

Abstract
Antibodies are indispensable tools for biomedicine and anticancer therapy. Nevertheless, their use is compromised by high production costs, limited stability, and difficulty of chemical modification. The design and preparation of synthetic polymer conjugates capable of replacing antibodies in biomedical applications such as ELISA, flow cytometry, immunocytochemistry, and immunoprecipitation is reported. The conjugates, named “iBodies”, consist of an HPMA copolymer decorated with low‐molecular‐weight compounds that function as targeting ligands, affinity anchors, and imaging probes. We prepared specific conjugates targeting several proteins with known ligands and used these iBodies for enzyme inhibition, protein isolation, immobilization, quantification, and live‐cell imaging. Our data indicate that this highly modular and versatile polymer system can be used to produce inexpensive and stable antibody substitutes directed toward virtually any protein of interest with a known ligand.
Keywords: antibody mimetics, HPMA, molecular recognition, polymer conjugates, protein targeting
The discovery of antibodies specifically targeting biologically relevant molecules revolutionized the life sciences. Nevertheless, the use of antibodies has several disadvantages, such as limited stability, difficulty of chemical modification, and difficulty targeting proteins in mouse models. To address these limitations, researchers have developed alternative molecular recognition tools capable of replacing antibodies in biomedical research applications (antibody mimetics). These include affibodies,1 designed ankyrin repeat proteins (DARPins),2 and aptamers.3 Recently, chemically synthesized small molecules that fulfill some functions of antibodies have been described.4 Polymers capable of molecular recognition of other molecules were described several decades ago. These molecularly imprinted polymers (MIPs) are based on structure complementarity with the target molecules and are used as molecular biosensors and binders.5
We set out to develop novel antibody mimetics based on N‐(2‐hydroxypropyl)methacrylamide (HPMA) copolymer conjugates decorated with three different low‐molecular‐weight ligands. We chose water‐soluble and biocompatible HPMA copolymers since they have been used for the development of drug delivery vectors, imaging agents, and polymer drugs.6 The HPMA copolymers are multivalent synthetic macromolecules that carry a number of reactive groups that enable covalent attachment of various ligands such as fluorescent probes, therapeutics, proteins, and oligonucleotides.7
Specific recognition, immobilization, and imaging of a protein of interest require a specific targeting ligand (e.g., an inhibitor), an affinity anchor, and an imaging probe attached to the polymer backbone (Figure 1 a). As an affinity tag, we chose biotin because it can be readily used for high‐affinity immobilization on a variety of commercially available resins (Figure 1 b). As an imaging probe, we chose the fluorophore ATTO488 (Figure 1 c). The choice of targeting ligand depends on the protein of interest; we developed and tested these antibody mimetics with glutamate carboxypeptidase II, HIV‐1 protease, pepsin, and His‐tagged proteins (Figure 1 d–g). To reflect functional similarity to antibodies and the use of inhibitors as targeting ligands, we propose the name iBodies for these polymer conjugates. All prepared iBodies were characterized in terms of chemical identity and the content of individual ligands, and their characteristics are summarized in Table 1 and Table S2 in the Supporting Information. We assume statistical distribution of the attached moieties, since each ligand terminates with the same sterically unhindered reactive moiety (‐CH2CH2NH2). The synthesis and characteristics of the conjugates are described in detail in the Supporting Information.
Figure 1.
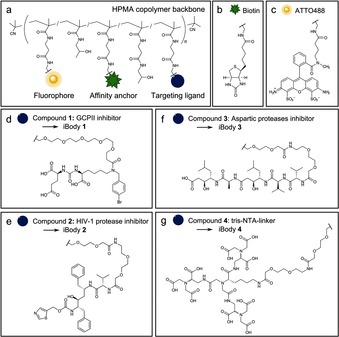
Structures of the iBodies and their functional modules. a) HPMA copolymer decorated with functional molecules. b) The affinity anchor biotin. c) ATTO488 fluorophore. d) GCPII inhibitor. e) HIV‐1 protease inhibitor. f) A class‐specific inhibitor of aspartic proteases. g) A nitrilotriacetic acid (NTA)‐based ligand for binding the His‐tag.
Table 1.
