

Abstract
Plant‐derived diterpenoids serve as important pharmaceuticals, food additives, and fragrances, yet their low natural abundance and high structural complexity limits their broader industrial utilization. By mimicking the modularity of diterpene biosynthesis in plants, we constructed 51 functional combinations of class I and II diterpene synthases, 41 of which are “new‐to‐nature”. Stereoselective biosynthesis of over 50 diterpene skeletons was demonstrated, including natural variants and novel enantiomeric or diastereomeric counterparts. Scalable biotechnological production for four industrially relevant targets was accomplished in engineered strains of Saccharomyces cerevisiae.
Keywords: biosynthesis, biocatalysis, combinatorial biosynthesis, cyclizations, terpenes
Diterpenoids are a diverse class of structurally complex natural products with broad therapeutic applications. Plants provide the major source of these chemicals and present a rich reservoir of potentially new pharmaceuticals. Exploitation of this diversity is severely limited by difficulties in extracting pure compounds from natural sources on an industrial scale. Alternatives include semisynthesis from naturally occurring precursors, plant cell cultures, and chemical synthesis.1 Stereospecific synthetic routes for complex diterpenoids often remain commercially impracticable owing to their low efficiency.2 Combinatorial metabolic engineering of microbial or plant hosts, using the enzymes of the biosynthetic pathways, offers an alternative environmentally benign access to structurally complex diterpenoids with specific and novel stereochemistry, which often defines the therapeutic value.3 The carbon skeletons of bi‐ and polycyclic labdanes and clerodanes are formed in a two‐step enzyme reaction catalyzed by bifunctional class I/II diterpene synthases (diTPSs) or pairs of distinct monofunctional diTPSs defined by their catalytic activity as class I or class II enzymes.4 Class II diTPSs catalyze the initial cyclization of (E,E,E)‐geranylgeranyl diphosphate (GGPP) into bicyclic diphosphate intermediates with a characteristic decalin core (Scheme 1). The structure and specific stereochemistry at the C9 and C10 carbon atoms of the intermediate are determined by the class II diTPS catalyzed reaction. The subsequent class I diTPS reaction completes the formation of the diterpene skeleton through dephosphorylation of the class II product and specific rearrangements, additional cyclizations, or water addition, thereby resulting in the introduction of additional chiral centers (Scheme 1).4b
Scheme 1.

The modular nature of diterpene biosynthesis. Overview of class II and class I diTPSs and their products. The decalin core is shown in bold. Zm=Zea mays; Cf=Coleus forskohlii; Ss=Salvia sclarea; Os=Oryza sativa; Mv=Marrubium vulgare, Tw=Tripterygium wilfordii.
We have recently identified a diverse suite of 40 class II and class I diTPSs from ten plant species, each harboring structurally exceptional diterpenoids.5 In plants, specific pairs of class I and class II diTPSs form distinct diterpene skeletons, which underpins the structural diversity of native diterpenoids.6 To explore the inherent metabolic potential and plasticity of modular combinations of diTPSs, we systematically paired diTPSs through co‐expression in heterologous hosts. Specifically, we probed the substrate and product plasticity for every combination of eleven class II and nine class I diTPSs representative of diverse plant species and potential functionalities. Eight functionally uncharacterized diTPSs were included in this approach (Table S1 in the Supporting Information).
We report herein the stereoselective formation of more than 50 labdane‐ and clerodane‐type skeletons through the assembly of combinations of diTPS pairs. These skeletons are labelled throughout (1–47) according to their Kovats retention indices (Figure S1–4 and Table S2 in the Supporting Information). The a and b suffixes indicate (+) or ent configurations, respectively, as shown by chiral GC–MS analysis, NMR, and optical rotation measurements of selected purified compounds. Three examples illustrate the potential of this approach for control of the stereochemical configuration of diterpenes through specific selection of class II diTPSs and for extension of the known structural repertoire of skeletons accessible by biosynthesis through combination with class I diTPSs. A list of all of the diterpenes produced is given in Table S2.
DiTPS combinations were tested by Agrobacterium tumefaciens mediated transient expression in Nicotiana benthamiana and the results were supported by in vitro assays with recombinant diTPS expressed and purified from E. coli for each type of class II diTPS. To demonstrate fermentation‐based diterpene production, selected diTPS combinations were introduced into engineered Saccharomyces cerevisiae to yield target molecules at a scale relevant for industrial consideration.
Comparison of substrate and product profiles showed that the class I diTPSs show distinct degrees of substrate and product plasticity with regards to the stereochemistry and hydroxylation pattern of the diphosphate substrate. Specifically, in a total of 12 combinations, a subset of six class I diTPSs converted the two stereoisomers of labda‐13‐en‐8‐ol diphosphate [(+)‐8‐LPP and ent‐8‐LPP, Figure 1].
Figure 1.
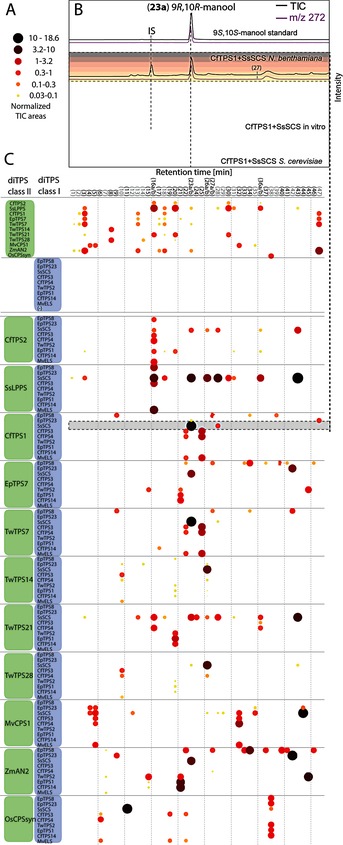
The diterpene chemoscape. A) Levels of diterpenes accumulating in the N. benthamiana/Agrobacterium system (n=3). B) GC–MS chromatograms of extracts from the N. benthamiana control, CfTPS1/SsSCS expressed in N. benthamiana or in S. cerevisiae, the in vitro assay, and an authentic standard of (9S,1(0S‐manool (23 a). Total ion current (TIC) and extracted‐ion chromatogram (EIC) levels are not to scale. C) 2D representation of diterpenes detected in N. benthamiana expressing either a single diTPS or combinations of diTPSs from the two classes (y axis). Diterpenes are sorted on the x axis by their Kovats retention indices. Cf=C. forskohlii; Ss=S. sclarea; Mv=M. vulgare; Ep=Euphorbia peplus; Os=O. sativa; Tw=T. wilfordii; Zm=Z. mays. Diterpenes in samples expressing solely class II diTPSs are likely the result of endogenous phosphatase activity and non‐specific rearrangement (Figure S7).6a, 7
The discrete formation of three stereoisomers of the diterpene manoyl oxide highlights how biosynthetic routes can be stereoselectively controlled (Figure 2). (13R)‐(+)‐manoyl oxide (16 a), a precursor for the diterpenoid pharmaceutical forskolin,6a was formed from (+)‐8‐LPP by the combination of Salvia sclarea SsLPPS with Marrubium vulgare MvTPS5 at 3.7 times higher levels than the native combination of CfTPS2 and CfTPS3 from Coleus forskohlii, which may be relevant for future production. Stereoselective access to manoyl oxide stereoisomers is a prerequisite for the biotechnological production of forskolin derivatives with novel stereochemical configurations. In combination with the class II diTPS TwTPS21 from Tripterygium wilfordii, CfTPS3 catalyzed the stereoselective formation of (13R)‐ent‐manoyl oxide (16 b). Combination of the class I diTPS TwTPS2 with CfTPS14 or Euphorbia peplus EpTPS1, which are unable to convert (+)‐8‐LPP, enabled the stereoselective biosynthesis of the diastereomer (13S)‐ent‐manoyl oxide (20 b; Figure 2 and Figure S16). The ent‐configuration of compounds 16 b and 20 b is consistent with a function of the class II diTPS TwTPS21 in providing specifically (5R,8S,9S,10S)‐labda‐13‐en‐8‐ol diphosphate (ent‐8‐LPP), a potential intermediate of bioactive diterpenoids with this stereochemistry found in nature yet unknown in biosynthetic routes.8
Figure 2.
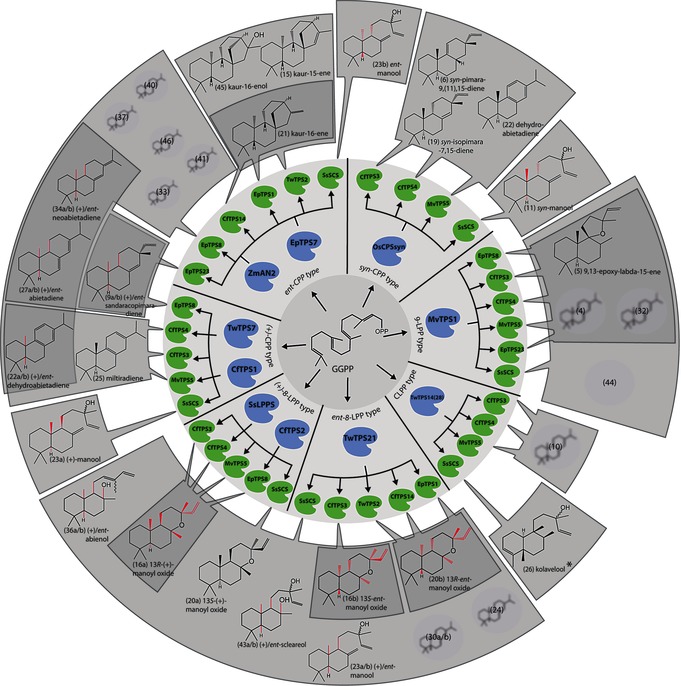
Combinatorial wheel. Formation of diterpenes by natural and engineered diTPS combinations. Bonds in a specified stereochemical configuration formed through stereoselective controlled synthesis are highlighted in red. Bonds in a specified stereochemical configuration for which there is no biosynthetic access to the other configuration are shown in black. The a and b suffixes indicate (+) and ent configurations, respectively, as dictated by the class II diTPS. Diterpenes with unknown structure are shaded in grey. * indicates clerodane diterpenes.
The formation of structurally distinct bicyclic diterpenes, such as sclareol, which is used in high‐end fragrance manufacture, is catalyzed in S. sclarea by SsLPPS and SsSCS.9 Antibiotic activities have been reported for a compound of this class, (+)‐manool,10 which is also recognized as a starting material for the semisynthesis of a wide range of diterpenoids with pharmaceutical properties.11 SsSCS was used as a representative of class I diTPSs that, following rearrangement, facilitate neutralization of the carbocation through water quenching at C13 to give regiospecific introduction of a hydroxy group.9 When offered the three known stereoisomers of copalyl diphosphate (CPP), SsSCS formed products with the stereochemistry preserved at the chiral centers C9 and C10, as defined by the class II diTPSs. Specifically, the pairing of SsSCS with diTPSs forming ent‐CPP, (+)‐CPP, or syn‐CPP, resulted in the stereoselective formation of ent‐manool (23 b), (+)‐manool (23 a), and syn‐manool (11), respectively, both in N. benthamiana and in vitro (Figure 2, Figure S16, and Tables S4, S5). Upon identification of the new combination of diTPSs that efficiently yields 23 a, CfTPS1 and SsSCS were codon optimized and stably integrated into the genome of a strain of S. cerevisiae engineered for diterpene production to afford 23 a at 310 mg L−1 (Table S9), thereby effectively tripling previously reported titers.12
The highly efficient conversion of bicyclic diphosphate intermediates by SsSCS proved instrumental in elucidating the function of an uncharacterized class II diTPS. The combination of TwTPS14 with SsSCS produced a compound consistent with a diTPS function new to plants and only recently reported in the bacterium Herpetosiphon aurantiacus.13 Purification and NMR analysis established the structure as kolavelool (26), from the distinct class of clerodane‐type diterpenes (Table S6).4b These diterpenes have been reported from a range of plant species and form the skeleton of salvinorin A, a lead drug candidate for the treatment of mental disorders and substance dependence,14 yet the biosynthetic route remains unknown. Similarly, coupling of two functionally uncharacterized class II diTPSs with the nine class I diTPSs and comparison of the product profiles with those of established enzymes enabled the classification of EpTPS7 and TwTPS7 as ent‐CPP synthases (Figure 2). Furthermore, combining ent‐CPP synthases with EpTPS8 or EpTPS23 resulted in the formation of ent‐neoabietadiene and ent‐dehydroabietadiene, respectively (Figure S14), diterpene scaffolds not previously reported in nature. Likewise, the TwTPS21/SsSCS combination yielded ent‐sclareol, which constitutes the first biosynthetic route to this specific stereoisomer.
Several of the combinations provided skeletons of high‐value diterpenoids, for example, miltiradiene (25), the backbone of cardioprotectives (tanshinones)15 and antioxidants (carnosoic acid),16 16 a and 23 a as precursors for the drug forskolin,6a and the synthetic ambergris substitute (−)‐ambrox, which is used in high‐end fragrances.17 For stereoselective production of these targets, expression‐optimized variants of CfTPS1/CfTPS3, CfTPS2/CfTPS3, and TwTPS21/EpTPS1 were introduced stably into the genome of a S. cerevisiae strain engineered for diterpene production. Production titers between 250–370 mg L−1 (Table S9) underline the potential for scale‐up production by fermentation.
In summary, we show that engineered diTPS combinations can be used to expand the biosynthetically accessible repertoire of diterpene skeletons with superior stereochemical control. Our approach meets a long‐standing desire for control over the stereochemical configuration of synthesized diterpenes, which remains a challenge in formal chemical synthesis. With chiral molecules representing more than half of approved drugs worldwide, collectively valued at over $200 billion in 2008,18 our findings make an important contribution to accelerate drug development and demonstrate how synthetic biology can complement chemical synthesis for the production of stereochemically complex small molecules.
Experimental Section
Generation of expression constructs, expression in N. benthamiana, S. cerevisiae and E. coli, extraction analysis of products, and structural elucidation by NMR were done as previously described and are given in supplementary material and methods.6a, 19
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Reuben Peters (Iowa State University) for providing ZmAN2 and OsCPSsyn and for discussions, Dr. C. E. Olsen for initial NMR analysis, Drs. J.‐É. Bassard and A. M. Heskes for discussion and experimental assistance, Dr. N. J. Clausen (Technical University of Denmark) for aid in the visualization of the diterpene chemoscape. This work was supported by a VILLUM research center of excellence (B.L.M.), the Center for Synthetic Biology supported by the UCPH Excellence Program for Interdisciplinary Research (Bj.H. and B.L.M.), the Novo Nordisk Foundation (Bj.H.), The Danish Council for Strategic Research (Bj.H.), an ERC Advanced Grant to BLM (ERC‐2012‐ADG_20120314), an NSERC Discovery Grant to J.B., and UC Davis, College of Biological Sciences funds to P.Z.
J. Andersen-Ranberg, K. T. Kongstad, M. T. Nielsen, N. B. Jensen, I. Pateraki, S. S. Bach, B. Hamberger, P. Zerbe, D. Staerk, J. Bohlmann, B. L. Møller, B. Hamberger, Angew. Chem. Int. Ed. 2016, 55, 2142.
References
- 1.
- 1a. Paddon C. J., et al., Nature 2013, 496, 528; [DOI] [PubMed] [Google Scholar]
- 1b. Wilson S. A., Roberts S. C., Plant Biotechnol. J. 2012, 10, 249; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Holton R. A., Kim H. B., Somoza C., Liang F., Biediger R. J., Boatman P. D., Shindo M., Smith C. C., Kim S., J. Am. Chem. Soc. 1994, 116, 1599; [Google Scholar]
- 1d. Jørgensen L., McKerrall S. J., Kuttruff C. A., Ungeheuer F., Felding J., Baran P. S., Science 2013, 341, 878. [DOI] [PubMed] [Google Scholar]
- 2. Appendino G., Angew. Chem. Int. Ed. 2014, 53, 927; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 943. [Google Scholar]
- 3.
- 3a. Chen F., Tholl D., Bohlmann J., Pichersky E., Plant J. 2011, 66, 212; [DOI] [PubMed] [Google Scholar]
- 3b. Pateraki I., Heskes A., Hamberger B. in Biotechnology of Isoprenoids, Vol. 148 (Eds.: J. Schrader, J. Bohlmann), Springer International Publishing, 2015, p. 107. [Google Scholar]
- 4.
- 4a. Zerbe P., Bohlmann J., Trends Biotechnol. 2015, 33, 419; [DOI] [PubMed] [Google Scholar]
- 4b. Peters R. J., Nat. Prod. Rep. 2010, 27, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zerbe P., et al., Plant Physiol. 2013, 162, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Pateraki I., et al., Plant Physiol. 2014, 164, 1222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Cyr A., Wilderman P. R., Determan M., Peters R. J., J. Am. Chem. Soc. 2007, 129, 6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zerbe P., Chiang A., Dullat H., O'Neil-Johnson M., Starks C., Hamberger B., Bohlmann J., Plant J. 2014, 79, 914. [DOI] [PubMed] [Google Scholar]
- 8. Gray C. A., Rivett D. E., Davies-Coleman M. T., Phytochemistry 2003, 63, 409. [DOI] [PubMed] [Google Scholar]
- 9. Caniard A., Zerbe P., Legrand S., Cohade A., Valot N., Magnard J.-L., Bohlmann J., Legendre L., BMC Plant Biol. 2012, 12, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moreira M. R., et al., Rev. Bras. Farmacogn. 2013, 23, 870. [Google Scholar]
- 11.
- 11a. van Wyk A. W. W., Lobb K. A., Caira M. R., Hoppe H. C., Davies-Coleman M. T., J. Nat. Prod. 2007, 70, 1253; [DOI] [PubMed] [Google Scholar]
- 11b. Salazar F. J., Villamizar J. E., J. Chem. Res. 2013, 37, 63; [Google Scholar]
- 11c. Salazar F. J., Villamizar J. E., J. Chem. Res. 2013, 37, 1. [Google Scholar]
- 12. Ignea C., Ioannou E., Georgantea P., Loupassaki S., Trikka F. A., Kanellis A. K., Makris A. M., Roussis V., Kampranis S. C., Metab. Eng. 2015, 17, 91. [DOI] [PubMed] [Google Scholar]
- 13. Nakano C., Oshima M., Kurashima N., Hoshino T., ChemBioChem 2015, 16, 772. [DOI] [PubMed] [Google Scholar]
- 14. Kutrzeba L., Dayan F. E., Howell J. L., Feng J., Giner J.-L., Zjawiony J. K., Phytochemistry 2007, 68, 1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo J., et al., Proc. Natl. Acad. Sci. USA 2013, 110, 12108.23812755 [Google Scholar]
- 16. Brückner K., et al., Phytochemistry 2014, 101, 52. [DOI] [PubMed] [Google Scholar]
- 17. Schenk H. R., Gutmann H., Jeger O., Ruzicka L., Helv. Chim. Acta 1954, 37, 543. [Google Scholar]
- 18. Caldwell J., Hum. Psychopharmacol. 2001, 16, S67. [Google Scholar]
- 19. Bach S. Spanner, Bassard J.-E., Andersen-Ranberg J., Emil Møldrup M., Toft Simonsen H., Hamberger B. in Methods of Molecular Biology: Plant isoprenoids, Vol. 1153 (Ed.: J. M. Walker), Springer, Heidelberg, 2014, p. 330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary