

Abstract
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), is a rare and understudied cancer with a dismal prognosis. SCCOHT's infrequency has hindered empirical study of its biology and clinical management. However, we and others have recently identified inactivating mutations in the SWI/SNF chromatin remodeling gene SMARCA4 with concomitant loss of SMARCA4 protein in the majority of SCCOHT tumors.1–4 Here we summarize these findings and report SMARCA4 status by targeted sequencing and/or immunohistochemistry (IHC) in an additional 12 SCCOHT tumors, 3 matched germlines, and the cell line SCCOHT-1. We also report the identification of a homozygous inactivating mutation in the gene SMARCB1 in one SCCOHT tumor with wild-type SMARCA4, suggesting that SMARCB1 inactivation may also play a role in the pathogenesis of SCCOHT. To date, SMARCA4 mutations and protein loss have been reported in the majority of 69 SCCOHT cases (including 2 cell lines). These data firmly establish SMARCA4 as a tumor suppressor whose loss promotes the development of SCCOHT, setting the stage for rapid advancement in the biological understanding, diagnosis, and treatment of this rare tumor type.
Keywords: BRG1, chromatin remodeling, small cell carcinoma of the ovary hypercalcemic type (SCCOHT), SMARCA4, SMARCB1, SNF5, ovarian cancer, SWI/SNF, tumorigenesis
Abbreviations
- SCCOHT
small cell carcinoma of the ovary, hypercalcemic type
- IHC
immunohistochemistry
- LOH
loss of heterozygosity
- SWI/SNF
SWItch/Sucrose NonFermentable
- AT/RT
atypical teratoid/rhabdoid tumor
- MRT
malignant rhabdoid tumor
- MRTK
malignant rhabdoid tumor of the kidney
SCCOHT: A Rare, Lethal, and Complex Cancer
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), is a rare and deadly ovarian cancer that predominantly affects young women. Fewer than 500 cases have been described in the literature since the disease was first characterized and it accounts for less than 1% of all ovarian cancer diagnoses.5-8 While the average age of diagnosis for most ovarian cancers is 63 years, SCCOHT primarily arises in young women at an average age of 24 years5,9 Histologically, SCCOHT is characterized by sheets of small rounded cells with hyperchromatic nuclei, single nucleoli, minimal cytoplasm and brisk mitotic activity. Roughly half of tumors contain variable numbers of larger cells with a luteinized or rhabdoid appearance. Though the presence of follicle-like spaces is diagnostically informative, other nonspecific morphologic and immunohistochemical features render the diagnosis challenging to establish.5,6,10 Indeed, until the discoveries outlined in this addendum, no specific immunohistochemical markers existed. Although SCCOHT is often diagnosed at an early stage, little evidence exists to support treatment selection and the prognosis is dismal with 2-year survival being less than 35%.5,6 SCCOHT's early age of onset and aggressive clinical course clearly establish a pressing need for innovations in management of this disease.
Inactivating SMARCA4 Mutations in SCCOHT
Prior to the discoveries described below, SCCOHT's molecular etiology was understood primarily according to its clinical pathology and expression profile. No mutations had been identified by targeted sequencing of candidate genes such as KRAS, BRAF, BRCA1, BRCA2, and TP53 and the genome was seen to be predominantly diploid by comparative genomic hybridization.11-13 Now, next-generation sequencing analyses from independent laboratories have reframed our biological understanding of SCCOHT by revealing that nearly all tumors harbor inactivating, often bi-allelic, mutations in the chromatin-remodeling tumor suppressor gene SMARCA4.1–4 We previously sequenced tumor or germline DNA from 12 SCCOHT cases and the patient-derived BIN-67 cell line, identifying inactivating mutations in 9 of these samples.1 We also found 15 of 18 cases with loss of SMARCA4 protein expression by immunohistochemistry. We now report the SMARCA4 status of an additional 12 SCCOHT tumors, 3 matched germlines, and the cell line SCCOHT-114, bringing the total number of cases analyzed in our hands to 24. This analysis was performed by PCR amplification of all coding exons of the SMARCA4 gene using DNA extracted from formalin-fixed paraffin-embedded (FFPE) blocks followed by Sanger sequencing in addition to immunohistochemistry against SMARCA4 and SMARCB1 as previously described.1 In total, we have now identified 19 of 24 sequenced tumors with SMARCA4 mutations and 16 of 19 stained tumors with loss of SMARCA4 protein (Table 1).
Table 1.
SMARCA4 mutations identified in DNA from SCCOHT patients and cell lines
| Sample ID | Publication | Age at diagnosis (years) | SMARCA4 mutations | IHC | ||
|---|---|---|---|---|---|---|
| Germline | Tumor | SMARCA4 | SMARCB1 | |||
| SCCO-001 | New case | 22 | N/A | p.Ala161Val | Negative | Positive |
| p.Ala532fs | ||||||
| SCCO-004 | New case | 32 | None | p.Val204fs | Negative | Positive |
| SCCO-005 | New case | 18 | None | p.Asp1299fs | N/A | N/A |
| SCCO-006 | New case | 32 | None | p.Trp764fs | Negative | Positive |
| p.Gly836* | ||||||
| SCCO-007 | New case | 25 | N/A | p.Gln331* | Negative | Positive |
| p.Ile542fs | ||||||
| SCCO-009 | New case | 27 | N/A | p.Tyr1050fs | Negative | Positive |
| SCCO-011 | New case | 30 | N/A | Homozygous p.Arg1189* | Negative | Positive |
| SCCO-016 | New case | 12 | N/A | Homozygous p.Arg1329fs | Negative | Positive |
| SCCO-018 | New case | 5 | N/A | None | Positive | Negative+ |
| SCCO-019 | New case | 27 | N/A | p.Phe844fs | Negative | Positive+ |
| SCCOHT-1 | New case | Tumor cell line | N/A | p.Pro1180fs p.Arg1077* | Negative+ | N/A |
| SCCO-002 | Ramos et al. | 26 | None | None | Negative | Positive |
| SCCO-008 | Ramos et al. | 9 | p.Arg979* | N/A | N/A | N/A |
| SCCO-010 | Ramos et al. | 6 | None | None2+ | Positive | Negative |
| SCCO-012 | Ramos et al. | 21 | N/A | None | Negative | Positive |
| SCCO-014 | Ramos et al. | 33 | N/A | p.Glu667fs | N/A | N/A |
| p.Leu1161fs | ||||||
| SCCO-015 | Ramos et al. | 27 | N/A | p.Arg1189* | N/A | N/A |
| SCCO-017 | Ramos et al. | 10 | p.Gly241fs | Homozygous p.Gly241fs2+ | Negative | Positive |
| DAH23 | Ramos et al. | 30 | N/A | c.2438+1_2438+2insTGA | Negative | N/A |
| DAH456 | Ramos et al. | 39 | None | None | Positive | Positive |
| DAH457 | Ramos et al. | 23 | N/A | p.Arg1093* | N/A | Positive |
| DG1006 | Ramos et al. | 34 | None | p.Glu952fs | Negative | N/A |
| p.Ser1591fs | ||||||
| DG1219 | Ramos et al. | 37 | None | c.3168+1>A | Negative | N/A |
| BIN-67 | Ramos et al. | Tumor cell line | N/A | c.2438+1G>A | Negative | Positive |
| c.2439–2A>T | ||||||
+This tumor was not previously stained for this marker
2+The tumor for this case was not previously sequenced
Across all published studies to date and including the new data reported here, nearly 100 mutations have been identified in SMARCA4 (Fig. 1) in 64 of 69 SCCOHT cases including 2 cell lines (Table 1 and Supplementary Table 1).1–4 With the exception of 3 missense mutations, all other SMARCA4 mutations identified in SCCOHT are truncating, frameshift, deletion, or splice-site mutations. Two of the 3 missense mutations were found in SMARCA4-negative tumors bearing a second inactivating SMARCA4 mutation, while in one case the tumor harbored the missense mutation p.Gly1080Asp and loss of heterozygosity (LOH) alongside SMARCA4 protein retention.3 Bi-allelic inactivation of SMARCA4 in SCCOHT is common either through the presence of 2 mutations or a single mutation and LOH at the SMARCA4 locus.3,4 In keeping with these findings, immunohistochemistry has revealed loss of SMARCA4 protein in 54 of 61 SCCOHT tumors and cell lines presumably due to nonsense-mediated decay as has been shown in several cases.1–4 However, a number of SMARCA4 negative cases carry heterozygous nonsense mutations and 2 cases have been shown to lack SMARCA4 protein with no identified sequence, copy number, or methylation alterations in the SMARCA4 gene (Table 1 and Supplementary Table 1).1-4 Mechanisms leading to gene inactivation in SMARCA4-negative tumors with heterozygous or unidentified gene alterations remain to be elucidated.
Figure 1.