The composition and basic characteristics of the iBodies.
| Conjugate (M n) | Target | No. of inhibitor moieties | No. of ATTO488 units | No. of biotin units | K i [nm] |
|---|---|---|---|---|---|
| iBody 1 (148 800)[a] | GCPII | 19 | 7 | 51 | 0.0043±0.0005 |
| iBody 2 (40 600)[a] | HIV‐1 protease | 6 | – | 7 | 7.9±0.5 |
| iBody 3 (42 100)[a] | Aspartic proteases | 7 | – | 9 | 18.9±0.2[b] |
| iBody 4 (135 800)[a] | His‐tag sequence | 12 | 7 | 46 | 3.5±0.1[c] |
| iBody 5 (108 100)[a] | – | – | 6 | 41 | – |
| iBody 6 (37 800)[a] | – | – | – | 8 | – |
[a] M n=number‐average molecular weight. For full molecular characteristics of the conjugates, see Table S2–S4 and Figure S8 in Supporting Information. [b] K i value was determined for wild‐type HIV‐1 protease. [c] For iBody 4, a dissociation constant K D is shown instead of an inhibition constant K i.
For developing the technique, we chose human glutamate carboxypeptidase II (GCPII; also known as prostate‐specific membrane antigen or PSMA). GCPII is a transmembrane metallopeptidase that is strongly expressed in the brain and prostate; its expression is markedly increased in prostate carcinoma.8
The polymer conjugate targeting GCPII (iBody 1, Figure S1 in the Supporting Information) contains a tight‐binding GCPII inhibitor as a targeting ligand. To avoid steric hindrance between GCPII and the polymer molecules, the previously described inhibitor (called compound 22 a,9 Table S1) was modified with a PEG5 linker to yield compound 1 (Figure 1 d). As a negative control, we prepared the corresponding conjugate lacking the GCPII inhibitor (iBody 5; Figure S2).
The utility of iBody 1 for the inhibition, binding, and visualization of GCPII was tested with a variety of common biochemical methods in which antibodies are normally used: immunoprecipitation and affinity pull‐downs, flow cytometry, immunocytochemistry, and ELISA.
First, we tested whether iBody 1 inhibits the enzymatic activity of GCPII and determined K i values for compound 1 and iBody 1 (Figure 2 a). Interestingly, attachment of several molecules of compound 1 to the HPMA copolymer led to a significant drop in the K i value [K i(compound 1)=2.0 nm vs. K i(iBody 1)=4.3 pm]. This nearly three‐order‐of‐magnitude decrease in K i can be explained by the synergic effect of several inhibitor molecules on the polymer scaffold and increased local inhibitor concentration (19 inhibitor molecules per polymer chain).
Figure 2.
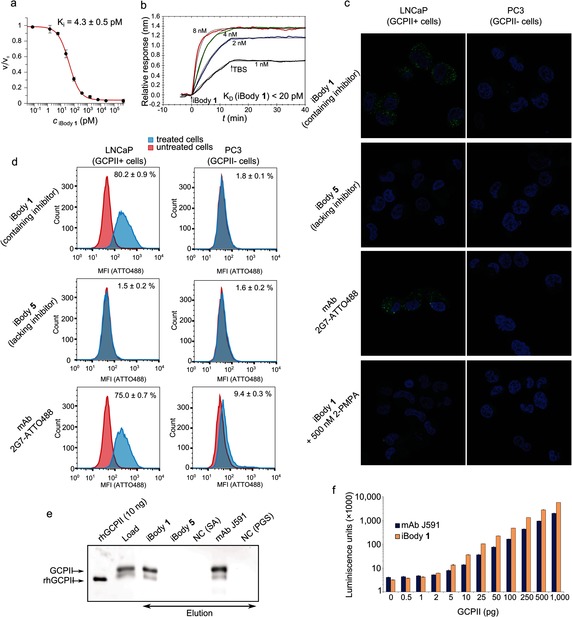
Application of iBody 1, which targets glutamate carboxypeptidase II (GCPII). a) The inhibition potency of iBody 1 in terms of GCPII hydrolytic activity. b) SPR analysis of iBody 1 binding to immobilized GCPII (K D<20 pm). c) Confocal microscopy of cells positive (LNCaP) and negative (PC3) for GCPII expression. Cells were stained with iBody 1; to compare iBody staining with antibody staining, the anti‐GCPII monoclonal antibody (mAb) 2G710 labeled with ATTO488 (2G7‐ATTO488) was used. Binding of iBody 1 to LNCaP cells can be blocked by using 2‐PMPA, a specific GCPII inhibitor. d) Flow cytometry analysis of LNCaP and PC3 cells incubated with iBody 1, iBody 5 (which lacks the targeting module), or 2G7‐ATTO488. e) Western blot analysis of affinity isolation of GCPII from LNCaP cell lysate using iBody 1 or the anti‐GCPII mAb J591. iBody 5, blank Streptavidin Agarose (NC (SA)), and blank Protein G Sepharose (NC (PGS)) were used as negative controls. rhGCPII is a recombinant extracellular GCPII. Load=LNCaP cell lysate. f) Sandwich ELISA with the anti‐GCPII capture mAb 2G7 and either the biotinylated anti‐GCPII mAb J591 or iBody 1 used as the detecting agent.
The binding of iBody 1 to GCPII was further evaluated by surface plasmon resonance (SPR), which revealed an extremely high association rate and a low dissociation rate (Figure 2 b). The dissociation constant (K D<20 pm) is comparable to that of the tightest‐binding anti‐GCPII antibodies available.
To analyze the utility of iBody 1 for visualizing GCPII‐expressing cells, we incubated LNCaP cells (a cell line that endogenously expresses GCPII) and PC3 cells (which do not express GCPII) with iBody 1. As controls, we used iBody 5 (which lacks a GCPII inhibitor) and a fluorescently labeled anti‐GCPII monoclonal antibody 2G710 (mAb 2G7‐ATTO488). iBody 1 and the anti‐GCPII mAb bound specifically to LNCaP cells and not to PC3 cells, while iBody 5 did not bind to either LNCaP or PC3 cells. iBody 1 and the mAb were taken up into the cells through internalization of membrane‐embedded GCPII as expected.11 Additionally, binding of iBody 1 to LNCaP cells was blocked by 2‐(phosphonomethyl)pentanedioic acid (2‐PMPA), a specific GCPII inhibitor (Figure 2 c).
To analyze the potential use of iBodies in flow cytometry, LNCaP and PC3 cells were first incubated with iBody 1, iBody 5, or 2G7‐ATTO48810 and then analyzed by flow cytometry. The results indicate that both iBody 1 and the anti‐GCPII mAb specifically recognize GCPII‐expressing cells (LNCaP) and enable their separation from GCPII‐negative cells (PC3). Moreover, iBody 1 and iBody 5 exhibited very low non‐specific binding to PC3 cells, which was approximately 5‐fold lower than that of mAb 2G7‐ATTO488 (Figure 2 d).
Furthermore, we used iBody 1 to isolate GCPII from cell lysates. iBodies 1 and 5 were immobilized on Streptavidin Sepharose resin, and GCPII was pulled down from LNCaP cell lysate (Figure 2 e). An anti‐GCPII mAb (J59112) bound to Protein G Sepharose was used as a positive control. As shown by subsequent western blot analysis, the amount of GCPII immunoprecipitated when using iBody 1 was comparable to that obtained with mAb J591. These findings suggest that iBodies could also be useful for the isolation of target proteins from complex biological materials such as tissue lysates or blood samples.
Finally, we used iBodies for the quantitative detection of GCPII. We employed a sandwich ELISA in which the detecting anti‐GCPII mAb was replaced with iBody 1. The detection limit was as low as 0.5 pg GCPII; furthermore, the signal was linear over a nearly three‐order‐of‐magnitude concentration range (Figure 2 f).
To demonstrate versatility of iBodies, we also developed iBodies targeting the aspartic proteases HIV‐1 and pepsin. HIV‐1 protease plays a crucial role in viral replication13 and a number of specific inhibitors are available. As aspartic proteases, both HIV‐1 protease and pepsin are also efficiently inhibited by the peptidic inhibitor pepstatin A.14
The polymer conjugate targeted towards HIV‐1 protease (iBody 2; Figure S3) contains compound 2, a specific HIV‐1 protease inhibitor derived from the commercially available inhibitor ritonavir (Figure 1 e), as a targeting ligand. The analogous iBody 3 (Figure S4) contains compound 3 (Figure 1 f), a derivative of the class‐specific aspartic protease inhibitor pepstatin A, as a targeting ligand. As a negative control, we prepared the corresponding conjugate lacking an inhibitor (iBody 6; Figure S5). We successfully used iBody 2 and iBody 3 to pull down HIV‐1 protease from an LNCaP cell lysate spiked with HIV‐1 protease (Figure S7a).
Besides HIV‐1 protease, another aspartic protease, pepsin, was also successfully pulled down from LNCaP lysate (spiked with pepsin) by using iBody 3 (Figure S7b), thus showing the general applicability of this iBody. In both experiments, iBody 6 (which contains only biotin and no inhibitor) was used as a negative control. This iBody was unable to pull down either HIV‐1 protease or pepsin from an LNCaP cell lysate (Figure S7a,b).
Finally, the use of iBodies is not limited to enzymes. Theoretically, iBodies can be designed to target any molecule of interest for which a ligand is known. To demonstrate this general principle, we set out to target proteins containing a polyhistidine tag (His‐tag), the most common affinity tag used for protein purification and visualization. Because the His‐tag is bound by nickel‐loaded nitrilotriacetic acid (NTA) derivatives, we prepared tris‐NTA connected to a linker15 (compound 4, Figure 1 g) and subsequently iBody 4 (Figure S6). iBody 4 was decorated with the fluorophore ATTO488 for visualization and biotin for both visualization and immobilization. As a negative control, iBody 5, which bears only the fluorophore ATTO488 and biotin, was used.
We compared the sensitivity and specificity of iBody 4 with that of commercially available peroxidase‐conjugated anti‐polyhistidine antibody by western blot. iBody 4 exhibited slightly increased sensitivity and equal specificity in comparison to the commercial antibody (Figure 3 a, b). When using iBody 4, we detected as little as 100 pg of the His‐tagged DNA damage protein 1 (Ddi1‐His). Additionally, we specifically pulled down the protein from an E. coli lysate (Figure 3 c). To quantify the binding, we analyzed the interaction between iBody 4 and Ddi1‐His by SPR, which indicated K D=3.5 nm (Figure 3 d).
Figure 3.
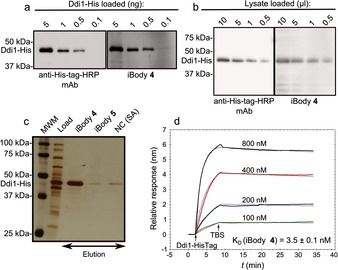
Application of iBody 4, which targets His‐tagged proteins. a) Comparison of iBody 4 and anti‐polyhistidine antibody sensitivity for the visualization of purified Ddi1‐His by western blot. b) Comparison of iBody 4 and the anti‐polyhistidine antibody for the visualization of Ddi1‐His in a cell lysate by western blot. c) Affinity isolation of Ddi1‐His by using iBody 4. iBody 5 (which does not possess the tris‐NTA ligand), and blank resin (NC) were used as negative controls. MWM=molecular‐weight marker. d) Binding of Ddi1‐His to immobilized iBody 4 as analyzed by SPR.
Our approach has two major advantages. First, the system is remarkably modular: any compound, functional group, or tag for any specific purpose can be added to form the final polymer conjugate. For the targeting of a specific protein with an iBody, simple replacement of the targeting moiety (the inhibitor) is sufficient to yield a new specific polymer conjugate. To some extent, this resembles the concept of molecularly imprinted polymers (MIPs). MIPs rely on lock‐and‐key interaction with the target, which makes them more suitable for the extraction of target compounds/proteins, even though the targeting of proteins on the cell surface has been reported.16 Second, the system is truly versatile: a single iBody can be used for several methods, as we have shown for GCPII. One potential limitation of the system is the need for a ligand that specifically binds to the target protein. Moreover, for attachment to the polymer backbone, the ligand must be modified with a linker that does not significantly compromise its binding affinity. Nonetheless, if a potent ligand is known and the attachment of the linker does not lead to a dramatic loss of potency, the preparation of a specific iBody is rather straightforward.
In summary, we have developed inexpensive, stable, and modular synthetic conjugates called iBodies for use as antibody mimetics. The presented data demonstrate that the prepared iBodies targeting various proteins of interest are efficient substitutes for the corresponding antibodies in standard immunochemical methods. Overall, iBodies offer an inexpensive, non‐animal‐based alternative for antibodies in biochemical methods involving the isolation and visualization of proteins, cells, and tissues.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Jana Starková and Karolína Šrámková for their technical support, Marco Colombatti for the D2B antibody, Michal Svoboda for expression of Ddi1‐His and Stanislava Matějková for ICP‐OES analysis. This work was supported by Grant No. P208‐12‐G016 (Center of Excellence) from the Grant Agency of the Czech Republic and InterBioMed project LO 1302 from the Ministry of Education of the Czech Republic.
P. Šácha, T. Knedlík, J. Schimer, J. Tykvart, J. Parolek, V. Navrátil, P. Dvořáková, F. Sedlák, K. Ulbrich, J. Strohalm, P. Majer, V. Šubr, J. Konvalinka, Angew. Chem. Int. Ed. 2016, 55, 2356.
Contributor Information
Dr. Vladimír Šubr, Email: subr@imc.cas.cz
Dr. Jan Konvalinka, Email: konval@uochb.cas.cz
References
- 1. Nord K., Gunneriusson E., Ringdahl J., Stahl S., Uhlen M., Nygren P. A., Nat. Biotechnol. 1997, 15, 772. [DOI] [PubMed] [Google Scholar]
- 2. Binz H. K., Stumpp M. T., Forrer P., Amstutz P., Pluckthun A., J. Mol. Biol. 2003, 332, 489. [DOI] [PubMed] [Google Scholar]
- 3. Ellington A. D., Szostak J. W., Nature 1990, 346, 818. [DOI] [PubMed] [Google Scholar]
- 4. McEnaney P. J., Fitzgerald K. J., Zhang A. X., E. F. Douglass, Jr. , Shan W., Balog A., Kolesnikova M. D., Spiegel D. A., J. Am. Chem. Soc. 2014, 136, 18034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Hoshino Y., Koide H., Urakami T., Kanazawa H., Kodama T., Oku N., Shea K. J., J. Am. Chem. Soc. 2010, 132, 6644; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Wulff G., Angew. Chem. Int. Ed. Engl. 1995, 34, 1812; [Google Scholar]; Angew. Chem. 1995, 107, 1958. [Google Scholar]
- 6.
- 6a. Haag R., Kratz F., Angew. Chem. Int. Ed. 2006, 45, 1198; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1218; [Google Scholar]
- 6b. Kopeček J., Kopečková P., Adv. Drug Delivery Rev. 2010, 62, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Šubr V., Kostka L., Strohalm J., Etrych T., Ulbrich K., Macromolecules 2013, 46, 2100. [Google Scholar]
- 8.
- 8a. Lapidus R. G., Tiffany C. W., Isaacs J. T., Slusher B. S., Prostate 2000, 45, 350; [DOI] [PubMed] [Google Scholar]
- 8b. Robinson M. B., Blakely R. D., Couto R., Coyle J. T., J. Biol. Chem. 1987, 262, 14498. [PubMed] [Google Scholar]
- 9. Tykvart J., Schimer J., Barinkova J., Pachl P., Postova-Slavetinska L., Majer P., Konvalinka J., Sacha P., Bioorg. Med. Chem. 2014, 22, 4099. [DOI] [PubMed] [Google Scholar]
- 10. Knedlík T., Navratil V., Vik V., Pacik D., Sacha P., Konvalinka J., Prostate 2014, 74, 768. [DOI] [PubMed] [Google Scholar]
- 11. Liu H., Rajasekaran A. K., Moy P., Xia Y., Kim S., Navarro V., Rahmati R., Bander N. H., Cancer Res. 1998, 58, 4055. [PubMed] [Google Scholar]
- 12. Liu H., Moy P., Kim S., Xia Y., Rajasekaran A., Navarro V., Knudsen B., Bander N. H., Cancer Res. 1997, 57, 3629. [PubMed] [Google Scholar]
- 13. Krausslich H. G., Ingraham R. H., Skoog M. T., Wimmer E., Pallai P. V., Carter C. A., Proc. Natl. Acad. Sci. USA 1989, 86, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eder J., Hommel U., Cumin F., Martoglio B., Gerhartz B., Curr. Pharm. Des. 2007, 13, 271. [DOI] [PubMed] [Google Scholar]
- 15. Huang Z., Hwang P., Watson D. S., Cao L., F. C. Szoka, Jr. , Bioconjugate Chem. 2009, 20, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Ma Y., Pan G. Q., Zhang Y., Guo X. Z., Zhang H. Q., Angew. Chem. Int. Ed. 2013, 52, 1511; [Google Scholar]; Angew. Chem. 2013, 125, 1551; [Google Scholar]
- 16b. Shinde S., El-Schich Z., Malakpour A., Wan W., Dizeyi N., Mohammadi R., Rurack K., Gjorloff Wingren A., Sellergren B., J. Am. Chem. Soc. 2015, 137, 13908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary