

Abstract
The zinc transporter protein ZIP13 plays crucial roles in bone, tooth, and connective tissue development, and its dysfunction is responsible for the spondylocheirodysplastic form of Ehlers-Danlos syndrome (SCD-EDS, OMIM 612350). We recently reported that the pathogenic mutations in ZIP13 reduce its functional protein level by accelerating the protein degradation via the VCP-linked ubiquitin proteasome pathway, resulting in the disturbance of intracellular zinc homeostasis that appears to contribute to SCD-EDS pathogenesis. Finally, we implicate that possible therapeutic approaches for SCD-EDS would be based on regulating the degradation of the pathogenic mutant ZIP13 proteins.
Keywords: corrector, degradation, pathogenic mutation, potentiator, SCD-EDS, SLC39A13, zinc transporter, ZIP13
Abbreviations
- SLC
Solute carrier
- SMAD
Sma and Mad related Family
- ZIP
Zrt/Irt-like protein
- ZnT
Zinc transporter
- CFTR
Cystic fibrosis transmembrane conductance regulator
- VCP
Valosin-containing protein
- HSP90
Heat shock protein 90
Zinc Transporters: ZIP and ZnT Families
Zinc (Zn) is an essential mineral in human health.1 In particular, Zn plays critical roles in the formation of connective tissues such as bone and skin, and in growth signaling, and Zn deficiency results in growth retardation, skin, and immunological problems.1-5 Zn deficiency was first discovered in Shiraz, Iran, where many people were suffering from similar symptoms, including severe growth retardation, anemia, rough and dry skin, and hepatosplenomegaly.1 However, when the patients went on a Zn-sufficient diet, their symptoms improved, demonstrating the importance of Zn in human health.1
In mammalian cells, Zn homeostasis is tightly maintained by Zn transporters, which mobilize Zn across the cell membrane.6,7 Based on their predicted membrane topology, they are divided into 2 major groups: the ZnT and ZIP families.6,7 The ZnT family is involved in Zn efflux from the cytosol, and is classified into 10 members including ZnT9, which lacks evidence of transporting Zn, and ZnT10, which was reported to mediate man- ganese transport in human cells. The X-ray structure of a bacterial ZnT homolog YiiP suggested that the ZnT family has a unique Y-shaped structure, with 6 transmembrane domains.8 Mutations in ZnT2 cause low Zn in human breast milk.9,10 ZnT3-null mice lack Zn in synaptic vesicles.11 ZnT8 is crucial for insulin-crystal formation in diabetes mellitus.12 ZIP family members, the first gates for Zn uptake into the cell, transport Zn into the cytosol, elevating the intracellular Zn level.6,7 ZIP family members have 7 or 8 putative transmembrane domains, but no X-ray structure is currently available. There are 14 ZIP family members in the human genome.6,7 The function, structure, and related diseases, however, for ZIP proteins are not well defined. The ZIP family members in animals are divided into 4 subfamilies according to their conserved sequence: the ZIPI, ZIPII, gufA, and LIV-1 subfamilies.7 The LIV-1 subfamily members consist mostly of mammalian Zn transporters; they represent about half of the known human Zn transporters, and contain a unique motif in their transmembrane domains, the HEXXH motif.7 In this paper, we focus on the LIV-1 subfamily member ZIP13 that is critically involved in SCD-EDS pathogenesis.
ZIP Family Zn Transporters and Human Diseases
ZIP family members play important roles in acquired pathogeneses.13 A recent report demonstrated that ZIP8-mediated Zn accumulation in chondrocytes induces the expression of metal-regulatory transcription factor 1 (MTF1), which causes osteoarthritis by up-regulating matrix-degrading enzymes.14 However, few human genetic diseases caused by mutations in ZIP family members have been reported.13 Until 2008, only one genetic disease associated with a ZIP member, acrodermatitis enteropathica, was known.13 Acrodermatitis enteropathica (AE; OMIM 201100) is a rare autosomal recessive disorder characterized by severe and generalized Zn-deficiency symptoms in infants, like periorificial and acral dermatitis, alopecia, and diarrhea.15,16 Mutations in ZIP4 are the main reason for AE.15 When the Zn level in the body is low, the N-terminal ectodomain of ZIP4 is cleaved off, and ZIP4 accumulates on the intestinal apical surface to take Zn in.17 Patients with AE show a low Zn level in their blood and suffer from clinical signs similar to patients with Zn deficiency.16 If they do not take additional Zn, they will die within 2 years.16 However, the health of these patients is dramatically improved by taking at least 1–2 mg Zn per kg body weight per day.15
Ehlers-Danlos syndrome (EDS) is an inherited disorder of connective tissue. 18 In 2008, a new type of EDS was reported.19,20 These patients exhibit both the common features of EDS, like heritable disorders of connective tissue, articular hypermobility, and skin hyperelasticity, and distinct physical signs, such as short stature, tapering fingers, wrinkled palms, and antimongoloid eye slant with a lack of periorbital tissue.19,20 We initially performed a mutation analysis for EDS-related genes in these patients, such as the genes for collagen 1 and 3, and lysyl hydroxylase, but failed to identify any mutations.19 However, we found that these patients’ symptoms were reminiscent of the Zip13-KO mouse phenotypes, which include growth retardation, kyphosis, osteopenia, abnormal cartilage development, craniofacial changes, and decreased dermal and corneal stromal collagen.19 SNP microarray, microsatellite mapping, and mutation analyses using samples from the patients’ family members revealed the pathogenic mutation to be glycine to aspartic acid at position 64 in the ZIP13 protein, which is encoded by the SLC39A13 gene.19 Another group identified a deletion mutation in the ZIP13 protein from patients with EDS features, and referred to the disease as a spondylocheiro dysplastic form of EDS.20 This new type of EDS is registered as spondylocheirodysplastic Ehlers-Danlos syndrome (SCD-EDS, OMIM 612350).
Pathogenic Mechanism of SCD-EDS
Analyses of Zip13-KO mice revealed that cells originating in the mesenchyme, including osteoblasts, chondrocytes, odontoblasts, and fibroblasts, show maturation defects that affect connective tissue development.19 Further molecular analyses using these cells revealed that the proper nuclear translocation of transcription factor SMADs, which respond to BMP and TGF-β and are critical for connective tissue development, was impaired in Zip13-KO cells, while their phosphorylation was unaffected.19 The Zn concentration in the serum and primary fibroblasts isolated from Zip13-KO mice was comparable with wild-type.19 Zn levels in the Golgi was up-regulated and while in the nucleus it was down-regulated in Zip13-KO cells.19 These findings were consistent with the observation that ZIP13 is normally expressed in the intracellular peri-nuclear Golgi region.19 Taken together, these results indicated that the intracellular Zn homeostasis is disrupted in Zip13-KO cells, and thus, that controlling intracellular Zn homeostasis may be a therapeutic strategy for SCD-EDS. To gain insights for improving the intracellular Zn homeostasis, we first characterized and analyzed the SCD-EDS pathogenic mutant ZIP13 proteins.21,22 We found that the pathogenic mutant ZIP13 proteins were promptly removed via the ubiquitin proteasome pathway.22 We next sought to identify molecules associated with the degradation pathways of the pathogenic mutant ZIP13 proteins, and found that inhibiting VCP or HSP90 caused the mutant proteins to accumulate within the cells (Fig. 1A).22
Figure 1.
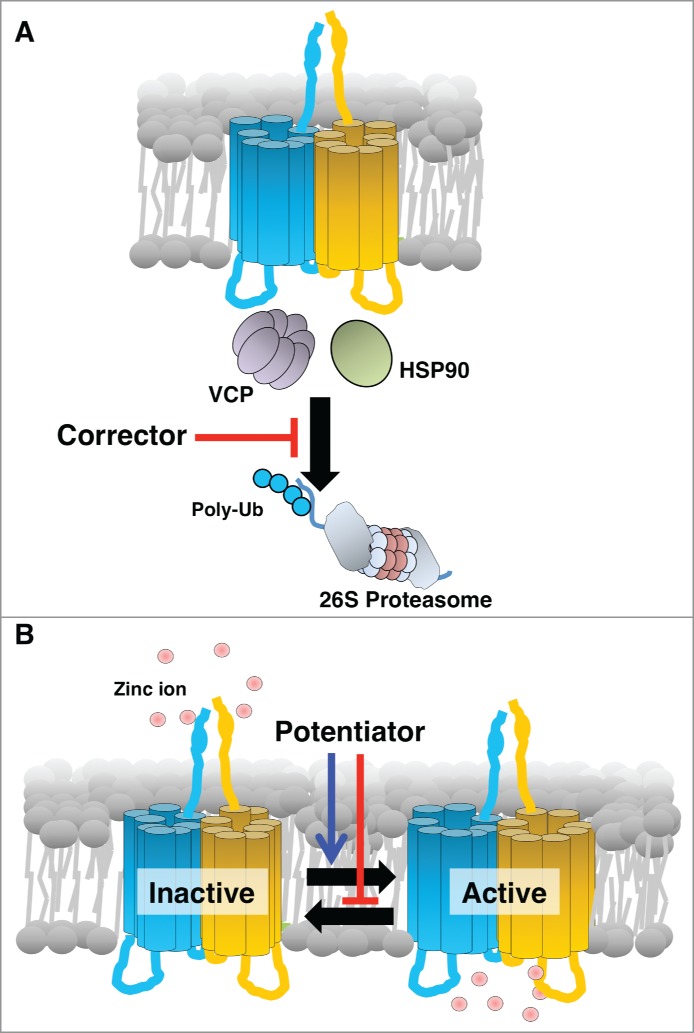
Schematic working mechanism of a “corrector” and a “potentiator” for a pathogenic mutant form of ZIP13. (A) ZIP13 “corrector” inhibits the coordinated actions of cofactors including the identified molecules, VCP and HSP90, involved in the unfolding and transport of mutant ZIP13 to the proteasome. As a result, the “corrector” causes an effective amount of functional ZIP13 protein to accumulate. (B) ZIP13 “potentiator” alters the equilibrium toward the active form, improving the intracellular Zn homeostasis by increasing Zn influx.
Understanding a pathogenic protein's degradation pathway is important for developing therapeutic approaches. All proteins have a defined lifetime; some last a short time, while others are long-lived. Similarly, pathogenic proteins are degraded and replaced with new pathogenic ones, if they are encoded in the genome. If a disease results from the accumulation of toxic proteins, the clinical research focuses on rapid removal of the pathogenic proteins by accelerating their degradation pathway. In contrast, if a disease is caused by a deficiency of functional proteins, and the pathogenic mutant proteins are still functional, clinical therapies seek to improve the lifetime and proper localization of the proteins. Developing a functional activator, called a “potentiator,” of a protein is an essential goal of pharmacological research. However, because relatively little is known about membrane protein degradation pathways compared to those for soluble proteins, and fewer direct functional analyses have been reported for membrane proteins, therapeutic approaches for diseases related to membrane transporters can be complicated and challenging.
Diseases Caused by Mutant Transporters: Clinical Trials and Therapies
How could SCD-EDS be treated? The answers might come from other transporter-related diseases. For example, cystic fibrosis (CF) is the most common autosomalrecessive genetic disorder; it is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes a chloride ion channel that mediates epithelial anion transport in the lung, liver, pancreas, digestive tract, and skin.23 CF is fatal, and many of the fatal mutations within the CFTR gene have been identified, and found to affect CFTR folding, stability, and channel gating.24 To control this disease, many researchers have focused on these 3 points.24 In fact, CFTR represents the largest research area on membrane protein-related folding and degradation, and various chaperones involved in CFTR's folding and degradation pathways have been identified.25 VX-809, which was screened from 164,000 small molecules, is the most promising investigational drug for CF.26 VX-809, a so-called CFTR “corrector,” targets the most common CFTR mutant protein, ΔF508, which exhibits impaired CFTR processing in the endoplasmic reticulum.27 VX-809 improves the processing of ΔF508 CFTR, allowing the mutant protein to acquire similar functional characteristics as normal CFTR.28
For SCD-EDS, we first confirmed that the proteasome inhibitors MG-132 and lactacystin could readily reverse the phenotypes of the pathogenic ZIP13 mutants.22 These inhibitors are normally toxic, activate various signaling pathways, and induce significant cell death. Bortezomib, the first therapeutic proteasome inhibitor to be used in humans, was approved in the US for treating multiple myeloma.29 Bortezomib successfully causes the pathogenic ZIP13 mutant proteins to accumulate at nanomolar ranges. However, this drug also exhibits high cell toxicity over the long term.22 Although researchers have developed many reversible proteasome inhibitors with different points of action and target cells, cell toxicity is still a barrier to their clinical use. Because proteasome-dependent degradation is a major pathway for eukaryotic protein degradation, and proteasomes are involved in various cellular processes, including cell growth, gene regulation, stress response, and apoptosis, it is not surprising that proteasome inhibitors are highly toxic to cells.30
Proteasomes are essential for protein degradation, but they do not act alone. For the degradation of membrane proteins, various molecules are recruited. Dozens of chaperones that act as folding factors to promote protein degradation have been reported, and there are diverse types of chaperone coordination, which depend on the target protein and mutation. Chaperones are attractive therapeutic targets, because they are not always essential for cell life, and some may specifically act on a pathogenic protein. Thus, to develop a drug for SCD-EDS, it might be useful to target a chaperone that is specifically involved in processing the pathogenic ZIP13 mutant protein, analogous to VX-809 for CFTR. For this approach, we initially focused on VCP and HSP90.22 Their inhibitors, DBeQ and 17AAG, readily induced the accumulation of pathogenic ZIP13 protein, and restored intracellular Zn homeostasis.22 This result indicated that clinical approaches for SCD-EDS might be accomplished by screening for “correctors." In CF research, chaperone-targeting inhibitors with lower toxicity are being developed and screened for clinical use. Similarly, the modification of known inhibitors, including DBeQ and 17AAG, to reduce their cell toxicity, might also lead to effective therapies for SCD-EDS (Fig. 1A).
Conclusions and Future Perspectives
We are beginning to understand the pathogenesis of SCD-EDS and possible clinical approaches for this disease. Therapeutic approaches for SCD-EDS based on regulating the degradation of the pathogenic mutant ZIP13 protein are being considered. Future clinical directions include “corrector” and “potentiator” (Fig. 1B), gene therapy, and stem cell-based therapy. Continuing detailed studies of the molecular and physiological pathogenic mechanisms underlying SCD-EDS will be important for eventually controlling this disease.
Disclosure of Potential Conflict of Interest
No potential conflict of interest were disclosed.
Acknowledgments
We thank our colleagues involved in the project, along with Dr. Taiho Kambe for his critical reading.
Funding
This project was supported by grants from KAKENHI, the Japan Society for the Promotion of Science, the Life Science Foundation of Japan, the Japan Osteoporosis Foundation, the Targeted Proteins Research Program, the RIKEN Junior Research Associate Program, and the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science, and Technology.
References
- 1. Prasad AS. Discovery of human zinc deficiency and studies in an experimental human model. Am J Clin Nutr 1991; 53:403-12; PMID:1989405 [DOI] [PubMed] [Google Scholar]
- 2. Kitamura H, Morikawa H, Kamon H, Iguchi M, Hojyo S, Fukada T, Yamashita S, Kaisho T, Akira S, Murakami M, et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat Immunol 2006; 7:971-7; PMID:16892068; http://dx.doi.org/ 10.1038/ni1373 [DOI] [PubMed] [Google Scholar]
- 3. Fukada T, Hojyo S, Bin BH. Zinc signal in growth control and bone diseases. In Zinc Signals in Cellular Functions and Disorders, Fukada T, Kambe T. (eds) Tokyo: Springer, 249-267 [Google Scholar]
- 4. Hojyo S, Miyai T, Fujishiro H, Kawamura M, Yasuda T, Hijikata A, Bin BH, Irié T, Tanaka J, Atsumi T, et al. Zinc transporter SLC39A10/ZIP10 controls humoral immunity by modulating B-cell receptor signal strength. Proc Natl Acad Sci U S A 2014; 111:11786-91; PMID:25074919; http://dx.doi.org/ 10.1073/pnas.1323557111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyai T, Hojyo S, Ikawa T, Kawamura M, Irie T, Ogura H, Hijikata A, Bin BH, Yasuda T, Kitamura H, et al. Zinc transporter SLC39A10/ZIP10 facilitates antiapoptotic signaling during early B-cell development. Proc Natl Acad Sci U S Am 2014; 111:11780-5; PMID:25074913; http://dx.doi.org/ 10.1073/pnas.1323549111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kambe T, Yamaguchi-Iwai Y, Sasaki R, Nagao M. Overview of mammalian zinc transporters. Cell Mol Life sci 2004; 61:49-68; PMID:14704853; http://dx.doi.org/ 10.1007/s00018-003-3148-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor KM, Nicholson RI. The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochimic Biophys Acta 2003; 1611:16-30; PMID:12659941; http://dx.doi.org/ 10.1016/S0005-2736(03)00048-8 [DOI] [PubMed] [Google Scholar]
- 8. Lu M, Chai J, Fu D. Structural basis for autoregulation of the zinc transporter YiiP. Nat Struct Mol Biol 2009; 16:1063-7; PMID:19749753; http://dx.doi.org/ 10.1038/nsmb.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chowanadisai W, Lonnerdal B, Kelleher SL. Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J Biol Chem 2006; 281:39699-707; PMID:17065149; http://dx.doi.org/ 10.1074/jbc.M605821200 [DOI] [PubMed] [Google Scholar]
- 10. Itsumura N, Inamo Y, Okazaki F, Teranishi F, Narita H, Kambe T, Kodama H, et al. Compound heterozygous mutations in SLC30A2/ZnT2 results in low milk zinc concentrations: a novel mechanism for zinc deficiency in a breast-fed infant. PloS one 2013; 8:e64045; PMID:23741301; http://dx.doi.org/ 10.1371/journal.pone.0064045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neuroscience 2000; 20:RC79; PMID:10807937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicolson TJ, Bellomo EA, Wijesekara N, Loder MK, Baldwin JM, Gyulkhandanyan AV, Koshkin V, Tarasov AI, Carzaniga R, Kronenberger K, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009; 58:2070-83; PMID:19542200; http://dx.doi.org/ 10.2337/db09-0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukada T, Kambe T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics 2011; 3:662-74.; PMID:21566827; http://dx.doi.org/ 10.1039/c1mt00011j [DOI] [PubMed] [Google Scholar]
- 14. Kim JH, Jeon J, Shin M, Won Y, Lee M, Kwak JS, Lee G, Rhee J, Ryu JH, Chun CH, et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014; 156:730-43; PMID:24529376; http://dx.doi.org/ 10.1016/j.cell.2014.01.007 [DOI] [PubMed] [Google Scholar]
- 15. Andrews GK. Regulation and function of Zip4, the acrodermatitis enteropathica gene. Biochem Soc Trans 2008; 36:1242-6; PMID:19021533; http://dx.doi.org/ 10.1042/BST0361242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maverakis E, Fung MA, Lynch PJ, Draznin M, Michael DJ, Ruben B, Fazel N. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol 2007; 56:116-24; PMID:17190629; http://dx.doi.org/ 10.1016/j.jaad.2006.08.015 [DOI] [PubMed] [Google Scholar]
- 17. Kambe T, Andrews GK. Novel proteolytic processing of the ectodomain of the zinc transporter ZIP4 (SLC39A4) during zinc deficiency is inhibited by acrodermatitis enteropathica mutations. Mol Cell Biol 2009; 29:129-39; PMID:18936158; http://dx.doi.org/ 10.1128/MCB.00963-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Byers PH, Murray ML. Ehlers-Danlos syndrome: a showcase of conditions that lead to understanding matrix biology. Matrix Biol 2014; 33:10-5; PMID:23920413; http://dx.doi.org/ 10.1016/j.matbio.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 19. Fukada T, Civic N, Furuichi T, Shimoda S, Mishima K, Higashiyama H, Idaira Y, Asada Y, Kitamura H, Yamasaki S, et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PloS one 2008; 3:e3642; PMID:18985159; http://dx.doi.org/ 10.1371/journal.pone.0003642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giunta C, Elcioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome–an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet 2008; 82:1290-305; PMID:18513683; http://dx.doi.org/ 10.1016/j.ajhg.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bin BH, Fukada T, Hosaka T, Yamasaki S, Ohashi W, Hojyo S, Miyai T, Nishida K, Yokoyama S, Hirano T. Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J Biol Chem 2011; 286:40255-65; PMID:21917916; http://dx.doi.org/ 10.1074/jbc.M111.256784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bin BH, Hojyo S, Hosaka T, Bhin J, Kano H, Miyai T, Ikeda M, Kimura-Someya T, Shirouzu M, Cho EG, et al. Molecular pathogenesis of Spondylocheirodysplastic Ehlers-Danlos syndrome caused by mutant ZIP13 proteins. EMBO Mol Med 2014; 6:1028-42; PMID:25007800; http://dx.doi.org/ 10.15252/emmm.201303809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059-65; PMID:2772657; http://dx.doi.org/ 10.1126/science.2772657 [DOI] [PubMed] [Google Scholar]
- 24. Fanen P, Wohlhuter-Haddad A, Hinzpeter A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int J Biochem Cell biol 2014; 52:94-102; PMID:24631642; http://dx.doi.org/ 10.1016/j.biocel.2014.02.023 [DOI] [PubMed] [Google Scholar]
- 25. Skach WR. CFTR: new members join the fold. Cell 2006; 127:673-5; PMID:17110327; http://dx.doi.org/ 10.1016/j.cell.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 26. Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol 2013; 9:444-54; PMID:23666117; http://dx.doi.org/ 10.1038/nchembio.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108:18843-8; PMID:21976485; http://dx.doi.org/ 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eckford PD, Ramjeesingh M, Molinski S, Pasyk S, Dekkers JF, Li C, Ahmadi S, Ip W, Chung TE, Du K, et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem Biol 2014; 21:666-78; PMID:24726831; http://dx.doi.org/ 10.1016/j.chembiol.2014.02.021 [DOI] [PubMed] [Google Scholar]
- 29. Curran MP, McKeage K. Bortezomib: a review of its use in patients with multiple myeloma. Drugs 2009; 69:859-88; PMID:19441872; http://dx.doi.org/ 10.2165/00003495-200969070-00006 [DOI] [PubMed] [Google Scholar]
- 30. Konstantinova IM, Tsimokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol 2008; 267:59-124; PMID:18544497; http://dx.doi.org/ 10.1016/S1937-6448(08)00602-3 [DOI] [PubMed] [Google Scholar]