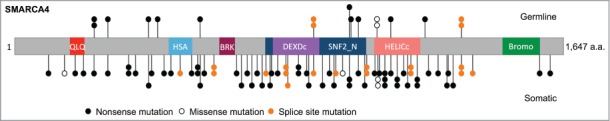
Schematic of SMARCA4 mutations in SCCOHT. SMARCA4 mutations identified in germline and tumor DNA from 62 SCCOHT patients, and in 2 SCCOHT cell lines (Case 103 from Jelinic et al. with exon deletion is not shown).1–4 QLQ, Gln, Leu, Gln motif; HSA, helicase/SANT-associated domain; BRK, brahma and kismet domain; DEXDc, DEAD-like helicase superfamily domain; HELICc, helicase superfamily C-terminal domain; Bromo, bromodomain.
Conversely, all but 4 SMARCA4-mutant SCCOHTs for which IHC has been performed also lack expression of SMARCA4 protein.1-4 These 4 SMARCA4-mutant, positive-staining tumors harbored either splice site or missense mutations or, in one case, an in-frame homozygous deletion of exons 25 and 26 that resulted in expression of an inactive protein product.3, 4 In our cohort, we also found 3 tumors that had no SMARCA4 mutations and showed retention of protein expression by IHC. Two of these cases, both associated with paraneoplastic hypercalcemia, lacked the protein SMARCB1, a SWI/SNF-associated tumor suppressor gene known to be frequently mutated in rhabdoid tumors (Supplementary Fig. 1). Targeted Sanger sequencing of the coding exons of SMARCB1 in these tumors revealed a novel homozygous frameshift mutation, p.Asn34fs, resulting from the deletion of 14 base pairs in exon 2 of SMARCB1 in SCCO-010, a large cell variant SCCOHT. This finding supports the hypothesis that SCCOHT may share an etiological link with rhabdoid tumors and that SMARCB1 inactivation can also promote the development of SCCOHT.1,15 The third case retained both SMARCA4 and SMARCB1 protein expression and may bear an as-yet unidentified SCCOHT driver gene mutation or may simply be a misdiagnosis.1 Overall, SMARCA4 is clearly a tumor suppressor inactivated by 2 hits in the majority of SCCOHTs, but several of the above exceptional cases provide clues to a more complex disease etiology.
Further supporting the prominence of SMARCA4's tumor suppressor role in SCCOHT, germline mutations have been identified in 17 SCCOHT cases, predominantly in younger patients.1,3,4 Such mutations have been found to segregate in 4 families in which all affected members whose tumors could be tested developed either a second inactivating mutation or LOH in the remaining wild-type allele.3 Alongside previous clinical descriptions of SCCOHT families, these mutations elucidate a heritable component to the disease and suggest that the broad age distribution of SCCOHT could reflect inherited versus acquired SMARCA4 mutations.11,16–21 SMARCA4 mutation also occurs in the absence of recurrent secondary genomic alterations and amidst relative karyotypic stability and, therefore, appears to be the primary driving event in SCCOHT tumorigenesis. The total number of somatic non-silent mutations detected by paired exome or whole-genome sequencing analysis in SCCOHT tumors and matched normal DNAs ranges from 2 to 12, reflecting a low mutation rate, similar to other pediatric tumors and tumors of non-self-renewing tissues.1,3,4,22 Among paired tumor and normal samples evaluated by exome, whole-genome, or panel-based sequencing, few secondary mutations in cancer genes were discovered and each such mutation (those in ASXL1, JAK3, NOTCH2, and WT1) occurred in only a single case.1,3,4 Overall, the low SCCOHT mutation rate, the nearly universal presence of inactivating SMARCA4 mutations in SCCOHT, the presence of these mutations in patient germlines and families, and the lack of recurrent secondary alterations in these tumors strongly suggest that loss of SMARCA4 is sufficient for SCCOHT initiation.
Biological, Diagnostic, and Therapeutic Implications of SMARCA4 Loss in SCCOHT
SMARCA4 is one of 2 mutually-exclusive ATPases of the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin-remodeling complex which was originally discovered to modulate mating type-switching and sucrose fermentation in yeast.23–25 This complex uses the energy of ATP hydrolysis to destabilize histone-DNA interactions and move, eject, or restructure nucleosomes, thereby regulating access to DNA of transcription, replication, and repair machinery.23,26 SWI/SNF subunits such as SMARCA4 have also been shown to interact with tumor suppressors such as p15INK4b, p16INK4a, p21CIP/WAF1, and hypophosphorylated RB to modulate cell cycle progression.27,28 Broadly, mutations in epigenetic regulators such as SWI/SNF members are enriched in many cancer types, particularly pediatric cancers in which as many as 30% of brain tumors and leukemias and 17% of solid tumors bear such mutations.29 This enrichment may be due to the pleiotropic effects these mutations exert on gene expression and cell differentiation programs. Many SWI/SNF subunits such as SMARCA4 have also been shown to interact with, or regulate, tumor suppressors with approximately 20% of cancers bearing mutations in these genes.30,31 SMARCA4 is one of the most commonly mutated subunits across cancer types, occurring at a frequency of about 4% in all cancers and arising regularly in non-small cell lung cancer, Burkitt's lymphoma, and medulloblastoma while also occurring occasionally in melanoma, pancreatic adenocarcinoma, ovarian clear cell carcinoma, and other tumor types.31 Loss of SMARCA4 is thought to lead to dependence on SMARCA2-bearing SWI/SNF complexes that induce gene expression changes driving oncogenic pro-survival and/or anti-apoptotic signaling.32,33 Elucidation of the impact of such mutations on SWI/SNF composition and downstream effects on expression programs and pathway regulation will shape future study of SCCOHT tumorigenesis and therapeutic vulnerability.
Given SCCOHT's complex histological appearance and the absence of known precursor lesions, the cellular origin of SCCOHT and its relationship to other tumor types remains unclear. SCCOHTs are characterized by poorly differentiated small tumor cells with scant cytoplasm and hyperchromatic nuclei, and the presence of follicle-like structures contained within sheets of cells.5 Despite SCCOHT's name, about half of tumors have populations of large cells with rhabdoid features.5 Indeed, there are many similarities between SCCOHT and atypical teratoid/rhabdoid tumors of the brain (AT/RTs) and malignant rhabdoid tumors (MRTs) of the kidney (MRTK). All 3 tumor types are linked to mutations in the SWI/SNF genes SMARCB1 (AT/RT, MRT, and now SCCOHT) or SMARCA4 (SCCOHT and AT/RT), all have diploid genomes and all occur in young or pediatric patients.5,34,35 Shared morphology and mutational spectra make a compelling case that SCCOHT may be a type of MRT.15 The strikingly similar morphology and genetics of rhabdoid tumors in 3 very different organs suggests either a common cell of origin or convergent morphologic evolution upon SMARCA4 or SMARCB1 loss (or both) although no MRT cell of origin has yet been identified.2,3,15,16 On the other hand, there is some histological evidence for a germ cell etiology for SCCOHT. In particular, a recent report identified immature teratoma in 2 SCCOHTs, one of which also contained foci of yolk-sac tumor.2 This finding agrees with Ulbright et al. who, in one of the earliest publications on SCCOHT in 1987,16 also suggested that SCCOHTs might be related to yolk-sac tumors based on presence of shared histopathological and ultrastructural features. Unfortunately, no clear origin has been indicated by mouse models of SMARCA4 mutation. The homozygous Smarca4 null genotype is embryonic lethal and, while 10% of heterozygotes spontaneously develop mammary tumors at 1 year, these tumors are molecularly heterogeneous, genomically unstable, and lack LOH at the Smarca4 locus and therefore do not contain a genomic landscape resembling that of SCCOHTs.36,37 Engineered SMARCA4 knockouts in putative precursor cells in vitro and in vivo are needed to shed light on SCCOHT histogenesis.
Among ovarian tumors, the loss of SMARCA4 protein appears to be highly specific for SCCOHT. Our assessment of 485 primary ovarian epithelial, sex cord-stromal, and germ cell tumors showed only 2 tumors (0.4%), both clear cell carcinomas, with negative SMARCA4 staining.1 Other ovarian tumors in the differential diagnosis of SCCOHT – undifferentiated carcinomas, adult and juvenile granulosa cell tumor, and germ cell tumors – all expressed SMARCA4 protein or were wild-type for the SMARCA4 gene.1,38 The expression status of SMARCA4 remains to be determined in several other primary and metastatic ovarian tumors in the differential diagnosis of SCCOHT including endometrioid stromal sarcoma, desmoplastic small round cell tumor, primitive neuroectodermal tumor, neuroblastoma, and others. However to date, the absence of SMARCA4 protein is highly sensitive and specific for SCCOHT and can be used to distinguish it from other ovarian tumors with similar histology to facilitate diagnosis.
SCCOHTs are extremely aggressive and refractory to treatment that most commonly includes surgical debulking followed by high-dose chemotherapy and/or radiation.5,9,20 Some evidence suggests that chemotherapeutic combinations including cisplatin or carboplatin, etoposide and vinca alkaloids may be associated with improved survival, yet patient outcomes are abysmal in most cases with a 65% recurrence rate and 2-year survival less than 35%.5,9,39 SCCOHT rarity limits the implementation of prospective clinical trials to guide effective treatment and its infrequency has also limited the study of its pathogenesis to uncover potential therapeutic vulnerabilities. Our finding that the majority of SCCOHTs contain SMARCA4 mutations amidst otherwise simple genomic backgrounds provides an opportunity to empirically develop effective treatment strategies with a high probability of impact for many of these patients. Given that this disease derives in virtually all cases from the loss of a tumor suppressor, the path to an effective small molecule may hinge on identification of a synthetic lethal target. To this end, a synthetic lethal dependence of SMARCA4-deficient cancers cells on SMARCA2 has recently been described in non-small cell lung cancer, ovarian and liver cancer cell lines.32,33 This dependence is likely due to SMARCA2's status as the only known alternative ATPase subunit of the SWI/SNF complex. However, preliminary SMARCA2 staining in 2 SCCOHT cell lines showed lack of protein in both cases (data not shown), suggesting that SCCOHT may lack the expression of both SMARCA2 and SMARCA4 and that investigation of other synthetic lethal partners is therefore warranted. Although it has been shown in other cancers such as non-small cell lung adenocarcinoma cell lines that the SWI/SNF core complex still forms in the absence of both SMARCA4 and SMARCA2,40 it remains to be determined whether this complex retains chromatin remodeling activity and whether targeting the residual complex can selectively kill SCCOHT cells. Of further importance will be identification of the mechanism inactivating the second SMARCA4 allele in cases in which only monoallelic mutations have been identified. Epigenetic lesions may present compelling targets for re-expression of SMARCA4 and/or SMARCA2. Ultimately, future progress in SCCOHT treatment will depend on expansion of the currently limited number of in vitro and in vivo model systems. The BIN-67 and SCCOHT-1 cell lines are the only such models to have yet been described and they have been implemented in few studies in vivo.13,14
The breakthrough identification of inactivating SMARCA4 mutations in almost all cases of SCCOHT is the first significant insight into the pathogenesis of the disease and offers the opportunity for genetic testing of family members at risk. The loss of the SMARCA4 protein is a highly sensitive and specific marker of the disease, highlighting its potential role as a diagnostic marker. Studies are currently in progress at our institutions to elucidate the cell of origin in hopes of better understanding the pathogenesis of this disease and to identify therapeutic vulnerabilities guiding clinical trials to further advance treatment options for patients with SCCOHT.
Materials and Methods
Samples
At TGen, all patients and their relatives signed consent forms according to IRB-approved and Health Insurance Portability and Accountability Act–compliant protocols. At the University of British Columbia, biospecimens were obtained from the Ovarian Cancer Research Program (OvCaRe) tissue bank in Vancouver, British Columbia, Canada; the University of Toronto in Toronto, Ontario, Canada; the Children's Oncology Group at Nationwide Children's Hospital in Columbus, Ohio, USA; and the Hospital de la Santa Creu i Sant Pau at the Autonomous University of Barcelona in Barcelona, Spain, using an IRB-approved protocol. All of the specimens were SCCOHT, with 4 cases (SCCO-009, SCCO-010, SCCO-017 and SCCO-019) classified as large cell variants of SCCOHT in their pathology reports. Cases of small cell carcinoma of pulmonary type were excluded from the study.
DNA extraction
FFPE DNA was extracted using Qiagen's All Prep DNA/RNA FFPE kit (Qiagen; Valencia, CA). Blood leukocytes (buffy coat) were isolated from whole blood by centrifugation at room temperature and resuspended in Buffer RLT plus. Samples were then processed for DNA isolation using the AllPrep kit (Qiagen). DNA was quantified using the Nanodrop spectrophotometer (Nanodrop; Wilmington, DE) on the basis of 260 nm/280 nm and 260 nm/230 nm absorbance ratios.
PCR amplification and Sanger sequencing analysis
PCR amplification of SMARCA4 was performed using previously published primers3 targeting 34 coding exons (the alternative exon 29 was not sequenced). Amplification of all SMARCB1 coding exons was performed using the following primers, some of which have been previously published41:
| Primer Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| Exon 1 | CTTCCGGCTTCGGTTTCCCT | GATGAATGGAGACGCGCGCT |
| Exon 2 | GTTGCTTGATGCAGTCTGCG | TTCATGACATAAGCGAGTGG |
| Exon 3 | GATGTGCTGCATCCACTTGG | TTCAGAAAAGACCCCACAGG |
| Exon 4 | TTAGTTGATTCCTGGTGGGC | GAACTAAGGCGGAATCAGCA |
| Exon 5 | TGTGCAGAGAGAGAGGCTGA | CACGTAACACACAGGGGTTG |
| Exon 6 | CAATCTCTTGGCATCCCTTC | CAGTGCTCCATGATGACACC |
| Exon 7 | TGGGCTGCAAAAGCTCTAAC | AGTTTTGCAGGGAGATGAGG |
| Exon 8 | GGCCAAAGCTTTCTGAGGAT | CATGGGAGACTGGGAAAGGC |
| Exon 9 | CCCTGTAGAGCCTTGGGAAG | GTCCTTGCCAGAAGATGGAG |
Universal M13 tails were added to all primers. Each primer pair was mixed with 10 ng of genomic DNA and subjected to the following cycling parameters: 94°C for 2 min., 3 cycles at each temperature: 30 sec. at 94°C, 30 sec. at 60–57°C, 45 sec. at 72°C; 25 cycles: 30 sec. at 94°C, 30 sec. at 62°C, 45 sec. at 72°C; final extension of 5 min. at 72°C. All amplification reactions were performed using Platinum Taq DNA Polymerase #10966–034 (Life Technologies; Carlsbad, CA). PCR amplicons were sequenced using M13 forward and reverse primers at the Arizona State University's DNA Laboratory (Tempe, AZ).
Immunohistochemistry
Whole slide sections were prepared from paraffin blocks of formalin-fixed SCCOHT tumor cases and SCCOHT-1 cells. Unstained slides were processed using the Ventana Discovery Ultra system (Ventana Medical Systems), using a rabbit monoclonal antibody to SMARCA4 (BRG1; Abcam, ab110641; 1:25 dilution) and mouse monoclonal antibody to SMARCB1 (INI1; BD Transduction Laboratories, 612110; 1:50 dilution). The antibody to SMARCB1 was used to confirm the antigenic reactivity of the tumor cells and cell lines that were negative for SMARCA4 expression. Tumors were scored positive if any tumor cell nuclei showed moderate to strong (definite) positive nuclear staining. Tumors were scored negative when tumor cells showed no nuclear staining only if there was adequate nuclear staining of an internal positive control (endothelial cells, fibroblasts or lymphocytes). No cytoplasmic staining was seen for SMARCA4.
Disclosure of Potential Conflicts of Interest
No conflicts of interest were disclosed.
Acknowledgments
We particularly thank the families and patients who joined this IRB-approved study for their critical contributions. We further thank Scottsdale Healthcare for institutional support and clinical leadership in addition to that provided by many other physicians: Drs. Jaime Prat, Emanuela D’Angelo, Blaise Clark, Joseph Pressey, and Richard Roden. We also thank the faculty and staff at TGen of the Macromolecular Analysis & Processing Center (Drs. Hostetter and LoBello), and the Office of Research Compliance & Quality Management (Lora Nordstrom, Stephanie Althoff, and Stephanie Buchholtz); Children's Oncology Group for sample collection and Drs. Ralf Haas and Barbara Vanderhyden for establishing and sharing the SCCOHT-1 and BIN-67 cells, respectively.
Funding
This study was supported by grants from The Marsha Rivkin Center for Ovarian Cancer Research, The Anne Rita Monahan Foundation, The Ovarian Cancer Alliance of Arizona, The Small Cell Ovarian Cancer Foundation, and Foster and Lynn Friess. Further support was provided to Yemin Wang by the Michael Smith Foundation for Health Research and to Anthony N. Karnezis and David G. Huntsman by the Terry Fox Research Initiative New Frontiers Program in Cancer.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K, Yang Y, et al. . Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 2014; 46:427-9; PMID:24658001; http://dx.doi.org/ 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, Plisiecka-Halasa J, Szafron L, Podgorska A, Rzepecka IK, Konopka B, Budzilowska A, Rembiszewska A, et al. . Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and smarca4 gene inactivation. a pilot study. Pol J Pathol: Off J Pol Soc Pathol 2013; 64:238-46; http://dx.doi.org/ 10.5114/pjp.2013.39331. [DOI] [PubMed] [Google Scholar]
- 3. Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J, Rivera B. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014; PMID:24658002 [DOI] [PubMed] [Google Scholar]
- 4. Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014; PMID:24658004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type: a clinicopathological analysis of 150 cases. Am J Surg Pathol 1994; 18:1102-16; PMID:7943531; http://dx.doi.org/ 10.1097/00000478-199411000-00004 [DOI] [PubMed] [Google Scholar]
- 6. Clement PB. Selected miscellaneous ovarian lesions: small cell carcinomas, mesothelial lesions, mesenchymal and mixed neoplasms, and non-neoplastic lesions. Mod Pathol: Off J U S Can Acad Pathol Inc 2005; 18(Suppl 2):S113-29; http://dx.doi.org/ 10.1038/modpathol.3800313 [DOI] [PubMed] [Google Scholar]
- 7. Scully RE. Tumors of the ovary and maldeveloped gonads. Atlas of tumor pathology 2nd Series, Fascicle 16 Washington, DC: Armed Forces Institute of Pathology; 1979. [Google Scholar]
- 8. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: Cancer J Clin 2013; 63:11-30; PMID:23335087 [DOI] [PubMed] [Google Scholar]
- 9. Estel R, Hackethal A, Kalder M, Munstedt K. Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch Gynecol Obstet 2011; 284:1277-82; PMID:21298438; http://dx.doi.org/ 10.1007/s00404-011-1846-5. [DOI] [PubMed] [Google Scholar]
- 10. McCluggage WG, Oliva E, Connolly LE, McBride HA, Young RH. An immunohistochemical analysis of ovarian small cell carcinoma of hypercalcemic type. Int J Gynecol Pathol: Off J Int Soc Gynecol Pathol 2004; 23:330-6; http://dx.doi.org/ 10.1097/01.pgp.0000139644.38835.9d [DOI] [PubMed] [Google Scholar]
- 11. Martinez-Borges AR, Petty JK, Hurt G, Stribling JT, Press JZ, Castellino SM. Familial small cell carcinoma of the ovary. Pediatr Blood Cancer 2009; 53:1334-6; PMID:19621450; http://dx.doi.org/ 10.1002/pbc.22184 [DOI] [PubMed] [Google Scholar]
- 12. Stephens B, Anthony SP, Han H, Kiefer J, Hostetter G, Barrett M, Von Hoff DD. Molecular Characterization of a Patient's Small Cell Carcinoma of the Ovary of the Hypercalcemic Type. J Cancer 2012; 3:58; PMID:22315651; http://dx.doi.org/ 10.7150/jca.3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gamwell LF, Gambaro K, Merziotis M, Crane C, Arcand SL, Bourada V, Davis C, Squire JA, Huntsman DG, Tonin PN. Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics. Orphanet J Rare Dis 2013; 8:33; PMID:23433318; http://dx.doi.org/ 10.1186/1750-1172-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otte A, Gohring G, Steinemann D, Schlegelberger B, Groos S, Langer F, Kreipe HH, Schambach A, Neumann T, Hillemanns P, et al. . A tumor-derived population (SCCOHT-1) as cellular model for a small cell ovarian carcinoma of the hypercalcemic type. Int J Oncol 2012; 41:765-75; PMID:22581215 [DOI] [PubMed] [Google Scholar]
- 15. Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol 2014; 233:209-14; PMID:24752781; http://dx.doi.org/ 10.1002/path.4362 [DOI] [PubMed] [Google Scholar]
- 16. Ulbright TM, Roth LM, Stehman FB, Talerman A, Senekjian EK. Poorly differentiated (small cell) carcinoma of the ovary in young women: evidence supporting a germ cell origin. Hum Pathol 1987; 18:175-84; PMID:3026945; http://dx.doi.org/ 10.1016/S0046-8177(87)80336-2 [DOI] [PubMed] [Google Scholar]
- 17. Peccatori F, Bonazzi C, Lucchini V, Bratina G, Mangioni C. Primary ovarian small cell carcinoma: four more cases. Gynecol Oncol 1993; 49:95-9; PMID:8387062; http://dx.doi.org/ 10.1006/gyno.1993.1093 [DOI] [PubMed] [Google Scholar]
- 18. Lamovec J, Bracko M, Cerar O. Familial occurrence of small-cell carcinoma of the ovary. Arch Pathol Lab Med 1995; 119:523-7; PMID:7605168 [PubMed] [Google Scholar]
- 19. Longy M, Toulouse C, Mage P, Chauvergne J, Trojani M. Familial cluster of ovarian small cell carcinoma: a new mendelian entity? J Med Genet 1996; 33:333-5; PMID:8730291; http://dx.doi.org/ 10.1136/jmg.33.4.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Distelmaier F, Calaminus G, Harms D, Strater R, Kordes U, Fleischhack G, Gobel U, Schneider DT. Ovarian small cell carcinoma of the hypercalcemic type in children and adolescents: a prognostically unfavorable but curable disease. Cancer 2006; 107:2298-306; PMID:16998935; http://dx.doi.org/ 10.1002/cncr.22213 [DOI] [PubMed] [Google Scholar]
- 21. McDonald JM, Karabakhtsian RG, Pierce HH, Iocono JA, Desimone CP, Bayliff SL, Ueland FR. Small cell carcinoma of the ovary of hypercalcemic type: a case report. J Pediatr Surg 2012; 47:588-92; PMID:22424359; http://dx.doi.org/ 10.1016/j.jpedsurg.2011.12.004 [DOI] [PubMed] [Google Scholar]
- 22. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/ 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 2011; 21:396-420; PMID:21358755; http://dx.doi.org/ 10.1038/cr.2011.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genet 1984; 108:845-58; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stern M, Jensen R, Herskowitz I. Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol 1984; 178:853-68; PMID:6436497; http://dx.doi.org/ 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- 26. Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer 2004; 4:133-42; PMID:14964309; http://dx.doi.org/ 10.1038/nrc1273 [DOI] [PubMed] [Google Scholar]
- 27. Hendricks KB, Shanahan F, Lees E. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol 2004; 24:362-76; PMID:14673169; http://dx.doi.org/ 10.1128/MCB.24.1.362-376.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, Knudsen ES. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci 2000; 97:7748-53; PMID:10884406; http://dx.doi.org/ 10.1073/pnas.97.14.7748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun 2014; 5; PMID:24710217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013; 45:592-601; PMID:23644491; http://dx.doi.org/ 10.1038/ng.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS one 2013; 8:e55119; PMID:23355908; http://dx.doi.org/ 10.1371/journal.pone.0055119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oike T, Ogiwara H, Tominaga Y, Ito K, Ando O, Tsuta K, Mizukami T, Shimada Y, Isomura H, Komachi M, et al. . A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res 2013; 73:5508-18; PMID:23872584; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-4593 [DOI] [PubMed] [Google Scholar]
- 33. Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, Hahn WC, Roberts CW. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McKenna ES, Sansam CG, Cho YJ, Greulich H, Evans JA, Thom CS, Moreau LA, Biegel JA, Pomeroy SL, Roberts CW. Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol 2008; 28:6223-33; PMID:18710953; http://dx.doi.org/ 10.1128/MCB.00658-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, Junckerstorff R, Rosenblum M, Alassiri AH, Rossi S. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 2014; 128:453-6; PMID:25060813; http://dx.doi.org/ 10.1007/s00401-014-1323-x [DOI] [PubMed] [Google Scholar]
- 36. Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G. A Brg1 Null Mutation in the Mouse Reveals Functional Differences among Mammalian SWI/SNF Complexes. Mol Cell 2000; 6:1287-95; PMID:11163203; http://dx.doi.org/ 10.1016/S1097-2765(00)00127-1 [DOI] [PubMed] [Google Scholar]
- 37. Bultman S, Herschkowitz J, Godfrey V, Gebuhr T, Yaniv M, Perou C, Magnuson T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 2007; 27:460-8; PMID:17637742; http://dx.doi.org/ 10.1038/sj.onc.1210664 [DOI] [PubMed] [Google Scholar]
- 38. Witkowski L, Lalonde E, Zhang J, Albrecht S, Hamel N, Cavallone L, May ST, Nicholson JC, Coleman N, Murray MJ, et al. . Familial rhabdoid tumour 'avant la lettre'–from pathology review to exome sequencing and back again. J Pathol 2013; 231:35-43; PMID:23775540; http://dx.doi.org/ 10.1002/path.4225 [DOI] [PubMed] [Google Scholar]
- 39. Clement PB. Selected miscellaneous ovarian lesions: small cell carcinomas, mesothelial lesions, mesenchymal and mixed neoplasms, and non-neoplastic lesions. Mod Pathol 2005; 18:S113-S29; PMID:15492757; http://dx.doi.org/ 10.1038/modpathol.3800313 [DOI] [PubMed] [Google Scholar]
- 40. Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, et al. . Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A 2014; 111:3128-33; PMID:24520176; http://dx.doi.org/ 10.1073/pnas.1316793111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fruhwald MC, Hasselblatt M, Wirth S, Kohler G, Schneppenheim R, Subero JI, Siebert R, Kordes U, Jurgens H, Vormoor J. Non-linkage of familial rhabdoid tumors to SMARCB1 implies a second locus for the rhabdoid tumor predisposition syndrome. Pediatr Blood Cancer 2006; 47:273-8; PMID:16206192; http://dx.doi.org/ 10.1002/pbc.20526 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.