

Summary
Monoclonal antibodies are essential therapeutics and diagnostics in a large number of diseases. Moreover, they are essential tools in all sectors of life sciences. Although the great majority of monoclonal antibodies currently in use are of mouse origin, the use of human B cells to generate monoclonal antibodies is increasing as new techniques to tap the human B cell repertoire are rapidly emerging. Cloned lines of immortalized human B cells are ideal sources of monoclonal antibodies. In this review, we summarize our studies to the regulation of the replicative life span, differentiation, and maturation of B cells that led to the development of a platform that uses immortalization of human B cells by in vitro genetic modification for antibody development. We describe a number of human antibodies that were isolated using this platform and the application of the technique in other species. We also discuss the use of immortalized B cells as antigen‐presenting cells for the discovery of tumor neoantigens.
Keywords: germinal center, antibodies, BCL6, B cells, rabbit
This article is part of a series of reviews covering Immunoglobulins: from genes to therapies appearing in Volume 270 of Immunological Reviews.
Introduction
The year 2015 marks the 40th anniversary of a landmark paper that would have a huge impact on the emerging field of biotechnology 1. In this paper, Köhler and Milstein describe a protocol to immortalize antibody‐producing B cells by fusing them to myeloma cells. The resulting fusion product combined the antibody‐producing capacity of the original B cell or plasma cell and the immortality of the myeloma cells. Cloned lines of the fusion products were called hybridomas and produced relatively large amounts of antibodies, which were called monoclonal antibodies. The impact of the hybridoma technology cannot be underestimated. Nowadays monoclonal antibodies have found applications in virtually all branches of life sciences. The therapeutic potential of monoclonal antibodies was quickly recognized resulting in a large number of lifesaving therapeutic and prophylactic antibodies that are currently produced and marketed by the pharmaceutical industry.
The first generation of antibodies developed for clinical applications were mouse monoclonal antibodies as both the myeloma fusion partners and the B cells were of mouse origin. The first antibody approved for the clinic was OKT3, an antibody against the human CD3ε component of the T‐cell receptor complex 2. OKT3 was approved for treatment in transplantation and deleted T cells, thereby reducing the damaging effects of T cell‐mediated antigraft reactions. The clinical utility of OKT3 turned out to be limited by its immunogenicity in humans, among other problems, as OKT3 is a mouse antibody. Second‐generation therapeutic antibodies were chimeric antibodies in which the constant domains of the mouse immunoglobulin were replaced with those of human. Two examples of such chimeric antibodies are rituximab, an antibody against the B‐cell antigen CD20, which is used for therapy in B‐cell malignancies and various other diseases, and infliximab, an antibody against tumor necrosis factor (TNF)‐α used in inflammatory diseases such as Crohn's disease and rheumatoid arthritis. Advances in antibody engineering resulted in techniques to fully humanize the mouse antibodies by grafting the complementarity determining region (CDR)3 regions into the human immunoglobulin (Ig)G framework. Although humanization of mouse antibodies is still used, animals with human Ig genes are now being used by a growing number of companies to raise therapeutic antibodies. In parallel with the use of mice with human Ig genes and the technical advances with antibody humanization, other technologies have been developed to directly generate fully human antibodies. Phage display is one of those technologies. This method uses a library of bacteriophages that have been engineered to display antibody fragments on their surface. These bacteriophages are screened for binding to target proteins in order to identify the antibody fragments of interest (reviewed in 3). The technology has an advantage over animal‐based methods as human B cells can be used to make the Ig libraries representing the naturally occurring repertoire. However, phage display antibodies are frequently unstable because of their artificial pairing of heavy and light chains. Currently, only three antibodies have been approved for clinical use, amongst which the TNF‐α‐specific antibody adalimumab.
Other methods to generate fully human antibodies involve PCR or direct RNA sequencing of single plasma cells or B‐cell blasts. The latter can be selected by flow cytometry using labeled antigen. These methods require prior knowledge of the antigen and substantial frequencies of antigen‐specific B cells in the samples.
Immortalization of human B cells provides us with a method that copes with the difficulty to isolate rare antibodies provided that the technology to immortalize B cells is efficient. This includes capturing the complete B‐cell repertoire and the generation of stable immortalized B cells. Although fusion of B cells and myeloma cells has been the method of choice to immortalize antibody‐producing mouse B cells, there are several drawbacks that limit its applicability to isolate rare B cells. First is that the efficiency of immortalization is rather low. Only a very low proportion of the total mouse B‐cell repertoire is captured by the hybridoma technology and clones are often instable. Secondly, whereas the technology works well with mouse and rat B cells, attempts to generate hybridomas with human B cells had limited success. This is caused by the lack of good fusion partners for the human B cells. Recently, the success rate has been improved by the development of better fusion partners and technical advances in using electrofusion 4, 5.
Epstein–Barr virus (EBV) transformation is a commonly used way to immortalize human B cells 6. The frequency of transformation which was traditionally low was increased considerably, from <10% to >30% by using TLR9 agonist 7. Using this technology, the group of Lanzavecchia has generated a number of broadly reacting highly neutralizing antibodies against a variety of pathogenic viruses 8.
Clones of human B cells have also been obtained by expanding individual cells with CD40L and cytokines like interleukin (IL)‐2, IL‐4, IL‐10, and IL‐21 9. Although a number of interesting monoclonal antibodies have been generated (for instance, 10), this technology has its limitations as B cells expand transiently in this protocol. Therefore, this method offers only a limited window of opportunity for finding the desired antigen‐specific clone. One possible reason for the limited proliferative capacity of B cells stimulated by CD40L and cytokines is that the activated B cells differentiate to plasma cells in this setting. Plasma cells are terminally differentiated cells and unable to proliferate. The observation by our group that forced expression of BCL‐6 (which is known to inhibit terminal differentiation to plasma cells) in activated B cells enables these cells to continue proliferating in response to cytokines and CD40L is consistent with this hypothesis 11, 12. In this review, we will give an overview of our work on regulation of self‐renewal of B cells and how this knowledge led to the development of a convenient antibody discovery platform that is applicable to a wide range of species.
Regulation of self‐renewal of human B cells by STAT5 and BCL‐6
B cells develop and are selected in the germinal center (GC) 13 structures in secondary lymphoid organs which arise as a consequence of antigen stimulation. GCs consist of two distinct areas called the dark (DZ) and light zones (LZ), respectively. The GC microenvironment contains a multiple of cell types that provide signals, including cytokines and chemokines that support proliferation, migration, and differentiation of B cells. Cytokines that support B‐cell differentiation in the GC include IL‐2, IL‐4 and IL‐21, all of which activate signal transducers of activation and transcription (STAT), particularly STAT3 and STAT5 14.
CD25+ B cells that expressed phosphorylated STAT5 were found in the LZ of GCs of human tonsils suggesting that STAT5 plays a role in B cells in humans 15. STAT5 is likely to be involved in the proliferation of human B cells as knockdown of STAT5 using RNA interference strongly decreased proliferation of B cells stimulated with CD40L in the presence of IL‐2 and IL‐4 15. To determine the exact role of STAT5 in B‐cell functions, we expressed a fusion of STAT5 with a mutated domain of the estrogen receptor (ER) and STAT3 in activated B cells and examined the response of these cells following incubation with 4‐hydroxy tamoxifen (4 HT). It was observed that addition of 4 HT strongly enhanced the proliferative response of B cells. Interestingly, ectopic expression of a constitutive active form of STAT5 resulted in long‐term in vitro proliferation of B cells cultured with CD40L and cytokines, whereas control transduced B cells proliferated only for a limited period of time. These results contradict those of studies in mouse models that have demonstrated that STAT5 is involved in early B‐cell development but not in B‐cell maturation. Deletion of STAT5 in B cells using CD19 CRE and floxed STAT5 alleles did not result in diminished antibody production 16. Also, STAT5‐deficient mouse B cells proliferate normally in response to IgM stimulation and IL‐4 16. Perhaps the growth‐promoting effect of IL‐4 in mice is exclusively mediated by STAT6, whereas in humans STAT5 may be involved in this process as well.
The continued expansion of human B cells by constitutive activation of STAT5 is most likely mediated by control of its target BCL‐6 because forced expression of BCL‐6 in human B cells also resulted in sustained proliferation of human B cells in response to cytokines and CD40L 15, 17. The effects of overexpression of active STAT5 in human B cells are however not identical to those of BCL‐6. Most notably, continued overexpression and activation of STAT5 eventually result in downregulation of Ig gene expression and other B cell markers, presumably because of epigenetic repression 18. STAT5‐overexpressing cells eventually acquire features of Hodgkin lymphoma cells 19.
BCL‐6 is highly expressed in GC B cells and studies in mouse have demonstrated that BCL‐6 is essential for the formation of GC 20. BCL‐6 functions to support proliferation and to inhibit differentiation of proliferating B cells to plasma cells in mice 20 and humans 11. BCL‐6 also allows activation‐induced cytidine deaminase (AID)‐mediated somatic hyper mutations (SHM) and class switch recombinations (CSR) which involves extensive DNA modifications by counteracting a DNA damage response. BCL‐6 regulates AID through repression of the microRNA, mir‐155 21. Plasma cells are characterized by the expression of a different set of transcription factors – the most important are BLIMP‐1 (encoded by PRDM1), which is essential for plasma cell differentiation 22, and XBP‐1, which is needed for the formation of the machinery to secrete large amounts of antibodies 23, 24. BCL‐6 and BLIMP1 crossregulate each other as BCL‐6 protein can bind to the PDRM1 locus and repress expression of BLIMP1, thereby inhibiting plasma cell differentiation 11, 25. The ratio of BCL‐6 and BLIMP1 is therefore one of the determining factors whether an activated B cell is poised to become a plasma cell or a memory cell type. The finding that forced expression of BCL‐6 inhibits plasma cell differentiation and allows for sustained proliferation of activated B cells was confirmed in other species including non‐human primates, rabbits, llamas, and mice.
Forced expression of BCL‐6 in activated human B cells induces a germinal center phenotype
Ex vivo isolated human memory B cells do not express BCL‐6 protein. It is therefore unlikely that BCL‐6 is needed for maintenance of a memory state of human B cells. In line with this, upon forced expression of BCL‐6 in activated peripheral blood B cells cultured with cytokines and CD40L these cells acquire features of GC B cells. More specifically, the BCL‐6‐overexpressing cells show similarities to plasmablasts as they produce immunoglobulin but also express B‐cell receptor (BCR) on the cell membrane 12.
Not only do BCL‐6 transduced peripheral blood‐derived memory B cells express cell surface antigens that are also found on GC B cells, they also express AID 12, 13. This enzyme mediates two important processes in GC B cells – SHM and CSR 26. AID is functional in BCL‐6‐expressing B cells as cloned lines of BCL‐6‐expressing human B cells show mutations in the IgG H and L chains of the monoclonal antibody accumulating over time. Intriguingly, however, CSR does not occur in the BCL‐6+ B cells indicating that SHM and CSR are differentially regulated. That CSR and SHM use different domains of AID and therefore can be uncoupled from SHM and gene conversion has been shown before. However, the mechanisms underlying the lack of CSR in B cells that undergo SHM is presently unknown. Taken together, BCL‐6 seems to be a master regulator conferring a GC phenotype and function to peripheral blood memory B cells.
IL‐21 is a strong inducer of human B‐cell maturation by inducing STAT3
Observations in patients suffering from an autosomal dominant hyper‐IgE syndrome (AD‐HIES) have established a critical role of STAT3 in the regulation of B‐cell maturation. AD‐HIES is caused by mutations in STAT3 resulting in expression of dominant negative STAT3 which reduces STAT3 function 27, 28. These patients show a high susceptibility to microbial pathogens due to deficiencies in the functions of a variety of immune cells. T‐cell‐dependent antibody production is strongly affected. Although STAT3 deficiency impairs the function of T follicular helper cells, thereby hampering B‐cell help 29, deficiencies in STAT3 function also intrinsically affect the capacity of B cells to differentiate into antibody‐secreting plasmablasts 30. There are several cytokines that can induce STAT3 in activated B cells including IL‐10 and IL‐21. Of those, IL‐21 is most likely the dominant STAT3 inducer in B cells as patients with IL‐21R mutations have deficiencies in antibody responses 31. Moreover, IL‐21 most strongly induces expression of PDRM1 and XBP‐1 in activated naive and memory B cells resulting in plasma cell differentiation and increased antibody production 11, 32. To study the effects of STAT3 activation in the absence of cytokine signaling, we followed the same approach as with STAT5, expressing a fusion of STAT3 with a mutated domain of ER in activated B cells and examined the response of these cells following incubation with 4 HT. These experiments revealed that activation of STAT3 itself increased expression of BLIMP1 and XBP1 similar to IL‐21, suggesting that these transcription factors are controlled by STAT3 11. However, whereas BCL‐6 was induced by IL‐21, it was not induced by STAT3. Because IL‐21 transiently induces STAT5 phosphorylation and BCL6 is a direct target of STAT5 in human B cells 15, it is likely that IL‐21 regulates BCL‐6 through STAT5. However, STAT5 and BCL‐6 are not sustained by IL‐21, and eventually B cells cultured with IL‐21 and CD40L differentiate terminally to plasma cells 11.
Immortalization of human B cells by overexpressing BCL‐6 and BCL‐XL
The knowledge we obtained in our studies of the regulation of human B‐cell maturation provided us with a convenient method to immortalize human B cells by genetic modification rather than by fusion to myeloma cells or by EBV‐mediated transformation. By forced expression of BCL‐6 into B cells by retrovirus‐mediated gene transfer, we prevent those cells from differentiating into plasma cells in a culture system with CD40L‐expressing mouse fibroblasts and cytokines. However, the pool of B cells expressing BCL‐6 expanded very slowly probably caused by the fact that many cells were dying. To offset cell death, we examined whether co‐expression of gene fragments encoding antiapoptotic molecules would prevent cell death of BCL‐6‐transduced cells. Indeed co‐expression of a variety of genes encoding BCL‐2 family members resulted in a strong inhibition of death of BCL‐6‐transduced cells. The best effect was observed with BCL‐XL but BCL‐2 and MCL1 were also effective (data not shown).
BCL‐6/BCL‐XL‐transduced B cells expand rapidly in response to a variety of cytokines including IL‐4, IL‐10, and IL‐21. The most robust proliferation was achieved with IL‐21. As mentioned in the previous paragraphs, BCL‐6‐expressing cells are inhibited in their differentiation into plasma cells but they are nonetheless capable of secreting significant amounts of antibodies. Besides the differences in effects on proliferation, we also observed differences in the capacities of IL‐4 and IL‐21 to induce production of antibodies. IL‐21 induced the highest antibody titers most likely because, in contrast to IL‐4, it activates STAT3 which promotes the secretion of antibodies.
The combination of BCL‐6 and BCL‐XL overexpression and the CD40L/IL‐21 culture system provides us with B cells that have several properties that enable the isolation of antigen‐specific antibodies (Fig. 1). B cell clones can be selected based on antibodies that are secreted. The culture supernatant can be used to test specific antibody properties, e.g. inhibition of viral infection, cell binding, antibody‐dependent cell‐mediated cytotoxicity, or complement‐dependent cytotoxicity. Using this direct functional screening approach, we identified the respiratory syncytial virus (RSV)‐specific antibodies D25, AM14, AM16, and AM23 12, cross‐neutralizing antibodies against Parecho virus 33, and the Staphylococcus aureus‐specific antibody rF1 34. Antibody secretion is maintained and stable during B‐cell culture allowing multiple rounds of cellular cloning and the ability to perform multiple screening assays during the selection process 35.
Figure 1.
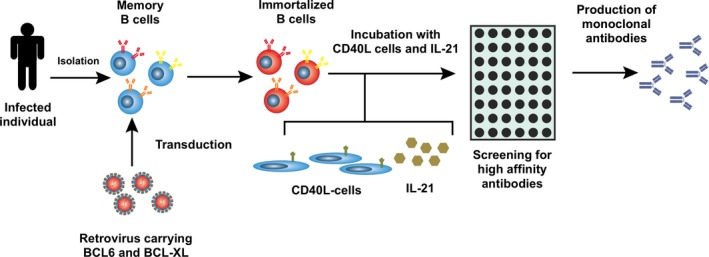
Generation of self‐renewing antibody‐producing B cells. B‐cells are isolated from selected individuals, stimulated, and transduced with a retrovirus containing BCL6 and BCL‐XL. Subsequently, transduced B cells are expanded on CD40L expressing fibroblasts in the presence of interleukin (IL)‐21. B‐cell clones are screened for production of desired antibodies.
High levels of BCR expression are detected on transduced cells compared to normal B cells 12. Fluorescently tagged antigens will bind to specific BCRs enabling the isolation of those specific B cells from a polyclonal pool using a flow cytometer. Labeled antigens that can be used include small peptides, monomeric proteins, protein complexes but also cell wall fractions and virus‐like particles. Additional rounds of antigen‐specific B‐cell selection can be done using different proteins to select for B cells that recognize a common conserved epitope on both antigens. We have successfully applied antigen‐specific B‐cell sorting for the isolation of tetanus toxoid antibodies 12, and broadly neutralizing influenza antibodies 36, 37, 38.
Isolation of monoclonal antibodies from immortalized human B cells and application in vaccine development
Antibodies against RSV
RSV infection is the main cause of lower respiratory tract disease. In the developing world RSV is the main viral etiology for severe pneumonia and bronchiolitis contributing considerably to the burden on health services. Especially premature neonates and children with bronchopulmonary dysplasia or congenital heart disease are at risk of pathological consequences of RSV infection 39, 40, 41. Nair et al. estimated that RSV accounted for ~3 million hospitalizations and between 66,000 and 199,000 deaths per year mainly occurring in developing countries 42.
There is currently no vaccine that confers protection against RSV but a humanized monoclonal antibody specific for the RSV F protein (palivizumab) is prophylactically administered to children at risk 43, 44. Early protection against RSV is important as lower respiratory tract infection early in life, especially in high‐risk infants, is associated with wheezing and asthma during infancy and later in life. RSV, but also other viral etiologies, may cause or enhance airway sensitization that could result in hyper‐responsive airways and asthma 45, 46, 47, 48, 49, 50, 51. Prophylactic treatment of children may therefore result in a significant reduction in wheezing days during the first year of life and the development of asthma later in life 45, 52. The high‐dose requirements of palivizumab make this medicine less useful for treatment of large groups of newborns and treatment is only possible for very young infants weighing less than 4 kg. Therefore, the search for other medicines and for more potent neutralizing antibodies continues.
We reasoned that humans exposed to RSV would be a superior source of neutralizing antibodies compared to mice. Using our B‐cell immortalization technology, we set out to identify RSV‐specific antibodies better or equally potent as palivizumab. In order to screen directly on neutralizing activity, we developed a sensitive high‐throughput neutralization assay using a fluorescent‐based detection method to distinguish and quantify infected from non‐infected cells. Using memory IgG+ B cells derived from peripheral blood from healthy individuals including daycare providers who should be frequently exposed to respiratory viruses, we isolated a series of B‐cell clones producing RSV F‐specific antibodies 12. Of these antibodies, D25, AM23, and AM14 neutralize RSV A and B strains with a potency 100‐fold greater than palivizumab. We demonstrated that these antibodies are specific for an F structural determinant that is not present on inactivated virus or viral lysates. The reason for this was unraveled when McLellan et al. determined the exact structure of the F protein by crystallography. It became clear that the structure targeted by the antibodies D25, AM23, and AM14 was the prefusion F trimer 53. The epitope on F recognized by D25 was annotated as the ø domain, which is an epitope located at the membrane‐distal apex of the RSV F glycoprotein, and thereby D25 locks the F protein in its prefusion state. AM14 is binding another quaternary epitope between 2 F protomers making it extremely specific for binding prefusion F trimers 54. In vitro, these F‐specific antibodies neutralize a broad selection of subgroup A and subgroup B RSV clinical isolates. The advantage of prefusion‐specific antibodies is that they only recognize F protein in its native conformation on virus particles and on infected cells and do not recognize free monomeric protein which is probably more abundantly present during infection and may serve as a sink for antibodies like palivizumab which recognize both pre‐ and postfusion F structures. Antibodies D25, AM23, and AM14 also have potent in vivo activity in cotton rats 12.
Altogether, antibodies like D25, which is now tested in vivo in premature infants under the name of MEDI8897, may be very useful in preventing RSV infection in the first months of life when given prophylactically immediately after birth. To give full protection after birth, the serum half‐life of MEDI8897 is prolonged by specific YTE mutations in the Fc tail thereby increasing its affinity for the FcRn receptor. This should keep serum antibody levels high enough to give protection for the whole RSV season 55. This was confirmed in a phase I study in which the YTE mutation resulted in an astonishing half‐life of the modified D25 antibody of 90 days. It is therefore likely that only one injection during the RSV season is sufficient to confer protection whereas four injections of palivizumab are needed to confer protection. This protection against RSV disease in early life is important to prevent serious damage to the lungs and recurrent wheezing as discussed above.
Broadly reactive highly neutralizing antibodies inform vaccine development
It was reported recently that the RSV‐neutralizing potency of serum depends mainly on the presence of prefusion F‐specific IgG 56, 57. Therefore, to protect infants and elderly later in life against lower respiratory tract disease caused by RSV, a vaccine is preferred that increases serum levels of RSV F prefusion‐specific antibodies. Extensive efforts are underway to develop and test RSV vaccines. However, in the most advanced trial induction of neutralizing palivizumab‐like antibodies was used as an endpoint, whereas it is now clear that such antibodies are inferior to prefusion F protein‐specific antibodies like D25 58. Recently, a prefusion F‐specific vaccine has been developed based on the knowledge of the epitope recognized by D25 under the assumption that such a vaccine should be able to preferentially elicit highly neutralizing prefusion F‐specific antibodies 53.
As mentioned in the following paragraph, we were also able to develop potent broadly reacting antibodies against the influenza stem region of group 2 influenza viruses 36, 37, 38, which should inform generation of an efficacious influenza virus vaccine similar to that done for group 1 influenza 59, 60. We have also generated broadly reactive highly neutralizing antibodies to human parechovirus 33, 61, a virus of the picornaviridea family, hCMV and HCV. It is likely that knowledge of the epitopes recognized by these antibodies will also inform vaccine development.
Antibodies against influenza virus
The emergence of highly pathogenic H5N1 avian influenza infections in humans since 1997 renewed the interest in influenza infection therapeutics and prevention, especially in neutralizing antibodies against influenza hemagglutinin (HA), a major glycoprotein on the cell surface of the virus. HA mediates viral binding by interacting with sialic acids on glycoproteins or glycolipids of the host cells, therefore antibodies that block this binding site prevent viral infection. Neutralizing antibodies are almost always directed against the HA protein. Influenza viruses rapidly mutate most of these antibody‐binding sides (known as antigenic drift) preventing long‐lasting immunity against the virus. Eighteen subtypes of HA have been identified which cluster in two groups 62 based on their sequence homology (group 1 comprises H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18 and group 2 comprises H3, H4, H7, H10, H14, and H15). Of note, H17 and H18 have been recently identified in bats and do not bind to sialic acids (reviewed in 63).
The isolation of the broadly neutralizing antibody C179 64 from H2N2‐immunized mice showed that influenza neutralizing antibodies can also target HA epitopes that are conserved in different influenza subtypes. Recently, such broad neutralizing antibodies were also found in humans vaccinated with an influenza virus vaccine. The search for human H5N1 neutralizing antibodies resulted in the isolation of the antibodies CR6261 65 and F10 66 that neutralized most group 1 influenza viruses. Although these antibodies are directed against HA and prevent viral infection, they do not block the sialic acid binding site located at the top of HA molecule (HA1) but inhibit the conformational changes needed for the fusion of the viral and endosomal membranes 66, 67. This is mediated by binding of the antibody to a conserved hydrophobic pocket in the stem domain of HA (HA2). Both human antibodies employ the heavy chain germline gene VH1‐69 and bind solely via the heavy chain; the light chain is not involved. Subsequent studies have identified the critical amino acids involved in the interaction and showed that a large proportion of these residues are germline encoded with only minimal contribution of mutated residues 68, 69.
As the C179, CR6261, and F10 antibodies only protect against group 1 influenza infection, we set out to isolate group 2 neutralizing antibodies. We first enriched for H3‐specific B cells by incubating the transduced polyclonal B cell pool from an influenza vaccinated donor with fluorescently labeled H3. B cells binding the labeled protein were isolated and cultured after which the antibody containing culture supernatants were tested for their cross‐binding potential for H3 and H7 HA. This procedure let to the identification by our group of antibodies 40C7, which was renamed CR8020 36, 37, and 55B4 (CR8043) 37, which were the first human broadly reacting group 2 influenza‐specific antibodies identified. Both antibodies bind a similar site in the HA stem region in close proximity to the viral membrane, more membrane proximal than the conserved epitope described for the group 1 antibodies. As for the broadly neutralizing group 1 antibodies, CR8020 and CR8043 do not inhibit viral infection by prohibiting host receptor binding but they block the conformational changes in the HA protein needed for viral membrane fusion and inhibit HA processing 36, 37. In an independent subsequent study we identified several other broadly neutralizing group 2‐specific antibodies (38 and data not shown). AT10‐002 is the most potent antibody and shows neutralizing capacity for all group 2 influenza subtypes tested 38. AT10‐002 binds a different epitope on the stem region of H3 than CR8020 because it reacts with a CR8020 escape mutant of the highly pathogenic H7N9 influenza virus.
FI6v3 70, 39.29 71 and CR9114 72 antibodies are the only antibodies reported to neutralize both group 1 and group 2 influenza A viruses. They bind the stem region of the HA molecule and protect mice from lethal influenza infection. With the exception of CR9114, all group 2 and pan‐influenza‐specific antibodies are found using screening systems utilizing life B cells and screening of the natively paired light and heavy chain antibodies. In agreement with this, the light chains of these antibodies have a very large contribution in target binding. In contrast, CR9114 is found using phage display, and target binding is mediated solely via heavy chain interactions.
Application of AID expression in immortalized B cells
A hallmark of the GC reaction is the occurrence of somatic hypermutations in the variable domains of the Ig locus in B cells. B cell clones with increased antigen‐affinity are selected to generate an effective antibody response 73. AID (encoded by AICDA) is one of the key enzymes that regulate somatic hypermutation and is expressed in GC B cells 26. Employing the activity of AID, one can select B cells that have mutations in the immunoglobulins they produce. Using the AID‐expressing chicken‐derived DT40 cell line and Ramos B cells 74, sub‐lines were generated which recognize several different antigens, even though these B cells express a single rearranged Ig gene. Using mammalian cell display, affinity maturation can be mimicked in vitro by co‐expressing antibody and AID in the HEK293 cell line. Following selection, clones with increased affinity or improved biophysical properties could be identified 75, 76.
In line with their GC‐like phenotype AICDA transcripts are expressed in BCL6/BCL‐XL‐transduced B cells in similar quantities as tonsil‐derived GC B cells 12. Sequence analysis of transduced B cells revealed that mutations are present in the Ig locus and that they are mainly localized in the CDRs and framework region‐3 indicating that the occurrence of these mutations was site‐directed and not random. The Ig variable mutation rate in transduced B cells is between 8.85 × 10−5 and 5.14 × 10−5 mutations per base pair per cell division, which is at the lower end of the estimated AID‐mediated mutation rate in vivo (1 × 10−3 to 1 × 10−5) 77. This ongoing somatic hypermutation does not lead to the loss specificity of the whole B‐cell pool as mutated B cells only form a very small part of the total population of cultured BCL6/BCL‐XL‐transduced B cells. To retrieve the small subset of mutated B cells within this pool, we stain the cells with labeled antigen and isolate those cells with either low or high binding avidity by flow cytometry (Fig. 2). This way we isolated subclones that produced antibodies with five to 10‐fold higher affinities than the parental antibody. It has been documented that AID expression is controlled by the basic helix‐loop‐helix transcription factor E47. The activity of this transcription factor can be inhibited by Id2 and Id3, which bind to E47 forming an inactive complex 78. As a consequence, overexpression of helix‐loop‐helix factor inhibitor of DNA binding‐3 (Id3) strongly reduced AICDA transcripts thereby providing a method to prevent AID‐induced mutations.
Figure 2.
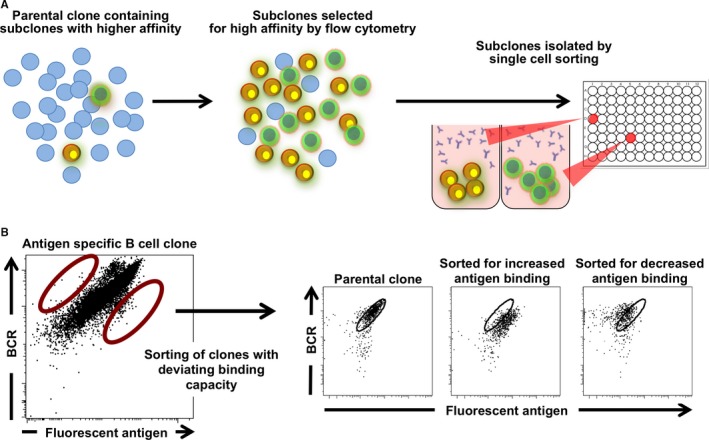
Ongoing somatic hypermutation in BCL 6/ BCL ‐ XL ‐transduced cells allows in vitro affinity maturation of B‐cell clones. (A) A small number of B cells within a large clonal B‐cell pool will acquire mutations in their antibody‐coding region due to activation‐induced cytidine deaminase (AID) activity. Using fluorescently labeled antigen, B cells with enhanced or decreased antigen binding can be selected using flow cytometry. Multiple rounds of sorting can be performed before single cell cloning to achieve optimal binding affinity. (B) Example of an antigen‐specific B cell clone from which subclones with deviating antigen binding capacity were selected. B cells were stained for B‐cell receptor (BCR) expression and subclones that showed increased or decreased antigen binding were obtained after single cell sorting using a flow cytometer and expanded.
In vitro affinity maturation of transduced B cells was done by correlating the BCR expression levels of a B‐cell pool to their antigen binding capacity and specifically selecting B cells that show binding patterns deviating from the median 35. Using this strategy, we could increase the affinity of one of our B cell clones at least sevenfold (from 35 nM to 5 nM) 35. Additionally, loss‐of‐binding mutants were informative for the residues involved in antigen binding, something that cannot be determined by sequence analysis alone. The ability to optimize antibody performance without the need for extensive sequence analysis and molecular cloning is one of the unique properties of the BCL6/BCL‐XL antibody discovery platform.
Application in other species
Due to tolerance to native human proteins the human B‐cell repertoire may not harbor all desired antibody (self) specificities. Therefore, to identify antibodies against human proteins, it is sometimes desirable to immunize animals to obtain such antibodies. Until recently monoclonal antibodies were almost exclusively derived from immunized mice or rats. We explored if immortalization by introduction of BCL‐6 and BCL‐XL could also be applied to non‐human species. B‐cell immortalization could be achieved for non‐human primates 12, mouse, llama, and rabbit B cells (Table 1). Particularly retroviral transduction of rabbit B cells was very efficient (90%) (Table 1 and Fig. 3) and immortalized rabbit cells grow very rapidly with an average doubling time of 18 h, compared to 25–29 h for human cells (Table 1 and Fig. 3). This allows the analysis of the complete B‐cell repertoire within a very short time period. Similar to transduced human B cells, rabbit B cells also secrete immunoglobulin and express BCR on their cell surface allowing antigen‐specific sorting and functional screens on secreted antibodies.
Table 1.
BCL6/BCL‐XL transduction efficiencies of non‐human B cells and the doubling time of the transduced cells
| Transduction efficiency (%) | Doubling time (h) | |
|---|---|---|
| Human | 60–80 | 25–29 |
| Non‐human primates | 5–20 | ND |
| Rabbit | 75–90 | 18–20 |
| Mouse | 60–80 | 30 |
| Llama | 15 | 27 |
ND, not determined.
Figure 3.
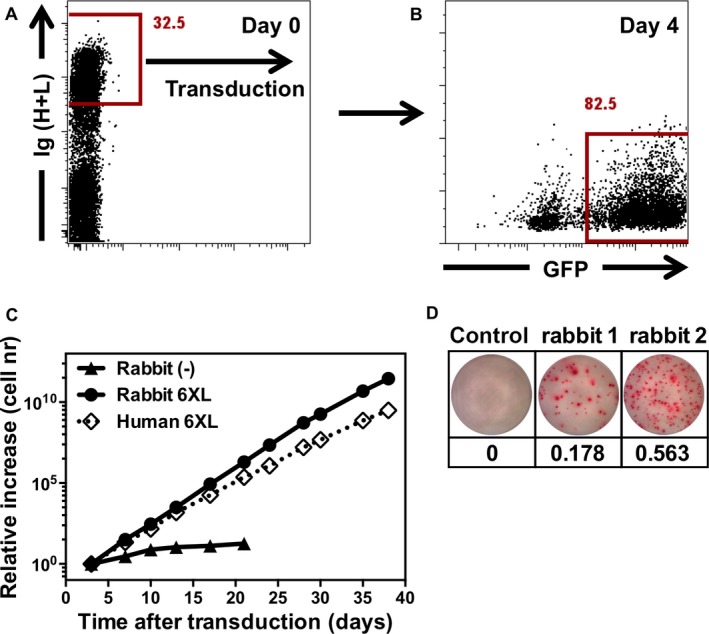
Transduction of antigen‐specific rabbit memory B cells. (A) Rabbit B cells were isolated from PBMCs based on immunoglobulin (Ig) expression, activated for 36–40 h on CD40L L‐cells with interleukin‐21 and transduced with a retroviral vector containing BCL6 and BCL‐XL. (B) Transduction efficiency based on green fluorescent protein expression 4 days after transduction. (C) Growth rates for rabbit B cells transduced with a retroviral vector containing BCL6 and BCL‐XL, non‐transduced rabbit B cells and human cells for comparison. (D) ELISpot analysis of the frequency of vaccine‐specific IgG B cells; B cells were incubated overnight in ELISpot plates coated with the influenza vaccine. Bound antibodies were visualized using an anti‐rabbit IgG antibody. Frequencies of vaccine‐specific cells are shown in percentages.
Discovery of monoclonal antibodies from rabbit memory B cells
Rabbit antibodies are of high interest as they outperform mouse antibodies in terms of affinity, diversity, and specificity for peptides and modified proteins. Also, rabbit antibodies can easily be selected for cross‐reactivity to mouse targets enabling their use in many human disease models. Finally, identification of antibodies reacting to both human proteins and their mouse counterparts enables the use of the same monoclonal in preclinical and clinical studies.
Current methods for obtaining rabbit monoclonal antibodies include (i) hybridoma formation by cell fusion with a rabbit plasmacytoma fusion partner developed from genetically modified rabbits (240E‐1) 79, (ii) phage display, (iii) in vitro differentiation into antibody‐producing cells followed by sequencing of individual B cells 80, and (iv) analysis of the polyclonal repertoire by comparison of affinity‐enriched serum IgGs to a database of IgG variable gene (V‐gene) sequences constructed by NextGen sequencing of mature B cells 81. Although these methods are well suited to identify antibodies that arise with a high frequency, identifying rare antibodies may prove time‐consuming and costly requiring extensive sequencing and recombinant expression efforts. For hybridoma formation, the low fusion efficiency and instability of clones challenges the identification of rare antibody‐producing cells. As indicated in the previous paragraph, the identification of rare B cells producing exceptional antibodies for instance with desired cross‐reactivities is not a problem when using BCL‐6 and BCL‐XL‐transduced rabbit B cells.
To assess if rabbit B cell immortalization can be used as a platform to obtain antigen‐specific rabbit monoclonal antibodies, we immunized rabbits with the human influenza vaccine Influvac that contains H1, H3, and influenza B. Already after two immunizations, vaccine‐specific transduced B cells could be detected with a high frequency (0.2 and 0.6%) (Fig. 3). To show that (as for human cells) both secreted antibodies and the cell surface expressed BCR could be used to identify specific clones, we seeded cells in mini‐cultures and in parallel single cell‐sorted B cells binding to fluorescently labeled H1, H3, and influenza B (Fig. 4). Subsequently, culture supernatants were analyzed for antibodies binding the different components of the influenza vaccine in ELISA.
Figure 4.
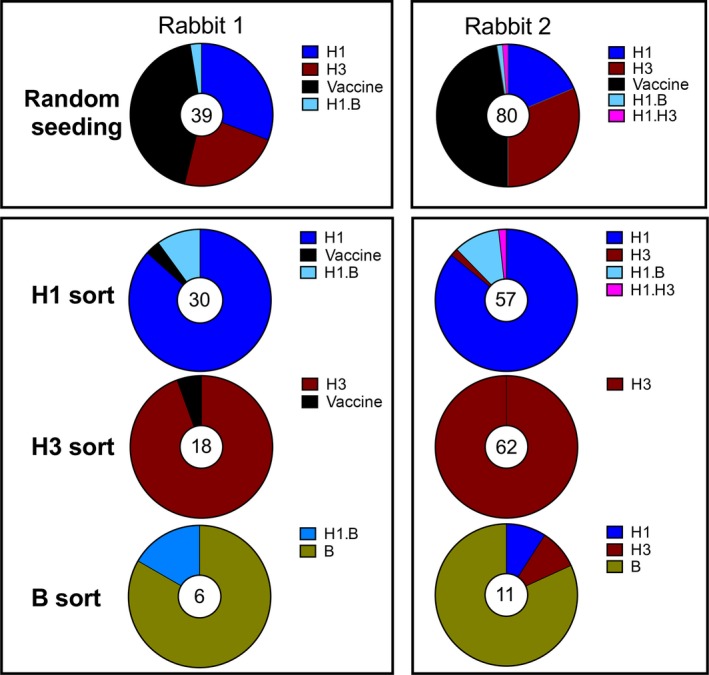
Analysis and isolation of antigen‐specific B cells. B cells from influenza H1\H3\influenza B‐vaccinated rabbits were seeded either randomly at 25 cells/well or sorted single cell using fluorescently labeled antigen. Supernatants were analyzed for binding to H1, H3, and influenza B in ELISA. The total number of positive wells is depicted in the middle of the graphs. Some clones showed reactivity to two hemagglutinin proteins.
Supernatants from the unbiased mini‐cultures contained antibodies directed to all three components of the vaccine and to the complete vaccine itself in both rabbits (Fig. 4). Analysis of the antibodies produced by the antigen‐sorted cells showed that the majority of the antibodies were directed against the component of the vaccine that was used for antigen‐specific sorting (Fig. 4). Thus antigen‐specific sorting allows focusing on the B‐cell population of interest. H3‐sorted clones were subsequently tested for binding to H7 virus‐infected cells to investigate if rabbit immunization resulted in the development of rare cross‐binding clones. Indeed we could identify one clone that showed cross‐binding to H3 and H7, corresponding to a frequency of 1 per 60,000. Due to the fast doubling time of the immortalized rabbit B cells over 200 HA‐specific, IgG+ B cell clones were obtained within 3 weeks of blood draw. As expected for rabbit antibodies, the affinities of individual antibodies were extremely high, up to 4 pM and the fact that the animals were not sacrificed allows for sequential sample taking and analysis. In a similar experiment where we were aiming to obtain peptide‐specific monoclonal antibodies, we obtained antibodies recognizing a FLAG tag or a peptide from the estrogen receptor with EC50s between 0.5 and 10 ng/ml in ELISA (data not shown). Summarizing these data, we conclude that immortalization of rabbit B cells offers a powerful platform to quickly obtain rabbit monoclonal antibodies.
Antigen presentation by immortalized B cells
It is well established that activated B cells that express high amounts of class II major histocompatibility complex (MHC) protein present antigen to T cells. Antigen is taken up by B cells via the BCR, processed, and peptides derived from the antigen are presented to the T cells. Thus, in physiological conditions, only antigens for which the B cells have a BCR are taken up, processed, and presented to T cells 82. Human B cells expanded with CD40L and IL‐4 have been shown to present antigenic peptides 83. Antigen‐specific BCL‐6/BCL‐XL‐transduced B cells are able to present native antigen to T cells as was also demonstrated for antigen‐specific EBV‐transformed B cells. We have used an influenza H1‐specific B cell clone capable of binding and internalizing intact H1, to show that native H1 but not H3 control protein was presented to a H1‐specific autologous T cell clone (data not shown).
As expected, the BCL‐6/BCL‐XL B cells present various antigenic peptides efficiently to autologous T cells independently of BCR specificity. This feature was successfully applied to discover neoantigens derived from highly malignant melanoma cells that were recognized by the T cells of the patient 84. These researchers first determined the spectrum of mutated proteins of a melanoma by exome sequencing and made a library encompassing peptides that contained the mutations. They then screened pools of peptides for their capacity to stimulate interferon‐γ production by autologous T cells. As antigen‐presenting cell, they used BCL6/BCL‐XL‐transduced B cells established from the patient. Importantly, these immortalized B cells did not activate autologous T cells in the absence of neoantigens. By contrast, EBV‐transformed B cells derived from patient's B cells stimulate EBV antigen‐specific T cells which mask the response to neoantigens 84 showing the importance of the BCL‐6/BCL‐XL‐transduced B cells in this strategy. Neoantigens could also be found by screenings using monocyte‐derived dendritic cells (DC) 85 but those DC cannot be expanded in vitro, in contrast to BCL‐6/BCL‐XL‐transduced B cells, limiting their use in large‐scale high‐throughput screening.
Conclusion
We describe here how activated B cells can be immortalized by a simple genetic modification. Nearly the complete B‐cell repertoire is captured using a highly efficient transduction of a retroviral construct harboring BCL6 and BCL‐XL. The immortalized B cells are stable and can be readily cloned, frozen, and thawed without loss of their antibody‐producing capacities. BCL6/BCL‐XL‐transduced B cells have a plasmablast‐like phenotype secreting antibodies in the supernatant and expressing the BCR on their surface, which makes them an excellent source for the discovery of monoclonal antibodies. Libraries encompassing hundreds of thousands of B cells can be established from individuals who produce exceptional antibodies, facilitating rapid antibody discovery and allowing investigators to search for rare B cells that make unique antibodies. We have discussed examples of broadly reacting highly neutralizing monoclonal antibodies against pathogenic viruses that were isolated from selected individuals. The method can also easily be applied to probe the B‐cell repertoire of cancer patients who respond favorably to immunotherapies for antibodies that recognize tumor antigens. We have already been able to identify antibodies from B cells of cancer patients that recognize new and unexpected targets on the cell surface of tumor cells. Future work in this area should provide us not only with a better understanding of the contribution of B cells to the immune response against malignant cells but will also allow the discovery of novel therapeutic antibodies.
The application of this method to other species than humans is just beginning to be explored. We efficiently generated rabbit monoclonal antibodies, which now offer us possibilities to tap the exceptional properties of these antibodies. It is also possible to make monoclonal antibodies from non‐human primates allowing analysis of the B‐cell repertoire against viruses like human immunodeficiency virus which are pathogenic for humans but not for non‐human primates.
Our work makes clear that expression of BCL‐6 by itself is sufficient to change a circulating B cell to a GC B cell and offers ample opportunities to study development and function of these cells in the GC. One example concerns the function of AID in BCL‐6/BCL‐XL‐transduced B cells. Surprisingly, SHM are observed in these cells but not CSR, indicating that yet to be identified signals are required for AID‐mediated CSR. Aside from the scientific questions that AID expression in BCL‐6/BCL‐XL‐transduced B cells raises, we could use this feature to generate subclones of antigen‐specific B cells that secrete antibodies with either higher or lower affinities than of the antibody produced by the parental clone, thereby offering a method to affinity mature antibodies without the need for extensive molecular engineering.
Finally, BCL‐6/BCL‐XL‐transduced B cells are excellent antigen‐presenting cells. They efficiently present peptides but intact proteins are processed only if they can bind to the BCR of the B cell clone. This feature allows mechanistic studies to the interaction of antigen‐specific B cells and T cells in humans and other species. Importantly, cultures of BCL6/BCL‐XL‐transduced B cells can be established from any cancer patient providing a convenient source of APC to discover neoantigens.
Acknowledgement
We thank our colleagues at AIMM Therapeutics for their input and continued support. All authors are employees of AIMM Therapeutics.
References
- 1. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256:495–497. [DOI] [PubMed] [Google Scholar]
- 2. Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979;206:347–349. [DOI] [PubMed] [Google Scholar]
- 3. Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011;29:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu X, Mcgraw P, House F, Crowejr J. An optimized electrofusion‐based protocol for generating virus‐specific human monoclonal antibodies. J Immunol Methods 2008;336:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith SA, Crowe JE. Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol Spectrum 2015;3:141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steinitz M, Klein G, Koskimies S, Makel O. EB virus‐induced B lymphocyte cell lines producing specific antibody. Nature 1977;269:420–422. [DOI] [PubMed] [Google Scholar]
- 7. Traggiai E, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med 2004;10:871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol 2013;31:705–742. [DOI] [PubMed] [Google Scholar]
- 9. Banchereau J, et al. The CD40 antigen and its ligand. Annu Rev Immunol 1994;12:881–922. [DOI] [PubMed] [Google Scholar]
- 10. Grandea AG, et al. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc Natl Acad Sci USA 2010;107:12658–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diehl SA, et al. STAT3‐mediated up‐regulation of BLIMP1 Is coordinated with BCL6 down‐regulation to control human plasma cell differentiation. J Immunol 2008;180:4805–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwakkenbos MJ, et al. Generation of stable monoclonal antibody‐producing B cell receptor‐positive human memory B cells by genetic programming. Nat Med 2010;16:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012;30:429–457. [DOI] [PubMed] [Google Scholar]
- 14. Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol 1998;16:293–322. [DOI] [PubMed] [Google Scholar]
- 15. Scheeren FA, et al. STAT5 regulates the self‐renewal capacity and differentiation of human memory B cells and controls Bcl‐6 expression. Nat Immunol 2005;6:303–313. [DOI] [PubMed] [Google Scholar]
- 16. Dai X, et al. Stat5 is essential for early B cell development but not for B cell maturation and function. J Immunol 2007;179:1068–1079. [DOI] [PubMed] [Google Scholar]
- 17. Shvarts A, et al. A senescence rescue screen identifies BCL6 as an inhibitor of anti‐proliferative p19(ARF)‐p53 signaling. Genes Dev 2002;16:681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandal M, et al. Epigenetic repression of the Igk locus by STAT5‐mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol 2011;12:1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheeren FA, et al. IL‐21 is expressed in Hodgkin lymphoma and activates STAT5: evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 2008;111:4706–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein U, Dalla‐Favera R. Germinal centres: role in B‐cell physiology and malignancy. Nat Rev Immunol 2008;8:22–33. [DOI] [PubMed] [Google Scholar]
- 21. Basso K, Dalla‐Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol 2010;105:193–210. [DOI] [PubMed] [Google Scholar]
- 22. Shapiro‐Shelef M, Lin KI, Mcheyzer‐Williams LJ, Liao J, Mcheyzer‐Williams MG, Calame K. Blimp‐1 is required for the formation of immunoglobulin secreting plasma cells and pre‐plasma memory B cells. Immunity 2003;19:607–620. [DOI] [PubMed] [Google Scholar]
- 23. Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP‐1. Nature 2001;412:300–307. [DOI] [PubMed] [Google Scholar]
- 24. Shaffer AL, et al. Blimp‐1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002;17:51–62. [DOI] [PubMed] [Google Scholar]
- 25. Tunyaplin C, Shaffer AL, Angelin‐Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl‐6 inhibits plasmacytic differentiation. J Immunol 2004;173:1158–1165. [DOI] [PubMed] [Google Scholar]
- 26. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000;102:553–563. [DOI] [PubMed] [Google Scholar]
- 27. Holland SM, et al. STAT3 mutations in the hyper‐IgE syndrome. N Engl J Med 2007;357:1608–1619. [DOI] [PubMed] [Google Scholar]
- 28. Minegishi Y, et al. Dominant‐negative mutations in the DNA‐binding domain of STAT3 cause hyper‐IgE syndrome. Nature 2007;448:1058–1062. [DOI] [PubMed] [Google Scholar]
- 29. Ma CS, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 2012;119:3997–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kane A, Deenick EK, Ma CS, Cook MC, Uzel G, Tangye SG. STAT3 is a central regulator of lymphocyte differentiation and function. Curr Opin Immunol 2014;28:49–57. [DOI] [PubMed] [Google Scholar]
- 31. Kotlarz D, et al. Loss‐of‐function mutations in the IL‐21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med 2013;210:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deenick EK, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody‐secreting plasma cells. J Exp Med 2013;210:2739–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Westerhuis BM, et al. Human memory B cells producing potent cross‐neutralizing antibodies against human parechovirus: implications for prevalence, treatment, and diagnosis. J Virol 2015;89:7457–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hazenbos WLW, et al. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence‐associated cell wall proteins. PLoS Pathog 2013;9:e1003653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kwakkenbos MJ, et al. Genetic manipulation of B cells for the isolation of rare therapeutic antibodies from the human repertoire. Methods 2013;65:1–7. [DOI] [PubMed] [Google Scholar]
- 36. Ekiert DC, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 2011;333:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Friesen RHE, et al. A common solution to group 2 influenza virus neutralization. Proc Natl Acad Sci USA 2014;111:445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagner K, et al. Bispecific antibody generated with sortase and click chemistry has broad antiinfluenza virus activity. Proc Natl Acad Sci USA 2014;111:16820–16825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. MacDonald NE, Hall CB, Suffin SC, Alexson C, Harris PJ, Manning JA. Respiratory syncytial viral infection in infants with congenital heart disease. N Engl J Med 1982;307:397–400. [DOI] [PubMed] [Google Scholar]
- 40. Meissner HC. Selected populations at increased risk from respiratory syncytial virus infection. Pediatr Infect Dis J 2003;22:S40–S44. discussion S44–S45. [DOI] [PubMed] [Google Scholar]
- 41. Thorburn K. Pre‐existing disease is associated with a significantly higher risk of death in severe respiratory syncytial virus infection. Arch Dis Child 2008;94:99–103. [DOI] [PubMed] [Google Scholar]
- 42. Nair H, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta‐analysis. Lancet 2010;375:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnson S, et al. Development of a humanized monoclonal antibody (MEDI‐493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis 1997;176:1215–1224. [DOI] [PubMed] [Google Scholar]
- 44. The IMpact‐RSV Study Group . Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high‐risk infants. Pediatrics 1998;102:531–537. [PubMed] [Google Scholar]
- 45. Blanken MO, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med 2013;368:1791–1799. [DOI] [PubMed] [Google Scholar]
- 46. Stensballe LG, et al. The causal direction in the association between respiratory syncytial virus hospitalization and asthma. J Allergy Clin Immunol 2009;123:e1. [DOI] [PubMed] [Google Scholar]
- 47. Kusel MMH, et al. Early‐life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol 2007;119:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med 2000;161:1501–1507. [DOI] [PubMed] [Google Scholar]
- 49. Jackson DJ, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med 2012;185:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. García‐García ML, et al. Human metapneumovirus bronchiolitis in infancy is an important risk factor for asthma at age 5. Pediatr Pulmonol 2007;42:458–464. [DOI] [PubMed] [Google Scholar]
- 51. Çalışkan M, et al. Rhinovirus wheezing illness and genetic risk of childhood‐onset asthma. N Engl J Med 2013;368:1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simoes EAF, et al. Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J Pediatr 2007;151:e1. [DOI] [PubMed] [Google Scholar]
- 53. McLellan JS, et al. Structure‐based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013;342:592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gilman MSA, et al. Characterization of a prefusion‐specific antibody that recognizes a quaternary, cleavage‐dependent epitope on the RSV fusion glycoprotein. PLoS Pathog 2015;11:e1005035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006;281:23514–23524. [DOI] [PubMed] [Google Scholar]
- 56. Ngwuta JO, et al. Prefusion F‐specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med 2015;7:309ra162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Magro M, et al. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc Natl Acad Sci USA 2012;109:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Glenn GM, et al. A randomized, blinded, controlled, dose‐ranging study of a respiratory syncytial virus recombinant fusion (F) nanoparticle vaccine in healthy women of childbearing age. J Infect Dis 2015; pii: jiv406. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 59. Impagliazzo A, et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015;349:1301–1306. [DOI] [PubMed] [Google Scholar]
- 60. Yassine HM, et al. Hemagglutinin‐stem nanoparticles generate heterosubtypic influenza protection. Nat Med 2015;21:1065–1070. [DOI] [PubMed] [Google Scholar]
- 61. Shakeel S, et al. Structural basis of human parechovirus neutralization by human monoclonal antibodies. J Virol 2015;89:9571–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Air GM. Sequence relationships among the hemagglutinin genes of 12 subtypes of influenza A virus. Proc Natl Acad Sci USA 1981;78:7639–7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mehle A. Unusual influenza A viruses in bats. Viruses 2014;6:3438–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okuno Y, Isegawa Y, Sasao F, Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol 1993;67:2552–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Throsby M, et al. Heterosubtypic neutralizing monoclonal antibodies cross‐protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 2008;3:e3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sui J, et al. Structural and functional bases for broad‐spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol 2009;16:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science 2009;324:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lingwood D, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 2012;489:566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pappas L, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 2014;516:418–422. [DOI] [PubMed] [Google Scholar]
- 70. Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza a hemagglutinins. Science 2011;333:850–856. [DOI] [PubMed] [Google Scholar]
- 71. Nakamura G, et al. An in vivo human‐plasmablast enrichment technique allows rapid identification of therapeutic Influenza a antibodies. Cell Host Microbe 2013;14:93–103. [DOI] [PubMed] [Google Scholar]
- 72. Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science 2012;337:1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McHeyzer‐Williams LJ, McHeyzer‐Williams MG. Antigen‐specific memory B cell development. Annu Rev Immunol 2005;23:487–513. [DOI] [PubMed] [Google Scholar]
- 74. Cumbers SJ, et al. Generation and iterative affinity maturation of antibodies in vitro using hypermutating B‐cell lines. Nat Biotechnol 2002;20:1129–1134. [DOI] [PubMed] [Google Scholar]
- 75. Bowers PM, et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci USA 2011;108:20455–20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McConnell AD, et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng Des Sel 2013;26:151–164. [DOI] [PubMed] [Google Scholar]
- 77. Peled JU, et al. MD. The biochemistry of somatic hypermutation. Annu Rev Immunol 2008;26:481–511. [DOI] [PubMed] [Google Scholar]
- 78. Sayegh CE, et al. E‐proteins directly regulate expression of activation‐induced deaminase in mature B cells. Nat Immunol 2003;4:586. [DOI] [PubMed] [Google Scholar]
- 79. Spieker‐Polet H, Sethupathi P, Yam PC, Knight KL. Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit‐rabbit hybridomas. Proc Natl Acad Sci USA 1995;92:9348–9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Seeber S, et al. A robust high throughput platform to generate functional recombinant monoclonal antibodies using rabbit B cells from peripheral blood. PLoS ONE 2014;9:e86184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wine Y, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc Natl Acad Sci USA 2013;110:2993–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lanzavecchia A. Antigen‐specific interaction between T and B cells. Nature 1985;314:537–539. [DOI] [PubMed] [Google Scholar]
- 83. Schultze JL, et al. CD40‐activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen‐specific T cells for adoptive immunotherapy. J Clin Invest 1997;100:2757–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Linnemann C, et al. High‐throughput epitope discovery reveals frequent recognition of neo‐antigens by CD4. Nat Med 2014;21:1–7. [DOI] [PubMed] [Google Scholar]
- 85. Tran E, et al. Cancer immunotherapy based on mutation‐specific CD4+ T cells in a patient with epithelial cancer. Science 2014;344:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]