies. Berlin, Heidelberg: Springer, 2008:183–219. [Google Scholar]
- 55. Kainer M, et al. Correlation between CD16a binding and immuno effector functionality of an antigen specific immunoglobulin Fc fragment (Fcab). Arch Biochem Biophys 2012;526:154–158. [DOI] [PubMed] [Google Scholar]
- 56. Woisetschläger M, et al. In vivo and in vitro activity of an immunoglobulin Fc fragment (Fcab) with engineered Her‐2/neu binding sites. Biotechnol J 2014;9:844–851. [DOI] [PubMed] [Google Scholar]
- 57. Jez J, et al. Significant impact of single N‐glycan residues on the biological activity of Fc‐based antibody‐like fragments. J Biol Chem 2012;287:24313–24319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leung KM, et al. A HER2‐specific modified Fc fragment (Fcab) induces antitumor effects through degradation of HER2 and apoptosis. Mol Ther 2015; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chaparro‐Riggers J, et al. Increasing serum half‐life and extending cholesterol lowering in vivo by engineering antibody with pH‐sensitive binding to PCSK9. J Biol Chem 2012;287:11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Igawa T, et al. Antibody recycling by engineered pH‐dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010;28:1203–1207. [DOI] [PubMed] [Google Scholar]
- 61. Traxlmayr MW, et al. Construction of pH‐sensitive Her2‐binding IgG1‐Fc by directed evolution. Biotechnol J 2014;9:1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bonvin P, et al. De novo isolation of antibodies with pH‐dependent binding properties. MAbs 2015;7:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lazar GA, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA 2006;103:4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heiss MM, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer 2010;127:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kienast Y, et al. Ang‐2‐VEGF‐A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF‐A and Ang‐2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res 2013;19:6730–6740. [DOI] [PubMed] [Google Scholar]
- 66. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015;20:838–847. [DOI] [PubMed] [Google Scholar]
- 67. Lewis SM, et al. Generation of bispecific IgG antibodies by structure‐based design of an orthogonal Fab interface. Nat Biotechnol 2014;32:191–198. [DOI] [PubMed] [Google Scholar]
- 68. Weidle UH, Kontermann RE, Brinkmann U. Tumor‐antigen‐binding bispecific antibodies for cancer treatment. Semin Oncol 2014;41:653–660. [DOI] [PubMed] [Google Scholar]
- 69. Schaefer W, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA 2011;108:11187–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ridgway JB, Presta LG, Carter P. ‘Knobs‐into‐holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996;9:617–621. [DOI] [PubMed] [Google Scholar]
- 71. Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol 1997;270:26–35. [DOI] [PubMed] [Google Scholar]
- 72. Merchant AM, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998;16:677–681. [DOI] [PubMed] [Google Scholar]
- 73. Davis JH, et al. SEEDbodies: fusion proteins based on strand‐exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel 2010;23:195–202. [DOI] [PubMed] [Google Scholar]
- 74. Gunasekaran K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem 2010;285:19637–19646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Strop P, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 2012;420:204–219. [DOI] [PubMed] [Google Scholar]
- 76. Moore GL, et al. A novel bispecific antibody format enables simultaneous bivalent and monovalent co‐engagement of distinct target antigens. MAbs 2011;3:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Von Kreudenstein TS, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. MAbs 2013;5:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Choi HJ, Kim YJ, Lee S, Kim YS. A heterodimeric Fc‐based bispecific antibody simultaneously targeting VEGFR‐2 and Met exhibits potent antitumor activity. Mol Cancer Ther 2013;12:2748–2759. [DOI] [PubMed] [Google Scholar]
- 79. Choi HJ, Seok SH, Kim YJ, Seo MD, Kim YS. Crystal structures of immunoglobulin Fc heterodimers reveal the molecular basis for heterodimer formation. Mol Immunol 2015;65:377–383. [DOI] [PubMed] [Google Scholar]
- 80. Traxlmayr MW, Wozniak‐Knopp G, Antes B, Stadlmayr G, Ruker F, Obinger C. Integrin binding human antibody constant domains–probing the C‐terminal structural loops for grafting the RGD motif. J Biotechnol 2011;155:193–202. [DOI] [PubMed] [Google Scholar]
- 81. Willuda J, et al. High thermal stability is essential for tumor targeting of antibody fragments: engineering of a humanized anti‐epithelial glycoprotein‐2 (epithelial cell adhesion molecule) single‐chain Fv fragment. Cancer Res 1999;59:5758–5767. [PubMed] [Google Scholar]
- 82. Shi L, et al. De novo selection of high‐affinity antibodies from synthetic fab libraries displayed on phage as pIX fusion proteins. J Mol Biol 2010;397:385–396. [DOI] [PubMed] [Google Scholar]
- 83. Kehoe JW, et al. Isolation and optimization for affinity and biophysical characteristics of anti‐CCL17 antibodies from the VH1‐69 germline gene. Protein Eng Des Sel 2014;27:199–206. [DOI] [PubMed] [Google Scholar]
- 84. Buchanan A, et al. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. MAbs 2013;5:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Traxlmayr MW, et al. Directed evolution of stabilized IgG1‐Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta 2012;1824:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Traxlmayr MW, et al. Construction of a stability landscape of the CH3 domain of human IgG1 by combining directed evolution with high throughput sequencing. J Mol Biol 2012;423:397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Traxlmayr MW, et al. Directed evolution of Her2/neu‐binding IgG1‐Fc for improved stability and resistance to aggregation by using yeast surface display. Protein Eng Des Sel 2013;26:255–265. [DOI] [PubMed] [Google Scholar]
- 88. Schaefer JV, Pluckthun A. Engineering aggregation resistance in IgG by two independent mechanisms: lessons from comparison of Pichia pastoris and mammalian cell expression. J Mol Biol 2012;417:309–335. [DOI] [PubMed] [Google Scholar]
- 89. Wozniak‐Knopp G, Stadlmann J, Ruker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS ONE 2012;7:e30083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wozniak‐Knopp G, Ruker F. A C‐terminal interdomain disulfide bond significantly stabilizes the Fc fragment of IgG. Arch Biochem Biophys 2012;526:181–187. [DOI] [PubMed] [Google Scholar]
- 91. Gong R, et al. Engineered human antibody constant domains with increased stability. J Biol Chem 2009;284:14203–14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci USA 2006;103:5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ponsel D, Neugebauer J, Ladetzki‐Baehs K, Tissot K. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules 2011;16:3675–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hasenhindl C, et al. Stability assessment on a library scale: a rapid method for the evaluation of the commutability and insertion of residues in C‐terminal loops of the CH3 domains of IgG1‐Fc. Protein Eng Des Sel 2013;26:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koide A, Jordan MR, Horner SR, Batori V, Koide S. Stabilization of a fibronectin type III domain by the removal of unfavorable electrostatic interactions on the protein surface. Biochemistry 2001;40:10326–10333. [DOI] [PubMed] [Google Scholar]
- 96. Xu L, et al. Directed evolution of high‐affinity antibody mimics using mRNA display. Chem Biol 2002;9:933–942. [DOI] [PubMed] [Google Scholar]
- 97. Parker MH, et al. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high‐affinity binding to vascular endothelial growth factor receptor two. Protein Eng Des Sel 2005;18:435–444. [DOI] [PubMed] [Google Scholar]
- 98. Lipovsek D, et al. Evolution of an interloop disulfide bond in high‐affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single‐domain camelid and shark antibodies. J Mol Biol 2007;368:1024–1041. [DOI] [PubMed] [Google Scholar]
- 99. Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. J Biol Chem 2011;286:27288–27293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gong R, et al. N‐terminal truncation of an isolated human IgG1 CH2 domain significantly increases its stability and aggregation resistance. Mol Pharm 2013;10:2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV‐1 neutralizing epitope and FcRn. PLoS ONE 2012;7:e42288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. North B, Lehmann A, Dunbrack RL Jr. A new clustering of antibody CDR loop conformations. J Mol Biol 2011;406:228–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Desiderio SV, et al. Insertion of N regions into heavy‐chain genes is correlated with expression of terminal deoxytransferase in B cells. Nature 1984;311:752–755. [DOI] [PubMed] [Google Scholar]
- 104. Furukawa K, Shirai H, Azuma T, Nakamura H. A role of the third complementarity‐determining region in the affinity maturation of an antibody. J Biol Chem 2001;276:27622–27628. [DOI] [PubMed] [Google Scholar]
- 105. Hasenhindl C, et al. Creating stable stem regions for loop elongation in Fcabs – insights from combining yeast surface display, in silico loop reconstruction and molecular dynamics simulations. Biochim Biophys Acta 2014;1844:1530–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Batori V, Koide A, Koide S. Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng 2002;15:1015–1020. [DOI] [PubMed] [Google Scholar]
- 107. Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol 2008;381:1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lloyd C, et al. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel 2009;22:159–168. [DOI] [PubMed] [Google Scholar]
- 109. Vaughan TJ, et al. Human antibodies with sub‐nanomolar affinities isolated from a large non‐immunized phage display library. Nat Biotechnol 1996;14:309–314. [DOI] [PubMed] [Google Scholar]
- 110. Knappik A, et al. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol 2000;296:57–86. [DOI] [PubMed] [Google Scholar]
- 111. Prassler J, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol 2011;413:261–278. [DOI] [PubMed] [Google Scholar]
- 112. Zemlin M, et al. Expressed murine and human CDR‐H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 2003;334:733–749. [DOI] [PubMed] [Google Scholar]
- 113. Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol 2006;357:100–114. [DOI] [PubMed] [Google Scholar]
- 114. Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol 2005;348:1153–1162. [DOI] [PubMed] [Google Scholar]
- 115. Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four‐amino‐acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci USA 2004;101:12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fellouse FA, et al. High‐throughput generation of synthetic antibodies from highly functional minimalist phage‐displayed libraries. J Mol Biol 2007;373:924–940. [DOI] [PubMed] [Google Scholar]
- 117. Hackel BJ, Ackerman ME, Howland SW, Wittrup KD. Stability and CDR composition biases enrich binder functionality landscapes. J Mol Biol 2010;401:84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hackel BJ, Wittrup KD. The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein Eng Des Sel 2010;23:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kiyoshi M, et al. Structural basis for binding of human IgG1 to its high‐affinity human receptor FcγRI. Nat Commun 2015;6:6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural basis for PRYSPRY‐mediated tripartite motif (TRIM) protein function. Proc Natl Acad Sci USA 2007;104:6200–6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schneider S, Zacharias M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol Immunol 2012;51:66–72. [DOI] [PubMed] [Google Scholar]
- 122. Saphire EO, et al. Crystal structure of a neutralizing human IGG against HIV‐1: a template for vaccine design. Science 2001;293:1155–1159. [DOI] [PubMed] [Google Scholar]