

Summary
Hapten‐binding antibodies have for more than 50 years played a pivotal role in immunology, paving the way to antibody generation (as haptens are very important and robust immunogens), to antibody characterization (as the first structures generated more than 40 years ago were those of hapten binders), and enabled and expanded antibody engineering technologies. The latter field of engineered antibodies evolved over many years and many steps resulting in recombinant humanized or human‐derived antibody derivatives in multiple formats. Today, hapten‐binding antibodies are applied not only as reagents and tools (where they still play an important part) but evolved also to engineered targeting and pretargeting vehicles for disease diagnosis and therapy. Here we describe recent applications of hapten‐binding antibodies and of engineered mono‐ and bispecific hapten‐binding antibody derivatives. We have designed and applied these molecules for the modulation of the pharmacokinetic properties of small compounds or peptides. They are also integrated as additional binding entities into bispecific antibody formats. Here they serve as non‐covalent or covalent coupling modules to haptenylated compounds, to enable targeted payload delivery to disease tissues or cells.
Keywords: digoxigenin, biotin, protein engineering, protein structure, targeted therapy
This article is part of a series of reviews covering Immunoglobulins: from genes to therapies appearing in Volume 270 of Immunological Reviews.
Introduction
More than 40 years ago, the structural composition of antibodies and functional consequences thereof became elucidated by crystallizing and analyzing hapten‐binding Fab fragments.
The first X‐ray structure of an antibody was revealed in 1973 when Poljak et al. published the structure at 2.8 Å resolution of the Fab fragment of the antibody NEW 1, 2. The structure of NEW determined initially, i.e. the first Fab structure available, was still that of a binding region without its ligand in its binding pocket. A structure of NEW with a ligand in its binding pocket (a vitamin K derivative) was subsequently provided. Shortly after publication of the NEW structure, Segal and coworkers solved the structure of the Fab fragment of the MCPC603 antibody with the cognate hapten phosphorylcholine in the binding pocket of the variable region 3, 4 (Fig. 1). These first descriptions of the NEW and MCPC603 antibody structures revealed not only the overall structural composition of antibody domains but also shed light on the modes of molecular interactions between antibodies and antigens 5, 6, 7. The structural information enabled also the definition of hypervariable, framework, and (initially by sequence homology) constant regions of antibodies. This information also provided rational explanations for the underlying molecular mechanisms of antigen recognition. Furthermore, the knowledge of structural composition of antibody binding regions, constant, variable, and hypervariable regions paved the way for many antibody engineering approaches that were emerging in subsequent years (and are still ongoing today). This includes also many aspects of antibody engineering 8, such as humanization of murine (and later on other species derived) antibodies 9, and the classification of antibody classes and subgroups 10. Finally, these structures paved the way for novel technologies that are widely applied such as optimized recombinant antibody fragment generation and expression 11, 12, display technologies 13, 14, as well as affinity maturation via modeling approaches and engineered V‐region stabilization technologies 15, 16, 17.
Figure 1.
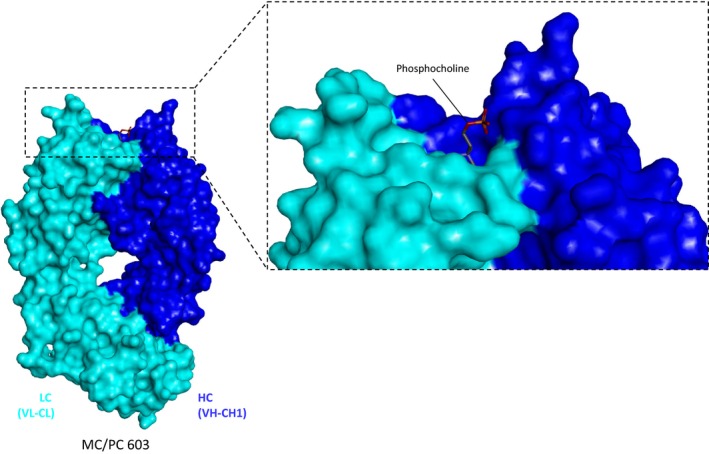
The first X‐ray structure of an antibody was that of the hapten‐binding antibody NEW without bound antigen ( PDB 7 FAB ). Shortly thereafter, the structure of the antibody MC/PC603 (shown here, PDB entry 2MCP 1984 by Padlan, Cohen & Davies) bound to its cognate hapten was solved. These structures demonstrated for the first time the molecular basis of antibody–antigen interactions. These and many more structures to come opened the field of rational structure‐based antibody engineering.
Up to date, more than hundred structures of hapten‐binding antibodies have been solved and are deposited in the public database PDB ( protein data bank, www.rcsb.org) 18. These structures provide comprehensive insights into the binding interactions of many different small compounds and their cognate antibodies. Hapten binders are not only the first antibodies for which high‐resolution structures were solved but they were also applied quite early as versatile analytical and experimental tools. In consequence, they are still widely applied as essential capturing, cross‐linking, or detection reagents in many diagnostic assays. Haptens are small chemically synthesized compounds (usually with a mass <1000 dalton) and most of them are stable and robust to handle. Because of that, hapten‐derived antibody–antigen technologies are frequently applied as model systems, for example in early proof‐of‐concept studies to demonstrate display or affinity maturation technologies. Hundreds of haptens and antibodies recognizing them are available, many of them covered by the database HaptenDB 19.
Hapten‐binding antibodies can serve in addition as versatile bridging elements between small chemically synthesized entities and large molecules that are produced recombinantly in bacterial or eukaryotic expression systems. More recently hapten‐binding antibodies are being also developed for targeting and pretargeting applications in disease diagnosis and therapy. Bispecific antibody (bsAb) derivatives that bind cell surface antigens as well as haptens can be applied for targeted or pretargeted therapies. Examples for that include approaches to specifically deliver to tissues or cells either small payloads such as fluorophores or chelated radioisotopes, or large payloads among them peptides, nanoparticles, nucleic acids, or modified proteins.
In this review, we summarize our experiences in generating, characterizing, and applying novel hapten‐binding antibody derivatives to modulate the pharmacokinetics of small compounds or peptides. We further address the application of hapten‐binding antibody derivatives as vehicles for targeted or pretargeted payload delivery.
Recombinant hapten‐binding antibodies and bispecific antibody derivatives
Over the past years, our lab has designed, engineered, generated, modified, and characterized a variety of hapten‐binding antibodies and ‐derived binding entities to serve as delivery vehicles for targeted therapy. Other derivatives of hapten‐binding antibodies were also designed and applied for the modulation or improvement of pharmacokinetic parameters of small chemically synthesized compounds and peptides.
Some of the antibodies that we used as ‘starting modules’ for protein design and engineering to develop delivery modules were originally developed by Boehringer Mannheim (now Roche Diagnostics in Penzberg, Germany). Examples for such molecules are digoxigenin‐binding or biotin‐binding antibodies 20, 21. They had been generated decades ago by immunizing animals followed by murine hybridoma technologies. The antibodies were originally generated as tools and reagents for diagnostics and molecular biology. Such antibodies, including those that bind digoxigenin or biotin are widely applied as capture or bridging reagents, for example for the detection of hapten‐incorporated or chemically labeled nucleic acids or peptides or proteins 20, 21.
Fig. 2 shows as an example the structure of one of our hapten‐binding antibodies. This ‘anti‐Bio’ antibody was crystallized and its structure solved together with bound cognate antigen biocytinamide 22. The antibody recognizes as antigen biotin which has been coupled to a payload, but it does not bind unmodified (free) biotin. Because of that, it can be applied to capture, deliver, or detect biotinylated payloads. This antibody and its derivatives can be applied in combination with and/or for the analysis of biological samples (and in vivo) even though these samples contain in many cases considerable amounts of biotin. An explanation for the non‐interference of free biotin with the binding of coupled biotin is provided by the structure and composition of its variable region: biotin binds like all haptens into a pocket that is formed by the V‐region CDRs of the antibody. The orientation of biotin in this pocket positions its carboxyl group to the outside of that pocket. The entry site of the binding pocket, in turn, contains at its surface two aspartate residues that present negative charges at their side chains. These negative charges are located in very close proximity to the biotin‐carboxyl group if one were to fit free biotin into the pocket 22. Thus, charge repulsion between the carboxyl group of biotin and the two aspartate side chains prevent the binding of free biotin to this antibody. In contrast, biotin‐payload conjugates have the carboxyl group of biotin ‘neutralized’ in terms of charge because conjugation occurs at and thereby alters the carboxyl group. Thus, biotin‐conjugates are not subject to charge repulsion. This effect explains the high specificity of the hapten‐binding antibody for coupled biotin.
Figure 2.
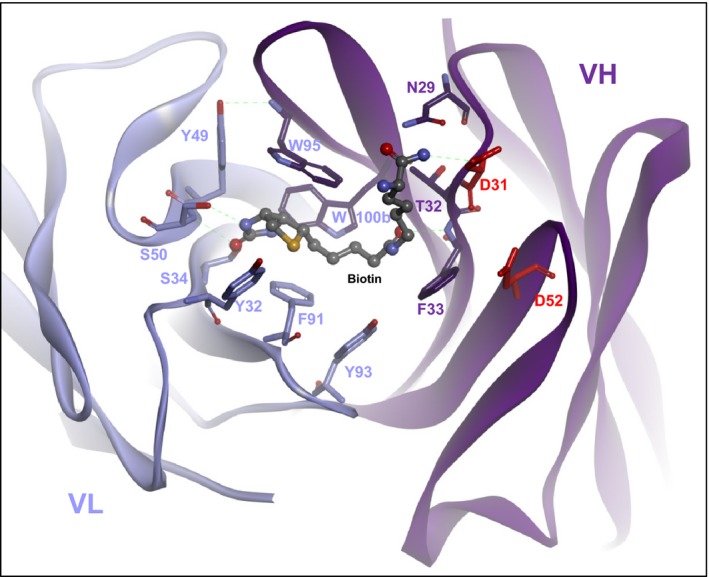
Structure of an antibody that specifically binds conjugated biotin (biocytinamide in this figure) but not free biotin ( PDB 4S1D). Note the negatively charged aspartate 31 and aspartate 52 of the VH domain lining the sides of the binding pocket. The negative charges of their side chains generate a charge repulsion which prevents entry of (COOH‐containing) free biotin. In contrast, biotin that is coupled to a payload and thereby is uncharged (in the same manner as biotcytinamide that was used to solve the structure) can enter and stay in the binding pocket without charge repulsion.
The conversion of the variable regions of hapten‐binding antibodies to vehicles for targeted payload delivery or PK modulation is based upon the generation of humanized ‘building blocks’ that retain antigen‐binding specificity and affinity. Furthermore, such entities need to be stable and preferentially producible by methods that are compatible with standard antibody production technologies. Because of that, most of our hapten‐binding and bispecific antibody derivatives are IgG‐derived and contain Fc regions. This permits the use or adaptation of standard upstream processing technologies (including expression in mammalian cells) and of downstream technologies (such as Protein‐A affinity chromatography) that are based on established production methods for therapeutic antibodies 23, 24, 25, 26, 27.
We have generated bsAbs of different composition, with different stoichiometries and geometries of their binding sites 28, 29, 30. Most formats that we generated contain one or two binding sites for cell targeting combined with one or two hapten‐binding sites (Fig. 3). A common challenge for the effective production of many formats is that antibodies are composed of different chains that need to assemble in a correct manner. Regular IgGs with one binding specificity contain two identical light chains and two identical heavy chains that assemble into one defined four‐chain molecule. Fab fragments by themselves are composed of just two different chains but, again, bispecific entities that contain at least two Fabs will be composed of four chains which must be assembled in a manner that generates only the fully functional form. Such Fab‐based bsAbs must have the correct heavy chain fragments assembled with their matching light chains. Thus, a common challenge is that bispecific antibody derivatives need to combine binding specificities that are encoded by different heavy chains and different light chains into one molecule, which still must be composed in a defined manner. Fig. 3 shows selected formats that we have applied for the generation of bispecific antibodies. These examples also demonstrate different protein engineering solutions to overcome the inherent chain association challenge: One design to generate stable bsAbs of defined composition and ‘benign behavior’ are tetravalent ‘2 + 2’ molecules composed of IgGs containing single‐chain Fv's added to the C‐ or N‐termini of their heavy or light chains. The principle of this format was first described by Coloma and Morrison 31. However, this basic format of these molecules was initially not fit for large scale production and in most cases not easy to handle because of the inherent instability and aggregation tendency of the attached single‐chain Fv modules. The aggregation propensity of single‐chain Fvs affected also bispecific Fv‐fusion proteins, i.e. bsAbs, and hence resulted initially in rather instable aggregation‐prone antibody derivatives. This problem was subsequently overcome by stabilization of the single‐chain Fv modules with interchain disulfide bonds between position 44 in VH and 100 in VL, a technology originally developed in the Pastan Lab at the NCI to generate stable Fv‐containing immunotoxins for cancer therapy 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43.
Figure 3.
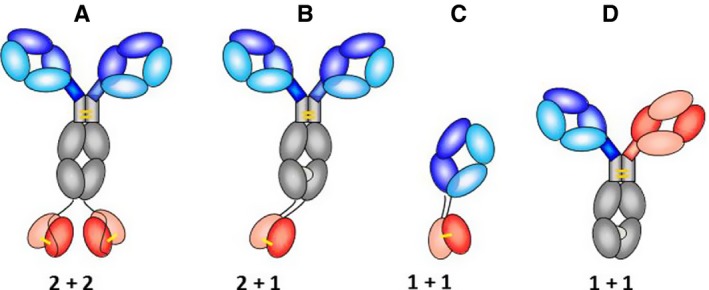
Engineered protein formats that have been applied to generate hapten‐binding bispecific antibodies. (A) 2 + 2 ‘Morrison‐Type’ bsAbs which are nowadays in most cases stabilized by interchain disulfides (indicated in yellow) in the attached scFvs. (B) 2 + 1 IgG‐derived bsAbs with knob‐into‐hole Fc regions and disulfide‐stabilized VH and VL, respectively, at the C‐termini of CH3. (C) Fab‐derived bsAb format that has a disulfide‐stabilized Fv attached to the C‐termini of CH1 or CL, respectively. (D) CrossMabs generate the desired H‐chain heterodimers via knob‐into‐hole mutations placed into their constant regions. They contain in addition one designed domain‐exchanged Fab arm to assure correct light chain pairing.
The combination of the Morrison‐format with disulfide stabilization of Fvs generated stable functional bispecific antibodies with low aggregation propensity which can be produced with procedures that were already established for the production of therapeutic antibodies 23, 24, 25, 26, 27, 44.
Another IgG‐based format of different composition and with different stoichiometry of their binding sites is IgGs that have one additional VH and one additional VL domain attached to the C‐termini of their H‐chains. This format does not require a peptide linker between the additional variable domains but instead connects the matching VH and VL domains by the above‐mentioned disulfide (between position H44 and L100 according to the Kabat nomenclature, 10). The thereby designed ‘2 + 1 bsAbs’ contain two regular Fabs as binding sites for one antigen and one additional binding site for another 45. In addition to attached single VH or VL domains, these molecules carry knobs or hole mutations 46, 47, respectively, in their H‐chains. This favors the formation of the desired H‐chain heterodimers because knob–knob or hole–hole homodimers are structurally incompatible and/or instable, hence forcing the generation of desired ‘knobs‐into‐holes’ heterodimers. The added VH and VL domains by themselves provide additional heterodimerization forces, by their own VH to VL association functionalities. The dual linkage of the additional Fv with two tethers can lead to restricted binding for some antigens, a phenomenon that has also been observed for dual variable domain (DVD)‐IgG bispecifics 48, 49, 50. This restriction, however, is observed primarily for large antigens and is not limiting the binding efficacy of haptens. Due to their small size, haptens can easily enter the space between the C‐termini of the CH3 domains and their binding pocket and are therefore not affected by the dual connection between the IgG and added variable region. In cases where the binding of larger antigens is restricted, full binding efficacy of such molecules can be unleashed by proteolytic processing of one linker (the other linker still tethers the fully functional Fv to the IgG 45. Other approaches that we applied for the generation of robust bispecific hapten‐binding antibodies of defined composition combine the H‐chain knobs‐into‐holes technology with Fab arm or VH–VL or CH1–CL domain exchange approaches. This generates ‘CrossMabs’ (Fig. 3 D) which are almost identical to IgGs in size, shape, and binding geometry, and can be produced by adaptation of standard procedures for therapeutic antibodies. In contrast to monospecific IgGs, however, they bind a separate antigen with each Fab arm 51, 52. Fab‐derived hapten‐binding bsAbs which do not contain Fc regions have also been generated for applications that desire ‘intermediate’ pharmacokinetic parameters (i.e. not as long as whole IgGs but not too rapidly cleared by renal excretion). This design of these bsAbs follows similar principles as that of IgG‐derived bsAbs, in many cases consisting of single‐chain disulfide‐stabilized variable antibody or separate disulfide‐stabilized domains fused to VH–Ch1 and/or L‐chains (Fig. 3).
Engineered hapten‐binding antibodies for covalent payload coupling
The tightness of the connection between hapten‐binding antibodies and haptenylated payloads is determined by on‐ and off‐rates of their binding regions. By default, this connection is not covalent for unmodified hapten‐binding antibodies. This permits the release of the hapten and hapten‐coupled payloads, which is desired for targeted therapy approaches that need to deliver payloads into cells (e.g. into the cytoplasm or nucleus). On the other hand, non‐covalent payload attachment is inherently associated with some degree of ‘non‐specific’ loss of payload (off reaction).
For some applications a very tight connection between antibody module and payload is desired. For such, we engineered and optimized a technology that connects haptenylated payloads by a covalent disulfide bond to the antibody. This linkage occurs in a hapten‐binding‐dependent manner by spontaneous redox shuffling reaction 22. Hapten‐binding antibodies were considered to be well suited for the engineering of a ‘bridgehead’ for covalent coupling of a payload. The reason is that hapten‐binding antibodies in general form pockets in which the small hapten is bound by the N‐ and C‐terminal parts of the CDRs, whereas the middle parts of the CDRs are often not involved in coordination of the hapten and are free for the introduction of functional residues. This feature can be deduced from crystal structures of haptens bound by their cognate antibodies. We called the group of antibody residues that are available for engineering without disturbing hapten binding the ‘rim region’. Such a region cannot be generally defined for antibodies binding to larger peptides or proteins, as all CDR residues are potentially involved in the interaction with the antigen, and ‘free’ residues need to be defined for each antibody–antigen pair by either structural or mutational studies. In contrast to that, we identified a ‘universal’ coupling position within the ‘rim region’ of anti‐hapten antibodies which should be amenable for engineering without further experimental characterization of the respective hapten‐binding antibody. This position was defined by structural analysis of several anti‐hapten antibodies (i.e. anti‐digoxigenin, ‐biotin, and ‐fluorescein antibodies) by applying a number of structural criteria:
The position must be in proximity to the binding pocket without interfering with hapten binding.
It must not compromise the structural integrity of the V‐domains (i.e. VH/VL interactions).
It must be solvent exposed and thus accessible for chemical reactions.
It must be structurally conserved in different hapten‐binding antibodies.
Six positions were found that fulfill all these criteria in the structures that were analyzed, of which one position on the heavy chain was chosen for further analysis. This position is called 52+2 which refers to Kabat numbering 10, meaning two residues C‐terminally from Kabat position 52 (our nomenclature cancels out an ambiguity in Kabat numbering, where structurally equivalent positions can have different numbers depending on the length of heavy chain CDR2).
The ‘universality’ of this position for payload coupling to any other hapten‐binding antibody is highly likely: we have analyzed via bioinformatics 140 different antibody–hapten complex crystal structures and found that in 139 of those structures, position 52+2 was part of the ‘rim region’ without having contact to atoms of the hapten molecules. Amino acids in position 52+2 were replaced by cysteines in the anti‐Dig, anti‐Biot, and anti‐Fluorescein (Fluo) antibodies 22 (Fig. 4). Purification of such modified antibodies from HEK293 cell culture supernatants showed that the cysteine was almost entirely oxidized by homocysteine present in the cell culture medium. However, analysis of binding kinetics by surface plasmon resonance also showed that Cys 52+2 (and with attached homocysteine) did not alter the binding of the haptens (Dig, Biot, Fluo) to the respective antibodies.
Figure 4.
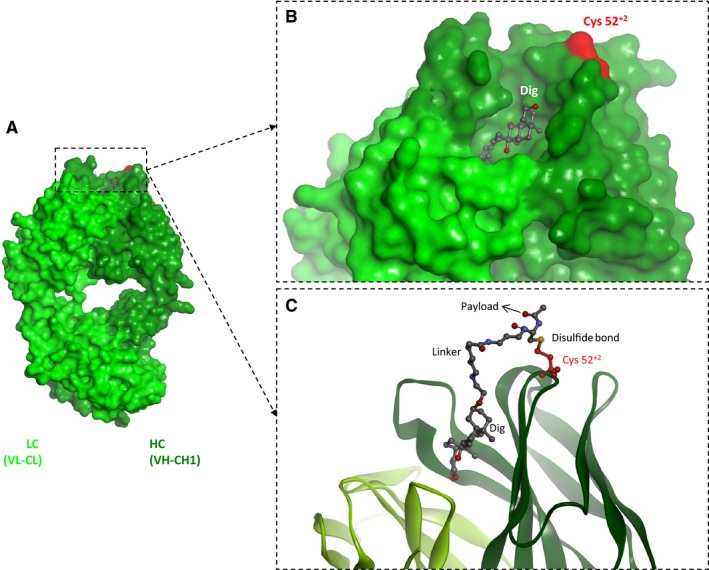
Engineered hapten‐binding antibodies enable covalent payload attachment via hapten‐positioned spontaneous disulfide shuffling. (A) Structure of the Fab fragment of a digoxigenin‐binding antibody (PDB 3RA7). (B) Position of the engineered cysteine in the rim region of hapten‐binding antibodies which is in close proximity to the binding pocket of most hapten binders yet does not interfere with antigen binding. (C) Hapten‐positioned spontaneous disulfide shuffling links thiol‐containing haptenylated payloads to the antibody.
Covalent attachment of a payload to such an antibody via a disulfide bond works by introduction of a cysteine‐containing linker between hapten and payload. The design of the linker also needs to fulfill several criteria. The most important one is that the linker must not be attached to a position on the hapten that is critical for hapten–antibody interactions. Thus, a hapten‐atom must be chosen for attachment that faces outwards of the binding pocket and is not coordinated by antibody residues. Such atoms can be identified in the absence of a dedicated antibody–hapten complex crystal structure, by analyzing the immunogen that was initially used for the generation of the respective antibody. Usually, small molecules like haptens are fused to carriers (e.g. KLH) to stimulate the immune response. Such hapten‐carrier conjugates will then raise antibodies which bind the hapten with the conjugation site facing outwards. Thus, the same hapten substrate that was used in the generation of the immunogen conjugate should be used for the design of the haptenylated payloads.
Furthermore, the cysteine‐containing linkers need to be of sufficient length and flexibility to allow for positioning of the cysteine for formation of a disulfide bond. In case of the digoxigenin‐payload, the linker had a length of about 21 Å when fully extended, which was about double the distance of the Cα‐atom of position 52+2 of the anti‐Dig‐antibody and C3 of digoxigenin, which is the atom to which the linker is attached. Of course, the linker between the cysteine and the payload must be sufficiently long to maintain functional properties of the payload and to avoid interference of the payload in productive disulfide bond formation.
It was shown that applying the described design principles for anti‐hapten antibodies and haptenylated payloads resulted in excellent yields of stable, covalent antibody‐payload conjugates in a simple and scaleable reaction that does not need a limited reduction of the antibody cysteines. Incubation of both components leads to a complexation of the hapten by the antibody, with the cysteine‐containing linker facing outwards, in spatial proximity of Cys 52+2 in the ‘rim region’. The payload cysteine brings in a reactive sulfhydryl group that attacks the disulfide bond between the Cys 52+2 and the homocysteine in a spontaneous redox shuffling reaction. The equilibrium of this reaction is in favor of the disulfide bond between Cys 52+2 and the payload cysteine, because the homocysteine will diffuse away once released from the rim region cysteine, whereas the payload is kept in proximity of Cys 52+2 by the antibody–hapten interaction. We could demonstrate that this technology generates site‐specific and defined covalent antibody‐payload complexes by a spontaneous disulfide shuffling reaction with high yield.
Hapten‐binding IgGs for modulation of pharmacokinetic properties of small molecules
The availability of hapten‐mediated antibody‐payload complexes (non‐covalent) and antibody‐payload conjugates (covalent via a disulfide bridge) opens up new options for PK modulation of pharmaceutically active payloads with low molecular weights. The problem with such compounds is a fast renal clearance that generally results in unfavorable pharmacokinetic (PK) properties. Thus, molecules that per se are highly active are cleared from the plasma in the range of minutes which results in an overall low therapeutic efficacy. Several solutions for this problem have been developed, like fusion of peptide sequences to serum albumin or to Fc regions of antibodies 53. These carriers confer long serum half‐lives because they bind to the neonatal Fc receptor (FcRn) and are thereby subjected to receptor‐mediated recycling and re‐release into the serum 54, 55. Alternatives to that are chemical modification with polyethylene glycol or hydroxylethyl starch (PEGylation or HESylation) 56, 57. Site‐directed covalent coupling of small entities to antibodies, e.g. via application of thiomab technologies 58, 59 or site‐directed coupling to other proteins have also been applied. One interesting additional technology (in some aspects similar to hapten‐binding principles) that also applies variable‐binding regions to recognize and thereafter covalently couple chemically generated compounds to antibodies is Covx‐bodies. These utilize ‘semi‐catalytic’ antigen‐binding regions to attach payloads to antibodies 60, 61.
Several aspects are of advantage upon using of hapten–antibody complexes for PK modulation. First, one is not limited to use ‘normal’ proteins or peptides containing natural amino acids, compared to technologies that are based on fusion protein approaches. Second, loss of activity that is often seen for PEGylated compounds can be minimized by careful design of the linker between the active compound and the hapten, that is the anchor point to the antibody. Third, immunogenicity of PEGylated or covalently coupled payloads is reduced in the hapten–antibody complexes, because the latter do not include unnatural chemical structures fixed to proteins that might be processed by antigen‐presenting cells.
In addition, antibody–hapten payload complexes can be produced in a very simple and scaleable reaction with a defined drug to antibody ratio. This can be achieved by just mixing the two components which form spontaneous and defined complexes. This may eliminate the need for subsequent and in many cases elaborate additional downstream purification processes.
The PK modulation properties of the hapten‐complexation approach with a pharmaceutically active compound were shown in an obese mouse model. These animals were treated with different NPY2 receptor agonists, known to be able to modulate the obese phenotype (provided active concentrations can be achieved for sufficient time) 62. Hoffmann and coworkers could demonstrate that the peptide PYY or Dig‐coupled PYY derivatives by itself had very limited potency (due to rapid elimination), but had improved potency when it was complexed with an anti‐digoxigenin antibody 63. Obese mice treated with the PYY‐Dig–antibody complexes showed reduced food intake even after 48 h, whereas peptide alone showed efficacy only in the first hour after treatment 63. Additionally, it was shown in a mouse‐PK model that the serum stability of a fluorescent Cy5‐payload fused to the hapten digoxigenin is significantly increased when complexed to the same anti‐Dig‐antibody in comparison to free digoxigenin‐Cy5.
The non‐covalent manner in which the payload is attached to the antibody gives the approach the characteristics of a sustained release principle. The reason is that stability of the antibody‐payload complexes is defined by the strength of the antibody–hapten interaction, with the off‐rate determining the release of the payload from the antibody paratope. Released payload can consequently move further as a separate entity with low molecular weight (e.g. for increased tissue penetration at a targeted site). One major difference to other sustained release principles however is the fact that the released payload can re‐bind the PK‐modulating hapten‐binding antibody in the circulation (driven by the on‐rate of the interaction). Thus, payloads can switch between the bound and free form, resulting in a ‘PK‐buffer’ principle, as opposed to standard sustained release formulations. The availability of various hapten‐antibody combinations with different binding kinetics (on‐ and off‐rates) and the possibility to modulate the binding kinetics of specific anti‐hapten antibodies by protein engineering makes this approach even more versatile to balance between ‘slow release’ and PK‐stable complexes.
The in vivo clearance of a haptenylated payload complexed by antibodies is faster than the clearance of the antibody itself (Fig. 5). This effect can be explained by the off‐rate driven release of the payload from the antibody, which makes the payload amenable for fast renal clearance. This implies that further improvement of PK properties of payloads is possible by a tighter attachment of the payload to the IgG. Indeed, it was shown that payloads that are covalently linked to antibodies by the hapten‐driven disulfide shuffling technology described above show in vivo serum half‐lives that are comparable to those of the IgG itself 22.
Figure 5.
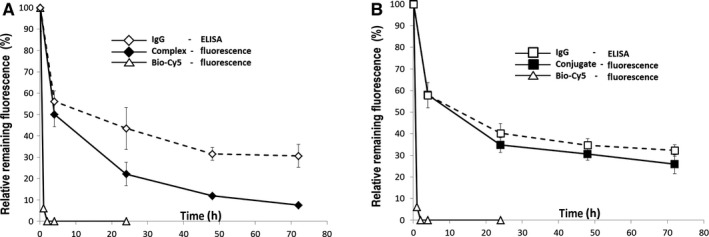
Modulation of the pharmacokinetic properties of a small compound by use of engineered hapten‐binding antibodies. Pharmacokinetic parameters were analyzed in a mouse model. A haptenylated fluorophore (BiotDig‐Cy5) was applied (intravenously) and its presence in serum at different time intervals detected by fluorescence measurements 22. The level of hapten‐binding antibody was determined by ELISA (detecting the Fc region). Due to its small size, the hapten‐fluorophore by itself is rapidly cleared from the serum. (A) Non‐covalent complexing with hapten‐binding antibodies prolongs the serum half‐life significantly. Differences between payload levels (fluorescence) and IgG levels reveal that the non‐covalently linked payload becomes released over time from the PK‐modulating antibody. (B) Covalent attachment via hapten‐mediated disulfide shuffling generates stable antibody conjugates with even further improved serum half‐life.
Interestingly, the engineered disulfide bridge seems to be reduced when the conjugates are delivered into cells by bispecific cell‐targeting antibodies. The fluorescent payload Biotin‐Cy5, which was conjugated to a bispecific anti‐biotin antibody was targeted to breast cancer cells by a second specificity against the internalizing cell surface carbohydrate antigen LeY. Confocal microscopy experiments showed that payload and antibody are separated over time upon internalization which can be explained by intracellular reduction of the disulfide bond between antibody and payload and consequent dissociation of the resulting complex.
In summary, the available hapten‐binding antibodies allow several options for PK modulation of haptenylated low molecular weight payloads: a sustained release‐like mechanism when hapten‐antibody complexes are used, long IgG‐like in vivo stability for covalent hapten–antibody conjugates, and an environment‐triggered release of payloads with hapten‐antibody conjugates targeted to internalizing receptors.
Hapten‐binding bsAbs for targeted and pretargeted payload delivery
Bispecific antibodies that bind haptens as well as cell surface antigens can be applied as vehicles to specifically deliver payloads to target cells. BsAbs that carry ‘unmodified’ hapten‐binding modules form non‐covalent complexes between delivery vehicles and payloads. These can separate upon antibody‐triggered internalization 44, 64. This confers intracellular payload release and can thereby facilitate the uptake and improve the activity of compounds whose molecular targets are located inside cells. Complexes of hapten‐binding antibodies that have payloads additionally stabilized by a designed disulfide bond are more stable in the circulation, minimizing undesired premature payload release. Their payloads can nevertheless be released by reduction of the disulfide bond inside cells 22.
Two general delivery principles can be applied to achieve specific targeting: (i) direct targeting of preformed antibody‐payload complexes or (ii) pretargeted payload delivery.
Fig. 6 shows two examples for delivery approaches with preformed complexes that we generated using bsAbs that harbor the digoxigenin‐ or biotin‐binding variable domains (see also Figs 2 and 3 for structures of the hapten‐binding entities). Silke Metz et al. 44 applied digoxigenin‐binding IgG‐derived tetravalent bispecific antibodies to target small compounds (Dig‐Cy5 shown in Fig. 6 or Dig‐Doxorubicine described by Metz et al. 44 to tumor cells. Cell surface targets that were addressed in that work were tumor antigens such as HER2, IGF1R, CD22, or LeY carbohydrate. The hapten‐binding (Dig‐binding) entity was a disulfide‐stabilized single‐chain Fv which was fused to the C‐termini of CH3 domains of the targeting antibodies. The digoxigeninylated payloads applied in these studies formed defined complexes with the bispecific antibodies in a 2:1 ratio as each bsAb contains two hapten‐binding sites. These preformed antibody‐payload complexes enabled antigen‐specific targeted delivery of payloads to tumor cells in vitro as well as in vivo.
Figure 6.
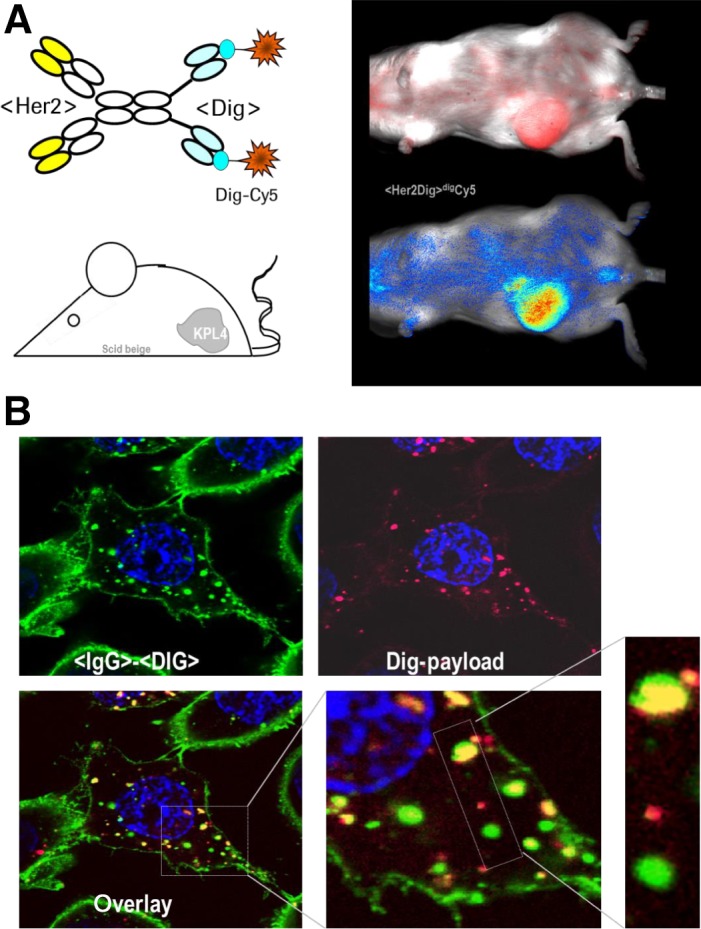
Hapten‐binding bsAbs for targeted payload delivery. (A) Dig‐coupled flourophore complexed with bsAbs that target tumor‐associated cell surface antigens (e.g. HER2) accumulate the payload at the tumors. (B) Hapten‐coupled nucleic acids (Dig‐payload) can be coupled to bsAbs and delivered to and into target cells where payload (red) and targeting vehicles (green) separate in vesicular compartments.
Schneider et al. described the application of Dig‐binding bispecific antibodies for targeted delivery of nucleic acids (siRNAs) to and into cells 64, 65. The delivery vehicles used in that study were composed in a similar manner as described above for tumor cell surface binding (internalizing) as well as Dig‐binding modules. Complexes of these bsAbs with hapten‐coupled small interfering RNA (siRNA) enable targeted delivery to tumor cells. Vehicles and payloads accumulated on target cells and internalized into vesicular compartments, however without releasing the nucleic acids into the cytosol. Lack of escape from endosomes initially prevented efficient siRNA‐mediated target gene knockdown. This bottleneck was subsequently overcome by packaging haptenylated siRNA into scaffold complexes (such as dynamic polyconjugates DPCs) 66 or into lipid‐based nanoparticles (LNPs) 67. Also, nanoparticles could be generated that contain haptens directly on their surfaces. This approach retained the cell surface antigen‐specific delivery to target cells or tissue, and not only delivered siRNAs to tumor cells but also (via the added endosome escape modules) into their cytoplasm. This – in turn – enabled effective and targeted siRNA‐specific messenger RNA knockdown. The principle of bispecific antibody mediated targeting of hapten‐coupled nanoparticles has subsequently been followed up by Tung et al. who generated and applied polyethylene glycol (PEG) binding bsAbs in a very similar manner for payload targeting 68, 69. PEG is a polymer that can be linked to various payloads including peptides, proteins, liposomes, and nanoparticles in the same manner as standard haptens. Antibodies that bind to defined units of PEG show therefore the same properties and similar application opportunities 69. Because of that, PEG‐binding bsAbs can successfully be applied for targeted delivery of PEGylated compounds. As many formulations contain PEG as integral part of nanoparticles, PEG‐binding bsAbs can be used for targeted nanoparticle delivery in a similar manner as nanoparticles are coupled to other haptens. Examples for such complexes are bsAb‐targeted nanoparticles containing doxorubicine which have demonstrated antibody‐dependent cytotoxicity toward target cells in vitro as well as in in vivo xenograft models 68.
Another principle for payload delivery via hapten‐binding bsAbs is ‘pretargeting’ 70, 71, 72, 73. In this concept, targeting vehicles and payloads are not combined prior to application. Instead, delivery (or capture) vehicles are applied without payload first to enable their distribution and binding to desired target sites. Subsequently, non‐bound targeting vehicles are cleared from circulation followed by administration of haptenylated payloads. These (small) payloads distribute rapidly throughout the body and become captured at the desired target sites by the hapten‐binding bsAbs. Any payload that is not captured becomes eliminated rapidly, in many cases by renal excretion. This in turn minimizes undesired systemic exposure and unspecific effects to non‐target tissues by non‐targeted payload. Antibody containing hapten‐binding pretargeting principles were initially generated by conjugating or fusing avidin/streptavidin modules to antibodies with the objective to capture and accumulate biotinylated (radioactive) payloads on target tissues such as tumors. Subsequently ‘standard’ hapten binders replaced the non‐human hapten‐capture modules avidin or streptavidin. This shall overcome potential immunogenicity issues associated with clinical applications of non‐human proteins. Such bispecific hapten‐binding antibody derivatives have shown functionality in pretargeted imaging and therapy approaches 70, 71, 72, 73.
Bispecific hapten‐binding antibodies as cell biology research tools
The precise coupling stoichiometry of haptenylated payloads to hapten‐binding antibodies combined with the feasibility to generate bispecific cell surface targeting derivatives makes such antibody–hapten systems attractive as versatile research tools. This includes various cell biology research applications, in particular in cellular quantitation processes 74. One example for that is the accurate determination of the number of proteins displayed on the surface and in intracellular vesicles of cells. Such measurements facilitate the modeling of cellular networks, the discrimination of cellular states, and enables also predictions (and subsequent modeling) of cell surface receptor saturation and/or potential target mediated drug disposition effects. Additionally, the knowledge of the amount of delivered antibody and thus payload to an intracellular vesicle (or the sum of vesicles per cell in a given time) helps to understand potency and dosing regimen requirements.
Quantitation approaches using flow cytometry are dependent on beads carrying a known number of fluorescent molecules which are used as reference standards. Beads are characterized by comparison to a calibrator solution of the fluorophore, resulting in a MESF value (molecules of equivalent soluble fluorophore) which is the common unit in referring to the number of cellular proteins 75. The precision of flow cytometry‐based methods usually suffers from the generation of an antibody with a well characterized labeling ratio. Most often a random lysine‐linker chemistry‐based coupling method is used resulting in a mixture of labeled antibodies which can result in loss of antigen binding, poor labeling reproducibility as well as shifted excitation and emission spectra. For such antibodies, an additional and thus error‐prone normalization step using beads which can bind a defined amount of antibody is required 76. Hapten‐binding antibodies circumvent these problems and in net effect reduce assay complexity, increase reproducibility and in its precision are at least as good as previously described methods 74, 75, 76. We have validated the method using different growth factor receptors (ErbB family and cMET) as cellular antigens. Formats which differ in valency, geometry of the position of their binding modules to others, and presence or absence of Fc part were used. Fluorescent labeling was achieved by using a DIG‐Cy5 conjugate. In theory and also experimentally confirmed, a monovalent high affine binder gives more precise results on antigen numbers than a format which is bivalent for the antigen. Labeled bispecific antibodies can be used in regular flow cytometry assay setups as they are not prone to fluorophore bleeding. Cell saturation curves demonstrate this, as shown for the cMET receptor expressed on the tumor cell line H1993. Histograms obtained under saturating antibody concentrations can then be used for comparison to reference beads to enable exact quantification. Additional normalization steps are not required thus allowing a quick and reliable determination of cellular antigen levels.
Using confocal microscopy, the presented approach can be expanded to encompass also the quantitation of endosomal antigens by measuring the amount of internalized antibody. By using a Cy5 calibrator solution antibody signals can be easily quantified (Fig. 7 shows the cell surface staining of cMET‐expressing H1993 cells). Upon internalization, intracellular vesicles are visible, the intensity of which can be matched to the Cy5 calibrator solution and the approach can be combined with z‐stacks to provide a volumetric analysis of delivered antibody in a given time frame. This information helps in understanding the amount of delivered payload, if for instance siRNA or cytotoxic compounds are delivered to aberrant cells. Thus, designed hapten‐binding bispecific antibodies, in combination with defined haptenylated fluorophores, can be applied as versatile reagents and tools in cell biology, for the understanding of cellular signaling events as well as drug delivery mechanisms.
Figure 7.
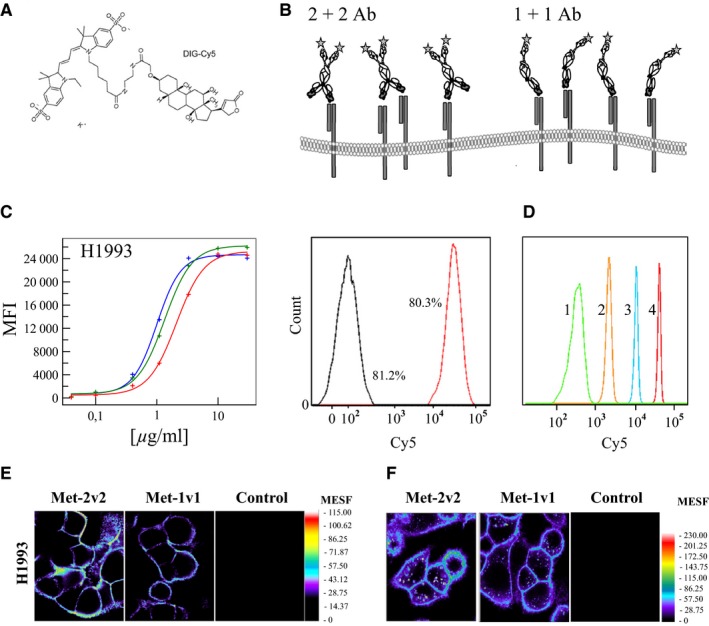
Cell‐surface targeting and hapten‐binding bispecific antibodies as research tools. (A) Chemical structure of the small molecule DIG‐Cy5. (B) Binding possibilities of a 1 + 1 and 2 + 2 format emphasizing that a 2 + 2 BsAb can also bind monovalently. (C) Concentration‐dependent analysis of BsAb cell surface binding to a growth factor receptor (cMET) in H1993 as well as histogram analysis at preset concentration. Red curve = 1 + 1 IgG, green = 1 + 1 Fab‐scFv, blue = 2 + 2 IgG) (D) Histogram of MESF calibration beads used for generating a calibration curve for quantitation. (E) Visualization of a growth factor receptor (cMET) on tumor cells by confocal microscopy using the indicated antibodies. (F) Analysis of cMET cell surface expression and internalized cMET in H1993. Cells were incubated with antibodies for 60 min at 37°C. Signals for Met2v2 were corrected for the number of Dig‐binding scFv per antibody.
Conclusions
Hapten‐binding antibodies have historically played and still possess a pivotal role in immunology, in particular in the antibody field. In fact, hapten‐binding antibodies and antibody fragments paved the way to antibody generation (because haptens are very important immunogens), to antibody characterization (the first structures were those of hapten binders), and finally expanded antibody engineering technologies that led the field into engineered therapeutic entities. Today, hapten‐binding antibodies are still widely applied as reagents and tools, and play an important role in the setup of analytical and/or diagnostic assays. More importantly, antibody derived hapten‐binding entities have also evolved over the past years to engineered delivery vehicles. These may play an important role in future targeting and pretargeting approaches in disease diagnosis and targeted therapy.
Acknowledgements
We thank Klaus Mayer for his valuable intellectual and operative contributions to the generation and characterization of engineered hapten‐binding antibody derivatives.
References
- 1. Poljak RJ, Amziel LM, Avey HP, Chen BL, Phizackerley RP, Saul F. Three‐dimensional structure of the Fab’ fragment of a human immunoglobulin at 2.8‐A resolution. Proc Natl Acad Sci USA 1973;70:3305–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amzel LM, Poljak RJ, Saul F, Varga JM, Richards FF. The three dimensional structure of a combining region‐ligand complex of immunoglobulin NEW at 3.5‐A resolution. Proc Natl Acad Sci USA 1974;71:1427–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Padlan EA, Segal DM, Spande TF, Davies DR, Rudikoff S, Potter M. Structure at 4.5 Å resolution of a phosphorylcholine‐binding fab. Nat New Biol 1973;245:165–167. [DOI] [PubMed] [Google Scholar]
- 4. Segal DM, Padlan EA, Cohen GH, Rudikoff S, Potter M, Davies DR. The three‐dimensional structure of a phosphorylcholine‐binding mouse immunoglobulin Fab and the nature of the antigen binding site. Proc Natl Acad Sci USA 1974;71:4298–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kabat EA. Proceedings. The structural basis of antibody specificity. Hoppe‐Seyler's Z Physiol Chem 1976;357:613–614. [PubMed] [Google Scholar]
- 6. Padlan EA. Structural implications of sequence variability in immunoglobulins. Proc Natl Acad Sci USA 1977;74:2551–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Padlan EA, Kabat EA. Modeling of antibody combining sites. Methods Enzymol 1991;203:3–21. [DOI] [PubMed] [Google Scholar]
- 8. Plueckthun A. Antibody engineering. Curr Opin Biotechnol 1991;2:238–246. [DOI] [PubMed] [Google Scholar]
- 9. Carter P, et al. Humanization of an anti‐p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA 1992;89:4285–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of immunological interest. National Institutes of Health Publication No 91‐3242. Bethesda, MD: National Institutes of Health, 1991. [Google Scholar]
- 11. Bird RE, et al. Single‐chain antigen‐binding proteins. Science 1988;242:423–426. [DOI] [PubMed] [Google Scholar]
- 12. Skerra A. Plueckthun A Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 1988;240:1038–1041. [DOI] [PubMed] [Google Scholar]
- 13. Barbas CF 3rd, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci USA 1991;88:7978–7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Little M, Breitling F, Duebel S, Fuchs P, Braunagel M. Human antibody libraries in Escherichia coli . J Biotechnol 1995;41:187–195. [DOI] [PubMed] [Google Scholar]
- 15. Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen‐binding affinity. Proc Natl Acad Sci USA 2000;97:10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thie H, Voedisch B, Dübel S, Hust M, Schirrmann T. Affinity maturation by phage display. Methods Mol Biol 2009;525:309–322. [DOI] [PubMed] [Google Scholar]
- 17. Glockshuber R, Schmidt T, Plueckthun A. The disulfide bonds in antibody variable domains: effects on stability, folding in vitro, and functional expression in Escherichia coli . Biochemistry 1992;31:1270–1279. [DOI] [PubMed] [Google Scholar]
- 18. Berman HM, et al. The protein data bank. Nucleic Acids Res 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singh MK, Srivastava S, Raghava GP, Varshney GC, Hapten DB. A comprehensive database of haptens, carrier proteins and anti‐hapten antibodies. Bioinformatics 2006;22:253–255. [DOI] [PubMed] [Google Scholar]
- 20. Kessler C. The digoxigenin:anti‐digoxigenin (DIG) technology – a survey on the concept and realization of a novel bioanalytical indicator system. Mol Cell Probes 1991;5:161–205. [DOI] [PubMed] [Google Scholar]
- 21. Holtke HJ, et al. The digoxigenin (DIG) system for non‐radioactive labelling and detection of nucleic acids – an overview. Cell Mol Biol 1995;41:883–905. [PubMed] [Google Scholar]
- 22. Dengl S, et al. Hapten‐directed spontaneous disulfide shuffling: a universal technology for site‐directed covalent coupling of payloads to antibodies. FASEB J 2015;29:1763–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grote M, Haas AK, Klein C, Schaefer W, Brinkmann U. Bispecific antibody derivatives based on full‐length IgG formats. Methods Mol Biol 2012;901:247–263. [DOI] [PubMed] [Google Scholar]
- 24. Croasdale R, et al. Development of tetravalent IgG1 dual targeting IGF‐1R‐EGFR antibodies with potent tumor inhibition. Arch Biochem Biophys 2012;526:206–218. [DOI] [PubMed] [Google Scholar]
- 25. Schanzer J, et al. Development of tetravalent, bispecific CCR5 antibodies with antiviral activity against CCR5 monoclonal antibody‐resistant HIV‐1 strains. Antimicrob Agents Chemother 2011;55:2369–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schanzer JM, et al. A novel glycoenginereed bispecific antibody format for targeted inhibition of EGFR and IGF‐1R demonstrating unique molecular properties. J Biol Chem 2014;289:18693–18706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haas AK, von Schwerin C, Matscheko D, Brinkmann U. Fluorescent citrine‐IgG fusion proteins produced in mammalian cells. mAbs 2010;2:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discovery Today 2015;20:838–847. [DOI] [PubMed] [Google Scholar]
- 29. Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U. The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genomics Proteomics 2013;10:1–18. [PubMed] [Google Scholar]
- 30. Weidle UH, Kontermann RE, Brinkmann U. Tumor‐antigen‐binding bispecific antibodies for cancer treatment. Semin Oncol 2014;41:653–660. [DOI] [PubMed] [Google Scholar]
- 31. Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol 1997;15:159–163. [DOI] [PubMed] [Google Scholar]
- 32. Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide‐stabilized Fv fragment. Proc Natl Acad Sci USA 1993;90:7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reiter Y, Brinkmann U, Webber KO, Jung SH, Lee B, Pastan I. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disulfide‐stabilized Fv. Protein Eng 1994;7:697–704. [DOI] [PubMed] [Google Scholar]
- 34. Reiter Y, Brinkmann U, Jung SH, Pastan I, Lee B. Disulfide stabilization of antibody Fv: computer predictions and experimental evaluation. Protein Eng 1995;8:1323–1331. [DOI] [PubMed] [Google Scholar]
- 35. Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide‐stabilized Fv fragments. Nat Biotechnol 1996;14:1239–1245. [DOI] [PubMed] [Google Scholar]
- 36. Rodrigues ML, et al. Development of a humanized disulfide‐stabilized anti‐p185HER2 Fv‐beta‐lactamase fusion protein for activation of a cephalosporin doxorubicin prodrug. Cancer Res 1995;55:63–70. [PubMed] [Google Scholar]
- 37. Reiter Y, et al. Improved binding and antitumor activity of a recombinant anti‐erbB2 immunotoxin by disulfide stabilization of the Fv fragment. J Biol Chem 1994;269:18327–18331. [PubMed] [Google Scholar]
- 38. Reiter Y, Kreitman RJ, Brinkmann U, Pastan I. Cytotoxic and antitumor activity of a recombinant immunotoxin composed of disulfide‐stabilized anti‐Tac Fv fragment and truncated Pseudomonas exotoxin . Int J Cancer 1994;58:142–149. [DOI] [PubMed] [Google Scholar]
- 39. Reiter Y, Pai LH, Brinkmann U, Wang QC, Pastan I. Antitumor activity and pharmacokinetics in mice of a recombinant immunotoxin containing a disulfide‐stabilized Fv fragment. Cancer Res 1994;54:2714–2718. [PubMed] [Google Scholar]
- 40. Reiter Y, Brinkmann U, Kreitman RJ, Jung SH, Lee B, Pastan I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 1994;33:5451–5459. [DOI] [PubMed] [Google Scholar]
- 41. Webber KO, Reiter Y, Brinkmann U, Kreitman R, Pastan I. Preparation and characterization of a disulfide‐stabilized Fv fragment of the anti‐Tac antibody: comparison with its single‐chain analog. Mol Immunol 1995;32:249–258. [DOI] [PubMed] [Google Scholar]
- 42. Bera TK, Onda M, Brinkmann U, Pastan I. A bivalent disulfide‐stabilized Fv with improved antigen binding to erbB2. J Mol Biol 1998;281:475–483. [DOI] [PubMed] [Google Scholar]
- 43. Brinkmann U, Chowdhury PS, Roscoe DM, Pastan I. Phage display of disulfide‐stabilized Fv fragments. J Immunol Methods 1995;182:41–50. [DOI] [PubMed] [Google Scholar]
- 44. Metz S, et al. Bispecific digoxigenin‐binding antibodies for targeted payload delivery. Proc Natl Acad Sci USA 2011;108:8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Metz S, et al. Bispecific antibody derivatives with restricted binding functionalities that are activated by proteolytic processing. Protein Eng Des Sel 2012;25:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ridgway JB, Presta LG, Carter P. ‘Knobs‐into‐holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996;9:617–621. [DOI] [PubMed] [Google Scholar]
- 47. Merchant AM, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998;16:677–681. [DOI] [PubMed] [Google Scholar]
- 48. Wu C, et al. Molecular construction and optimization of anti‐human IL‐1alpha/beta dual variable domain immunoglobulin (DVD‐Ig) molecules. mAbs 2009;1:339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu C, et al. Simultaneous targeting of multiple disease mediators by a dual‐variable‐domain immunoglobulin. Nat Biotechnol 2007;25:1290–1297. [DOI] [PubMed] [Google Scholar]
- 50. Gu J, Ghayur T. Generation of dual‐variable‐domain immunoglobulin molecules for dual‐specific targeting. Methods Enzymol 2012;502:25–41. [DOI] [PubMed] [Google Scholar]
- 51. Schaefer W, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA 2011;108:11187–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klein C, et al. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012;4:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kontermann RE. Strategies for extended serum half‐life of protein therapeutics. Curr Opin Biotechnol 2011;22:868–876. [DOI] [PubMed] [Google Scholar]
- 54. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007;7:715–725. [DOI] [PubMed] [Google Scholar]
- 55. Hutt M, Färber‐Schwarz A, Unverdorben F, Richter F, Kontermann RE. Plasma half‐life extension of small recombinant antibodies by fusion to immunoglobulin‐binding domains. J Biol Chem 2012;287:4462–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pasut G, Veronese FM. State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release 2012;161:461–472. [DOI] [PubMed] [Google Scholar]
- 57. Antonelli M, Sandroni C. Hydroxyethyl starch for intravenous volume replacement: more harm than benefit. JAMA 2013;309:723–724. [DOI] [PubMed] [Google Scholar]
- 58. Junutula JR, et al. Site specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008;26:925–932. [DOI] [PubMed] [Google Scholar]
- 59. Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site‐specific antibody drug conjugates for cancer therapy. mAbs 2014;6:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Doppalapudi VR, et al. Chemical generation of bispecific antibodies. Proc Natl Acad Sci USA 2010;107:22611–22616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Doppalapudi VR, et al. Chemically programmed antibodies: endothelin receptor targeting CovX‐Bodies Bioorg. Med Chem Lett 2007;17:501–506. [DOI] [PubMed] [Google Scholar]
- 62. Zac‐Varghese S, De Silva A, Bloom SR. Translational studies on PYY as a novel target in obesity. Curr Opin Pharmacol 2011;11:582–585. [DOI] [PubMed] [Google Scholar]
- 63. Hoffmann E, et al. PK modulation of haptenylated peptides via non‐covalent antibody complexation. J Controlled Release 2013;171:48–56. [DOI] [PubMed] [Google Scholar]
- 64. Schneider B, et al. Targeted siRNA delivery and mRNA knockdown mediated by bispecific digoxigenin‐binding antibodies. Mol Ther Nucleic Acids 2012;1:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thorey IS, Grote M, Mayer K, Brinkmann U. Hapten‐binding bispecific antibodies for the targeted delivery of siRNA and siRNA‐containing nanoparticles. Methods Mol Biol (Clifton, N.J.) 2016;1364:219–234. [DOI] [PubMed] [Google Scholar]
- 66. Rozema DB, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA 2007;104:12982–12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Frank‐Kamenetsky M, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA 2008;105:11915–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tung HY, et al. Selective delivery of PEGylated compounds to tumor cells by anti‐PEG hybrid antibodies. Mol Cancer Ther 2015;14:1317–1326. [DOI] [PubMed] [Google Scholar]
- 69. Kao CH, et al. One‐step mixing with humanized anti‐mPEG bispecific antibody enhances tumor accumulation and therapeutic efficacy of mPEGylated nanoparticles. Biomaterials 2014;35:9930–9940. [DOI] [PubMed] [Google Scholar]
- 70. Goldenberg DM, Chang CH, Rossi EA, McBride JW, Sharkey RM. Pretargeted molecular imaging and radioimmunotherapy. Theranostics 2012;2:523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van de Watering FC, Rijpkema M, Perk L, Brinkmann U, Oyen WJ, Boerman OC. Zirconium‐89 labeled antibodies: a new tool for molecular imaging in cancer patients. BioMed Res Int 2014;2014:203601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aarts F, et al. Pretargeted radioimmunoscintigraphy in patients with primary colorectal cancer using a bispecific anticarcinoembryonic antigen CEA X anti‐di‐diethylenetriaminepentaacetic acid F(ab’)2 antibody. Cancer 2010;116(Suppl):1111–1117. [DOI] [PubMed] [Google Scholar]
- 73. Kraeber‐Bodere F, et al. A pretargeting system for tumor PET imaging and radioimmunotherapy. Front Pharmacol 2015;6:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Panke C, et al. Quantification of cell surface proteins with bispecific antibodies. Protein Eng Des Sel 2013;26:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schwartz A, Gaigalas AK, Wang L, Marti GE, Vogt RF, Fernandez‐Repollet E. Formalization of the MESF unit of fluorescence intensity. Cytometry B Clin Cytom 2004;57:1–6. [DOI] [PubMed] [Google Scholar]
- 76. Lenkei R, et al. Performance of calibration standards for antigen quantitation with flow cytometry. Cytometry 1998;33:188–196. [DOI] [PubMed] [Google Scholar]