

Abstract
Background
Fight or flight heart rate (HR) increases depend on protein kinase A (PKA) and calmodulin kinase II (CaMKII) mediated enhancement of Ca2+ uptake and release from sarcoplasmic reticulum (SR) in sinoatrial nodal cells (SANC). However, the impact of specific PKA and CaMKII phosphorylation sites on HR is unknown.
Methods and Results
We systematically evaluated validated PKA and CaMKII target sites on phospholamban (PLN) and the ryanodine receptor (RyR2) using genetically modified mice. We found that knockin alanine replacement of RyR2 PKA (S2808) or CaMKII (S2814) target sites failed to affect HR responses to isoproterenol or spontaneous activity in vivo or in SANC. Similarly, selective mutation of PLN amino acids critical for enhancing SR Ca2+ uptake by PKA (S16) or CaMKII (T17) to alanines did not affect HR in vivo or in SANC. In contrast, CaMKII inhibition by expression of AC3-I has been shown to slow SANC rate responses to isoproterenol and decrease SR Ca2+ content. PLN deficiency rescued SR Ca2+ content and SANC rate responses to isoproterenol in mice with AC3-I expression, suggesting CaMKII affects HR by modulation of SR Ca2+ content. Consistent with this, mice expressing a superinhibitory PLN mutant had low SR Ca2+ content and slow HR in vivo and in SANC.
Conclusions
SR Ca2+ depletion reduces HR and SR Ca2+ repletion restores physiological SANC rate responses despite CaMKII inhibition. PKA and CaMKII do not affect HR by a unique target site governing SR Ca2+ uptake or release. HR acceleration may require an SR Ca2+ content threshold.
Keywords: sinoatrial node, heart rate, calcium/calmodulin-dependent protein kinase II, phospholamban, ryanodine receptor, protein kinase A
Introduction
Cardiac pacemaker cells control heart rate by generating spontaneous and repetitive action potentials. Action potential acceleration drives heart rate increases by a mechanism that relies on an intracellular Ca2+ release and uptake process that is coupled to inward cell membrane ionic currents. These cellular mechanisms are often described separately as a Ca2+ clock and a cell membrane clock 1. Cardiac pacemaker cells can accelerate heart rates in response to β adrenergic receptor agonist stimulation as part of the physiological fight-or-flight response. β adrenergic receptor agonists increase the activity of protein kinase A (PKA) and calmodulin kinase II (CaMKII), serine-threonine kinases, in cardiac pacemaker cells. Both CaMKII 2 and PKA 3 contribute to fight or flight heart rate increases in response to isoproterenol injection or spontaneous ambulatory activity by catalyzing phosphorylation of proteins important for regulating intracellular Ca2+ uptake and release from the sarcoplasmic reticulum (SR). However, to our knowledge, a systematic study of the effects of PKA and CaMKII phosphorylation on individual proteins that govern SR Ca2+ uptake and release in sinoatrial node (SAN) pacemaker cells is lacking.
SR Ca2+ release in SAN cells occurs through type 2 ryanodine receptors (RyR2). RyR2 Ca2+ release is enhanced by phosphorylation of S2808 (by CaMKII and PKA) 4, 5 and by S2814 (by CaMKII) 6. The increased RyR2 Ca2+ leak drives SAN cell membrane depolarizing inward current through the Na+/Ca2+ exchanger. The Na+/Ca2+ exchanger current (INCX) increases heart rate by hastening the late diastolic SAN cell membrane depolarization rate, causing higher frequency action potential triggering in SAN pacemaking cells 7. We measured heart rates and SAN automaticity in mice with RyR2 knockin mutations that disable PKA (S2808A) and CaMKII (S2814A) catalyzed phosphorylation at these sites.
SR Ca2+ uptake in SAN cells is governed by a protein complex that includes phospholamban (PLN) and the sarcoplasmic endoplasmic reticulum ATPase (SERCA2a) 1-3. PLN is a negative regulator of SERCA2a under basal conditions 8. PLN phosphorylation by PKA (at S16) or CaMKII (at T17) leads to loss of SERCA2a inhibition and increased Ca2+ transport from the cytoplasm to the SR. We studied mice and isolated SAN cells lacking PLN (Pln−/−), expressing mutant PKA-resistant PLN (S16A), CaMKII-resistant PLN (T17A) and mice with a superinhibitory PLN mutant, N27A, which constitutively suppresses SERCA2a even after PKA and CaMKII catalyzed phosphorylation 9-11.
Despite the important roles of PKA and CaMKII in activating the Ca2+ clock and promoting fight or flight heart rate increases, the relative importance of PKA and CaMKII sites on RyR2 and PLN are unknown; furthermore, the potential for any particular site to exert a controlling influence over fight or flight physiology in vivo and in vitro is untested. In order to address this knowledge gap, we used a cohort of genetically modified mouse models in which CaMKII and PKA phosphorylation sites were specifically ablated to interrogate the role of each site and determine if any of these SR protein sites exercises a decisive influence on heart rate responses to isoproterenol or activity.
Here we show that none of four well validated PKA or CaMKII phosphorylation sites on PLN and RyR2 are individually required for physiological rate increases in isolated SAN cells or in vivo in response to spontaneous activity or isoproterenol injection. Although no single phosphorylation site in our models had a controlling role in heart rate acceleration, we found that superinhibitory PLN mutants significantly lowered SR Ca2+ content and limited heart rate acceleration while PLN deficiency prevented reduction in SR Ca2+ content and normalized heart rate acceleration during CaMKII inhibition. Taken together, our data show that loss of any single site failed to affect heart rates and suggest that fight-or-flight heart rate increases are built from redundant PKA and CaMKII catalyzed phosphorylation target proteins. Our data are consistent with a concept where CaMKII inhibition slows heart rate by lowering the pool of SR Ca2+ in cardiac pacemaker cells.
Methods
Genetically modified mice
The cohort of mice used in this study is shown in Table 1. Pln−/−, S16A, T17A, N27A, and WT-PLN mice were obtained from MMRRC (Columbia, MO), but were originally developed by the Kranias group 9-11. All of these mice were engineered by transgenic expression of wild type (WT) and mutant PLN on a Pln−/− background. The lines were selected for similar transgenic expression levels of S16A, T17A, N27A and WT-PLN 10, 11. The littermate controls S16A(−), T17A(−) are Pln−/−. WT mice in the C57 background are designated (WT), and WT-Pln indicates controls where a WT Pln transgene was expressed in a Pln−/− background. The S2808A and S2814A mice were developed by knockin replacement of the designated alleles, as described 12, 13. Pln−/− mice were interbred with mice overexpressing CaMKII inhibitor peptide AC3-I 2 (AC3-IxPln−/−) and mice overexpressing scrambled control peptide AC3-C (AC3-CxPln−/−) for > 10 generations and the genetic identity of Pln−/− pups with transgenic expression of AC3-I or AC3-C was confirmed using PCR, as described 14.
Table 1.
Genetically modified mice
| MICE | MUTATIONS | NOTES | CONTROL | SOURCE |
|---|---|---|---|---|
| Pln −/− | Pln −/− | Background for all PLN mutant mice | WT*, WT-Pln | MMRRC |
| S16A | PLN S16A | TG replacement of S16A in the Pln−/− background | S16A(−), WT-Pln | MMRRC |
| T17A | PLN T17A | TG replacement of T17A in the Pln−/− background | T17A(−), WT-Pln | MMRRC |
| S16A(−) | Pln −/− | Littermates of S16A lacking TG S16A | MMRRC | |
| T17A(−) | Pln −/− | Littermates of T17A lacking TG T17A | MMRRC | |
| WT-Pln | WT-PLN | TG replacement of WT PLN in the Pln−/− background | MMRRC | |
| N27A | PLN N27A | TG replacement of N27A PLN in the Pln−/− background | WT*, N27A(−), WT-Pln | MMRRC |
| N27A(−) | Pln −/− | WT-Pln | MMRRC | |
| S2808A | S2808A | S2808A(−) | HV lab | |
| S2814A | S2814A | WT*, S2814A(−) | XW lab | |
| S2808A(−) | Littermates of S2808A with WT RyR2 | HV lab | ||
| S2814A(−) | Littermates of S2814A with WT RyR2 | XW lab | ||
| AC3-I X PLN−/− | AC3-I & PLN−/− | AC3-I(−) & PLN−/− | MEA lab | |
| AC3-C X PLN−/− | AC3-C & PLN−/− | AC3-C(−) & PLN−/− | MEA lab | |
| AC3-I(−) | littermate control lacking tg AC3-I | MEA lab | ||
| AC3-C(−) | littermate control lacking tg AC3-C | MEA lab |
WT mice in C57 background
Telemetry ECG and spontaneous activity recording
Surgical implantation of ECG and ambulatory activity-sensing and recording telemeters was conducted as described 15. (see supplemental methods for detail).
SAN cell isolation, action potential, If, INCX and SR Ca2+ content recordings
Isolation of single SAN cells from mice, action potential recordings, If and INCX current measurements from single SAN cell were performed as previously published 2, 16, 17. (see supplemental methods for detail).
Brifly, SAN cells were identified by their characteristic morphology (spindle or spider shape) and spontaneous activity. SAN cells were also identified electrophysiologically by typical spontaneous action potentials with slow depolarizing phase 4 and the hyperpolarization-activated current (If).
Spontaneous action potentials were recorded in current clamp (I=0) mode using the perforated (amphotericin B) patch-clamp technique on single SAN cell at 36±0.5 °C in Tyrode’s solution. The pipette was filled with (mM): 130 potassium aspartate, 10 NaCl, 10 HEPES, 0.04 CaCl2, amphotericin B 240 μg/ml. If was recorded in voltage clamp mode. Membrane potential was held at −35 mV, the voltage steps were applied for 5 s ranging from −125 mV to −45 mV in 10 mV increments, or vice versa.
INCX was recorded using whole-cell patch clamp technique at 36±0.5 ° C. The external solution contained the following (mM): 140 NaCl, 1.0 MgCl2, 5.0 Hepes, 2.5 CaCl2, 1.0 BaCl2, 10 glucose, and 10 μM nitrendipine, and 10 μM strophanthidin. The internal solution contained the following (mM): 110 CsCl, 10 NaCl, 5.0 glucose, 1.0 CaCl2, 0.4 MgCl2, 10 Hepes, and 20 TEA chloride, 5.0 EGTA. Membrane currents were elicited by a command ramp of 2 s from +80 mV to −120 mV. Holding potential was −80 mV. The protocol was applied every 10 s. Recordings were taken when five consecutive ramp recordings showed nearly identical currents. INCX was measured as the Ni-sensitive current. NiCl2 (5 mM) was added to define the fraction of current from NCX (the difference currents between total currents and post-Ni2+ currents from the same cell).
SR Ca2+ content was measured by integrating the INCX in response to a ‘spritz’ of caffeine, as described 2. Briefly, cells were loaded with Ca2+ by repetitive voltage command steps (300 ms, 0.5 Hz) from −80 to 0 mV at 36±0.5 °C. The bath solution contained (mM): 137 NaCl, 10 HEPES, 10 glucose, 1.8 CaCl2, 0.5 MgCl2, 25 CsCl. The intracellular pipette solution contained (mM): 120 CsCl, 10 TEA, 1.0 MgATP, 1.0 NaGTP, 5.0 phosphocreatine, 10 HEPES, Indo-1 0.2. After > 15 conditioning voltage command steps the SAN cell membrane was held at −80 mV and a caffeine (20 mM) spritz was locally applied. The resultant INCX was integrated and normalized to cell size.
Data analysis and statistics
Spontaneous action potential rates were analyzed using Clampfit 10. Arbitrary activity counts from the telemetry ECG recording system (Data Sciences International) were binned for analysis, with a bin width of 5 counts (see supplemental methods for detail). The mean heart rate was determined separately for each mouse and activity bin. One way ANOVA combined with Holm-Sidak test was used when more than two groups were compared. Student’s t-test was used when appropriate, paired t-test was used when comparing before and after treatment data on the same cells; the unpaired t-test was used for comparing data from two groups. Data were tested for normal distribution (Shapiro-Wilk (normality test) or Brown-Forsythe (equal variance test), as appropriate). A Signed Rank Test was used to generate a P value for non-parametric data. We rejected the null hypothesis for P<0.05.
Results
Loss of RyR2 S2808 or S2814 failed to affect heart rates in vivo
We studied mice with knockin mutations that selectively eliminated a validated PKA (S2808A) 5, 12 or CaMKII phosphorylation site (S2814A) 6, 13. The S2808A and S2814A mice had normal heart rate acceleration compared to WT control in response to isoproterenol injection (Fig 1A and B) or spontaneous activity (Fig 1C and D). These findings suggest that loss of RyR2 S2808 or S2814 alone are insufficient to impair the in vivo heart rate response to catecholamine or physiological stress related to spontaneous ambulatory activity.
Figure 1.
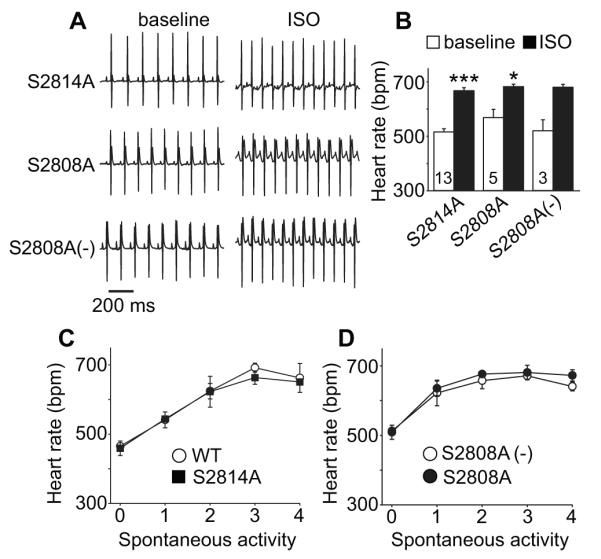
In vivo basal heart rate and heart rate responses to ISO or spontaneous activity were not altered in RyR2 mutant mice implanted with ECG and activity-sensing telemeters. A. Representative telemetry ECG traces (at baseline and after ISO injection) from S2814A, S2808A and littermate WT (S2808A (−)) mice, scale bar 200 ms. B. Summary of telemetry-recorded heart rates at baseline and in response to ISO stimulation in S2814A, S2808A and littermate control mice. The numerals indicate the number of mice in each experimental group here and in other figures. * p<0.05, ** p<0.01, *** p<0.001, paired Student‘s t-test, Error bars indicate s.e.m. C. Activity related heart rate change in S2814A and WT (C57) control mice, n=3-6. D. Activity related heart rate changes (see Methods) in S2808A and S2808A (−) littermate control mice, n=4- 7. The error bars indicate s.e.m. in all figures.
Loss of RyR2 S2808 or S2814 did not affect SAN cell rates
Heart rate is a complex integrated response where multiple inputs converge on SAN cells to drive basal and fight or flight heart rate responses 18. We found that SAN cells isolated from S2808A and S2814A mice had similar action potential rates at baseline, in the absence of catecholamine agonist stimulation, and after isoproterenol compared to SAN cells isolated from WT littermates (Fig 2A and B). These results showed that neither loss of S2808 nor S2814 on RyR2 was sufficient to disrupt SAN cell rate responses, suggesting that the observed preservation of in vivo heart rate responses in S2808A and S2814A was not the result of compensatory changes in SAN cell innervation. The results so far, using S2808A and S2814A knockin mice, showed that loss of these RyR2 target sites individually was inadequate to affect heart rate, suggesting that PKA and CaMKII cannot modulate heart rate by affecting SR Ca2+ release through post-translational modification of a single RyR2 site.
Figure 2.

SAN cell baseline beating rate and response to ISO were not altered in RyR2 mutant mice. A. Representative action potential recording traces (at baseline and after ISO application) from SAN cells isolated from S2814A (upper), S2808A (middle) and S2808A (−) littermate control (lower) mice. The short horizontal bars mark 0 mV. B. Summary data of SAN cell beating rates at baseline and response to increasing ISO concentration. n=6-14 cells/data point. C. Representative INCX current traces induced by caffeine triggered (20 mM) SR Ca2+ release (indicated by perfusion mark) in SAN cells isolated from the mice as indicated. The horizontal bars indicate 0 current level. D. SR Ca2+ content was increased in SAN cells isolated from the RYR mutation mice compared to SAN cells from littermate WT mice (C/F: Coulombs/Farad). Numbers of experimental SAN cells is indicated by numerals. *p <0.05, ** p<0.01 *** p<0.001 baseline vs ISO, unpaired Student’s t-test. + p<0.05, S2814A vs the other groups, One way ANOVA combined with Holm-Sidak test.
Loss of RyR2 S2814 increases SAN cell SR Ca2+ content
RyR2 phosphorylation by PKA and CaMKII can increase late diastolic SR Ca2+ leak 6, 12, 19, 20, potentially reducing SR Ca2+ content. We measured SAN cell SR Ca2+ content by integrating the INCX following caffeine–triggered SR Ca2+ release and found that S2814A SAN cells had relatively increased basal SR Ca2+ content compared to S2808A and controls (Fig 2 C and D). SAN cells isolated from all backgrounds exhibited a significant increase in SR Ca2+ content after isoproterenol (Fig 2 D). These data showed that loss of a PKA site (S2808) was insufficient to affect basal SAN cell SR Ca2+ content or the physiological increase in SR Ca2+ content after isoproterenol, but loss of the S2814 CaMKII site did result in a modest but significant increase in basal SAN cell SR Ca2+ content.
Loss of PLN maximized SR Ca2+ content without affecting heart rate
We next turned our attention to processes affecting SR Ca2+ uptake. PKA and CaMKII catalyze phosphorylation of PLN at single, distinct, extensively validated sites, leading to loss of negative regulation of SERCA2a by PLN and increased SR Ca2+ uptake 2, 3, 8.
As a first step, we assessed whether loss of PLN affects heart rate or SR Ca2+ content in SAN cells. We found that Pln−/− mice had heart rates that were similar to WT controls at rest and in response to isoproterenol (Fig 3A and B), similar to earlier descriptions 9, or in response to spontaneous activity (Fig 3C). SAN cells isolated from Pln−/− and WT-Pln mice also exhibited similar resting and isoproterenol-stimulated rates (Fig 3D), mirroring the in vivo heart rate responses to isoproterenol or activity. We next measured SR Ca2+ content in isolated SAN cells at baseline and after isoproterenol. The SR Ca2+ content in WT SAN cells was significantly increased over baseline by isoproterenol exposure. In contrast, SR Ca2+ content in Pln−/− SAN cells was replete under basal conditions and not further increased by isoproterenol (Fig 3E and F). We interpreted these findings as confirming that loss of PLN expression did not affect heart rate responses in vivo nor in isolated SAN cells but effectively released SERCA2a from inhibition, increasing basal SR Ca2+ content in SAN cells, similar to established actions in ventricular myocytes 21.
Figure 3.

In vivo heart rate responses to ISO and activity were not affected by Pln knock out or transgenic replacement with WT Pln. A. Representative telemetry ECG recording traces (at baseline and after ISO injection) from Pln−/− and WT-Pln mice. B. Summary ECG telemetry data at baseline and in response to ISO stimulation in WT-Pln and Pln−/− mice. *** p<0.001, paired Student’s t-test. C. Activity related heart rate changes in Pln−/− and WT-Pln mice, n=4-20. D. SAN cell beating rates at baseline and in response to ISO were not affected by Pln knock out. Summary data for SAN cell beating rates at baseline and in response to increasing ISO concentrations. n=8-14 cells/data point. E. Representative INCX current traces induced by caffeine triggered (20 mM) SR Ca2+ release (indicated by arrow) in SAN cells isolated from WT (C57) (left) or Pln−/− (right) mice. The horizontal bars indicate 0 current level. F. SR Ca2+ content was increased in SAN cells isolated from Pln−/− mice compared to SAN cells from WT (C57) mice (C/F: Coulombs/Farad). Numbers of experimental SAN cells is indicated by numerals. ** p<0.01 baseline vs ISO, + p<0.05, +++ p<0.001, Pln−/− vs WT (C57), unpaired Student’s t-test.
Expression of S16A or T17A did not impair heart or SAN cell rates
We next turned our attention to extensively characterized mice where Pln was transgenically replaced by WT Pln or mutant Pln lacking either the PKA (S16A) or CaMKII (T17A) sites 10. We found that mice transgenically expressing PLN S16A or T17A had normal heart rate acceleration in response to isoproterenol injection (Fig 4A and B); mice with transgenic expression of WT, S16A and T17A had equivalent rate responses to spontaneous activity (Fig 4C and D). SAN cells isolated from S16A and T17A mice had similar action potential rates at baseline and after isoproterenol compared to SAN cells isolated from mice with transgenic WT PLN expression (Fig 5A and B). We next checked SR Ca2+ content at baseline and in response to ISO in SAN cells isolated from S16A and T17A mice (Fig 5C and D). Neither PLN mutation affected SR Ca2+ content at baseline, but each mutation abolished the physiological increase in SR Ca2+ content after ISO (Fig 5D). These data suggest that PLN and PKA and CaMKII sites on PLN are individually dispensable for fight or flight heart rate responses in vivo and in isolated pacemaker cells.
Figure 4.
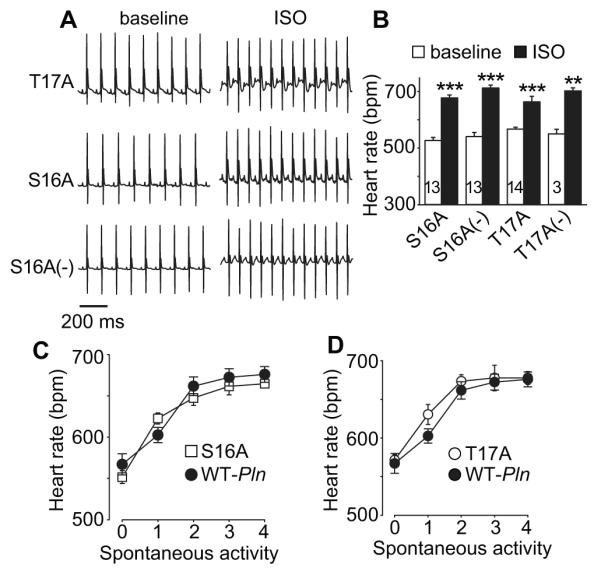
In vivo basal heart rate and response to ISO or activity were not altered in S16A or T17A mice. A. Representative ECG telemetry recording traces (at baseline and after ISO injection) from T17A (upper), S16A (middle) and S16A (−) (lower) mice. B. Summary data of ECG telemetry recorded heart rate at baseline and in response to ISO stimulation in S16A, T17A and their littermate control Pln−/− background mice (S16A(−) and T17A(−)). The number of mice used in the summary data is indicated by numerals in the corresponding bars. * p<0.05, ** p<0.01, *** p<0.001, paired Student’s t-test, Wilcoxon Signed Rank Test was used for T17A group data. C. Activity related heart rate changes in S16A and WT-Pln control mice, n=3-20. D. Activity related heart rate changes (see Methods) in T17A and WT-Pln control mice, n=4-20.
Figure 5.
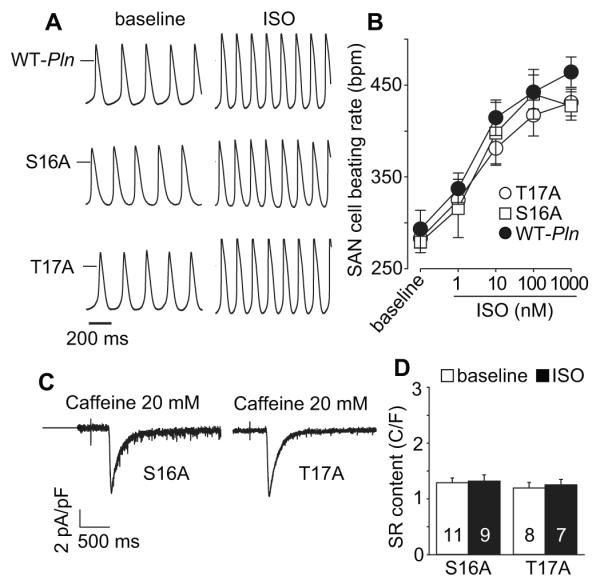
SAN cell baseline and ISO response rates were not altered in Pln mutant mice. A. Example action potential traces (at baseline and after ISO application) from SAN cells isolated from WT-Pln (upper), S16A (middle) and T17A (lower) mice. The horizontal bars mark 0 mV. B. Summary data for beating rates at baseline and in response to increasing ISO concentrations in SAN cells isolated from Pln mutant mice. n=5-8 cells/data point. C. Representative INCX current traces induced by caffeine (20 mM) triggered SR Ca2+ release (indicated by perfusion mark) in SAN cells isolated from the mice as indicated. The horizontal bars indicate 0 current level. D. SR Ca2+ content was not increased in SAN cells isolated from the PLN mutation mice. Numbers of experimental SAN cells are indicated by numerals.
SAN rate slowing by CaMKII inhibition required PLN
SAN cell CaMKII inhibition by expression of a CaMKII inhibitory peptide, AC3-I, compresses the dynamic range of heart rate acceleration by approximately 50% 2, however the mechanism of action for AC3-I expression in SAN cells to slow heart rate is incompletely understood. AC3-I expression in isolated SAN cells 2 and ventricular myocytes 22 reduces SR Ca2+ filling in response to isoproterenol (Fig 6A), suggesting the possibility that SAN SR Ca2+ repletion, above a threshold, is required for physiological rate acceleration and that the heart rate slowing actions of CaMKII inhibition are related, at least in part, to reduction in SR Ca2+ content below a critical threshold required for physiological heart rate increases.
Figure 6.
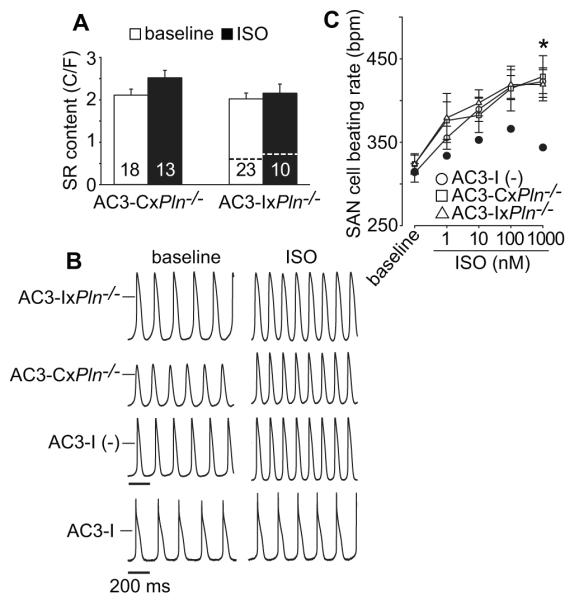
Increased SR Ca2+ content and restored ISO stimulated rate responses in Pln−/− mice with transgenic AC3-I expression. A. SR Ca2+ content in Pln−/− mice with transgenic expression of AC3-I and AC3-C. The number of SAN cells studied is indicated by numerals in the corresponding bars. Dotted line in right group of bars indicated SR Ca2+ content level and response to ISO in SAN cells isolated from AC3-I mice (Data re-draw of Figure 6C data from Wu et al., PNAS 2009). B. Example action potential recording traces (at baseline and after ISO application) from SAN cells isolated from AC3-IxPln−/−, AC3-CxPln−/− , AC3-I (−) and AC3-I mice as indicated. The horizontal bars mark 0 mV and the scale bar indicates 200ms. C. Summary data for SAN cell rates at baseline and in response to increasing ISO concentrations, n=7-10 cells/data point. Dotted curve (black filled circle) indicated ISO response in SAN cells isolated from AC3-I mice. (Data of AC3-I mice are re-draw of Figure 2B data from Wu et al., PNAS 2009). * p<0.05, One way ANOVA combined with Holm-Sidak test.
Based on the hypothesized association between SR Ca2+ content and SAN rate responses to isoproterenol, we next isolated SAN cells from Pln−/− mice interbred with AC3-I transgenic mice (AC3-IxPln−/−) or interbred with mice transgenically expressing an inactive control peptide, AC3-C (AC3-CxPln−/−) 14, 22 to determine if CaMKII inhibition affected SR Ca2+ content in the absence of Pln, potentially under conditions of replete SR Ca2+ content. The SR Ca2+ content was replete at baseline and not further increased by isoproterenol in SAN cells isolated from AC3-IxPln−/− and AC3-CxPln−/− mice (Fig 6A), indicating that reduced SR Ca2+ content in SAN cells expressing AC3-I was repaired by loss of PLN expression.
These interbred mice provided a new model to test if the relationship between AC3-I expression and reduced SR Ca2+ content was essential for slower SAN rates identified in AC3-I transgenic mice 2, 22, 23. We found that all SAN cells had similar resting and isoproterenol stimulated rates compared to WT and Pln−/− SAN cells (Fig 6B and C, Fig 3D). We compared these SAN cell rates with published rates from AC3-I SAN cells (Fig 6C), to demonstrate the ISO rate recovery response to Pln knock out. The interbred, AC3-IxPln−/− mice unexpectedly exhibited atrioventricular nodal conduction block that prevented a meaningful assessment of in vivo heart rates (Supplemental Fig 1). These data show that CaMKII inhibition does not slow isoproterenol-stimulated SAN cell rates in the absence of PLN and are consistent with the idea that CaMKII inhibition hinders heart rate acceleration by reducing SR Ca2+ content.
Heart rate slowing in mice with super inhibitory PLN
Based on our findings that the heart rate slowing from AC3-I expression was rescued in mice with a Pln−/− background, we next asked if a superinhibitory PLN mutant (N27A) that inhibits SERCA2a despite phosphorylation 11 would depress heart rate. We found that N27A mice had significantly reduced heart rates at baseline and in response to isoproterenol or spontaneous activity (Fig 7A-C). We next isolated SAN cells from N27A and Pln−/− littermate mice as well as WT-Pln mice to test if defective chronotropic performance was present in SAN cells. SAN cells isolated from N27A mice had significantly reduced resting rates and impaired isoproterenol rate responses compared to WT-Pln control and littermate Pln−/− SAN cells (Fig 5A, 6B, 7D and E). Finally, we found that SR Ca2+ content was reduced at baseline and was not significantly increased by isoproterenol in N27A SAN cells (Fig. 7F). We interpreted these findings to show that enhanced PLN-mediated SERCA2a inhibition could constrain SAN cell SR Ca2+ filling and reduce physiological heart rate responses. We summarized the SR Ca2+ content findings for all models, before and after ISO, to facilitate comparisons (supplementary Table).
Figure 7.

Reduced in vivo and SAN cell rates in N27A mutant mice. A. Example telemetry ECG recordings (at baseline and after ISO injection) from N27A mutant and WT-Pln mice, scale bar indicates 200 ms. B. Summary data for ECG telemetry recorded heart rates at baseline and in response to ISO stimulation in WT-Pln, N27A and littermate control Pln−/− mice. The number of mice used is indicated in the corresponding bars. *** p<0.001, baseline vs. after ISO, paired Student’s t-test. +++ p<0.001, N27A vs. WT-Pln or Pln−/−, ANOVA, holm-sidak test. C. Activity related heart rate changes in N27A and Pln−/− and WT-Pln control mice, n=4-20. D. SAN cells rates at baseline and in response to ISO were significantly reduced in SAN cells isolated from N27A mice. Example action potential recording traces (at baseline and after ISO application) from SAN cells isolated from WT-Pln (upper), Pln−/− (middle) and N27A (lower) mice. The horizontal bars mark 0 mV. E. Summary data for rates at baseline and in response to increasing ISO concentrations from SAN cells isolated from N27A, Pln−/− and WT-Pln mice. n=6-14 cells/data point. F. Reduced SR Ca2+ content at baseline and after ISO stimulation in SAN cells isolated from N27A mice. The summary SR Ca2+ content data at baseline and after ISO stimulation in SAN cells from WT (C57) and N27A mice. The number of SAN cells studied is indicated by numerals in the corresponding bars. The data for WT (C57) presented here are the same data in shown in Figure 3F. ** p<0.01 baseline vs ISO, +++ p<0.001, N27A vs WT, unpaired Student’s t-test.
HCN (If) currents and NCX currents were not altered by PLN deletion or mutation
We recorded If currents from SAN cells isolated from WT-Pln, Pln−/− and N27A mice (Fig 8A and B). There were no difference in If density between those groups. Similarly, we did not find significant differences in INCX current density between those groups (Fig 8C and D).
Figure 8.

HCN currents (If) and NCX currents (INCX) were not altered by PLN mutation. A. Representative If currents traces recorded from an SAN cell isolated from WT-Pln (upper) and Pln−/− mice (lower). B. Summary I-V relationship of If density from WT-Pln (n=6), Pln−/− (n=13) and N27A mice (n=6). C. Ramp voltage clamp protocol for recording INCX (upper) and an example INCX trace (lower). D. Summary data of INCX density at +80 mV (upper) and at −60 mV (lower) from SAN cells isolated from WT-Pln, Pln−/− and N27A mice.
Discussion
Heart rate is protected by partially redundant mechanisms
Given the importance of heart rate to survival it is not surprising that the SAN cellular mechanisms for maintaining and accelerating heart rate are reinforced by redundancy. However, certain components of the pacemaking machinery exert clear and quantifiable contributions to heart rate maintenance and/or acceleration because their removal or inactivation causes loss of basal heart rate or impairs heart rate increases. The drug ivabradine slows heart rate, at least in part, by antagonist actions on HCN4 24. Loss of the Na+/Ca2+ exchanger 25-27, inhibition of the mitochondrial Ca2+ uniporter 17 and application of toxins that prevent SR Ca2+ uptake (thapsigargin) 16, 28 and release (ryanodine) 16, 28 reduce or eliminate heart rate acceleration. These latter examples comprise some of the evidence in support of the Ca2+ clock concept for pacemaker cell automaticity 1. Mouse models of PKA 29 and CaMKII 2 inhibition by α myosin heavy chain promoter driven transgenic expression of kinase-inhibitory peptides maintain normal resting heart rates but exhibit selective loss of heart rate acceleration after isoproterenol injection. Based on the now established role of SR Ca2+ uptake and release to affect SAN rates and the role of PKA and CaMKII in regulating SR Ca2+, we determined whether individual loss of one of four established PKA or CaMKII sites on RyR2 or PLN could impact basal heart rate or heart rate acceleration.
RyR2 phosphorylation sites do not control heart rate individually
We found that loss of either RyR2 phosphorylation site was insufficient to affect basal heart rate or heart rate acceleration in response to isoproterenol injection or spontaneous activity. Furthermore, the behaviors of isolated SAN cells mirrored in vivo responses in that resting and isoproterenol accelerated automaticity was similar between mutant and control preparations. Our findings are partially confirmed by one study using the same S2808A mice 30 but different than another report showing S2808A mice have defective heart rate acceleration 31. While we are uncertain about the basis for the discrepant findings, we note that our study and the work by Shan et al. 31 used the same isoproterenol dosing for in vivo studies, although heart rates in our mice, implanted with ECG and activity telemeters, were measured in the absence of anesthesia. It is also possible that the lack of alignment between our results and the findings of Shan et al. 31 are due to undetermined differences in the mouse models. Taken together, we believe our new data constitute strong evidence that RyR2 S2808 and S2814 are individually dispensable for normal resting heart rates and for physiological heart rate responses. Furthermore, additional RyR2 phosphorylation sites, already known (e.g. S2030 32) or yet to be identified, may contribute to heart rate control. In contrast to the lack of effect on heart rate, our data suggest that the S2814 site is more important than S2808 for determining basal SR Ca2+ leak because S2814A SAN cells show elevated SR Ca2+ content compared to S2808A and WT counterparts.
A role for PLN in heart rate?
We found that Pln−/− mice and SAN cells isolated from Pln−/− mice have similar resting heart and automaticity rates compared to WT and transgenic controls. These findings are partially consistent with earlier studies showing Pln−/− and WT mice had equivalent baseline heart rates and responses to isoproterenol 9. We interpret these data to suggest that PLN plays a permissive role, perhaps to set SR Ca2+ content to a physiological set point, but is not required for basal or physiological heart rate responses. We hypothesize that phosphorylation at either S16 or T17 is not required to disinhibit SERCA2a sufficiently to sustain such a set point. In contrast, mice with superinhibitory PLN (N27A) had severely impaired resting, isoproterenol- and activity-stimulated heart rates, consistent with earlier reports 11, 16. These data show that extreme inhibition of SERCA2a activity does impact heart rate.
CaMKII inhibition does not affect heart rate in mice lacking PLN
SAN CaMKII activity is critical for sustaining the full dynamic range of heart rate under physiological stress but is dispensable for maintaining resting heart or SAN rates 2. We tested the idea that CaMKII actions to affect heart rate in SAN cells operated by a PLN or PLN-associated pathway by interbreeding AC3-I and Pln−/− mice. Surprisingly, loss of Pln allowed AC3-I expressing hearts and SAN cells to achieve a heart rate similar to WT. We interpret this result to suggest lowering of SR Ca2+ content in AC3-I transgenic mice may be a key component of the mechanism of heart rate slowing by CaMKII inhibition. We originally reported that SR Ca2+ content is reduced in AC3-I SAN cells 2 and here we show that AC3-I expression is not capable of reducing SAN cell SR Ca2+ content in the absence of PLN. We speculate that AC3-I expression reduces SR Ca2+ content below a set point required to sustain physiological rate increases and that loss of PLN supersedes the SR Ca2+ lowering consequences of AC3-I expression. Based on our published (Wu et al. 2, see Fig 6C) and new data (Fig 2D, Fig 3F, Fig 5D, Fig 6A and Fig 7F) we expect that SAN cell SR Ca2+ content less than 1.0 C/F, assessed as a caffeine releasable store and interrogated by integration of INCX, is insufficient to support physiological heart rate responses (Supplemental Table). It is also possible that CaMKII contributes to SR Ca2+ filling by actions at extra-PLN targets, such as mitochondria or novel sites on RyR2, PLN and/or RyR2 associated proteins.
Supplementary Material
WHAT IS KNOWN
Fight or flight heart rate increases depend on protein kinase A (PKA) and Ca2+/calmodulin kinase II (CaMKII) mediated enhancement of Ca2+ uptake and release from sarcoplasmic reticulum (SR) in sinoatrial nodal pacemaker cells (SANC).
CaMKII inhibition slows fight or flight heart rate increases.
The impact of individual PKA and CaMKII phosphorylation sites regulating SR Ca2+ uptake and release on heart rate modulation is unknown.
WHAT THE STUDY ADDS
We used a suite of genetically modified mice to evaluate PKA and CaMKII target sites on phospholamban (PLN), which modulates SR Ca2+ uptake, and the ryanodine receptor (RyR2), which mediates SR Ca2+ release, in SANC.
We found that ablation of individual RyR2 or PLN PKA or CaMKII target sites failed to affect heart rate in vivo or in SANC.
Our data suggest CaMKII inhibition slows heart rate by reducing SR Ca2+ content and that a minimum SR Ca2+ content is required in SANC for physiological fight or flight heart rate increases.
Acknowledgments
We thank William J. Kutschke and Jinying Yang for technical support, Shawn Roach for graphic contributions.
Funding Sources: This work was supported by National Institutes of Health (NIH) Grants R01-HL079031, R01-HL096652, R01-HL070250, and R01-HL113001 to M.E.A, R01-089598, R01-HL091947, R01-HL117641, and R41-HL129570 to X.H.T.W., and American Heart Association grant 13EAI-14560061 to X.H.T.W., R01-HL055438 and R01-HL120108 to HHV
Footnotes
Conflict of Interest Disclosures: None
Reference
- 1.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. 2010;106:659–673. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XH, Mohler PJ, Song LS, Anderson ME. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci USA. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 4.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- 5.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 7.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- 8.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 9.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 10.Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J Biol Chem. 2000;275:38938–38943. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 11.Zhai J, Schmidt AG, Hoit BD, Kimura Y, MacLennan DH, Kranias EG. Cardiac-specific overexpression of a superinhibitory pentameric phospholamban mutant enhances inhibition of cardiac function in vivo. J Biol Chem. 2000;275:10538–10544. doi: 10.1074/jbc.275.14.10538. [DOI] [PubMed] [Google Scholar]
- 12.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 13.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Shintani A, Grueter C, Zhang R, Hou Y, Yang J, Kranias EG, Colbran RJ, Anderson ME. Suppression of dynamic Ca(2+) transient responses to pacing in ventricular myocytes from mice with genetic calmodulin kinase II inhibition. J Mol Cell Cardiol. 2006;40:213–223. doi: 10.1016/j.yjmcc.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–3288. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. doi: 10.1038/ncomms7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA. 2015;112:9129–9134. doi: 10.1073/pnas.1504705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008;88:919–982. doi: 10.1152/physrev.00018.2007. [DOI] [PubMed] [Google Scholar]
- 19.Wehrens XHT, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: A critical mediator of heart failure progression. Proc Natl Acad Sci USA. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Chu G, Kranias EG, Bers DM. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am J Physiol. 1998;274:H1335–H1347. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- 22.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 23.Khoo MS, Kannankeril PJ, Li J, Zhang R, Kupershmidt S, Zhang W, Atkinson JB, Colbran RJ, Roden DM, Anderson ME. Calmodulin kinase II activity is required for normal atrioventricular nodal conduction. Heart Rhythm. 2005;2:634–640. doi: 10.1016/j.hrthm.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 24.Roubille F, Tardif JC. New therapeutic targets in cardiology: heart failure and arrhythmia: HCN channels. Circulation. 2013;127:1986–1996. doi: 10.1161/CIRCULATIONAHA.112.000145. [DOI] [PubMed] [Google Scholar]
- 25.Gao Z, Rasmussen TP, Li Y, Kutschke W, Koval OM, Wu Y, Wu Y, Hall DD, Joiner ML, Wu XQ, Swaminathan PD, Purohit A, Zimmerman K, Weiss RM, Philipson KD, Song LS, Hund TJ, Anderson ME. Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ Res. 2013;112:309–317. doi: 10.1161/CIRCRESAHA.111.300193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrmann S, Lipp P, Wiesen K, Stieber J, Nguyen H, Kaiser E, Ludwig A. The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc Res. 2013;99:780–788. doi: 10.1093/cvr/cvt154. [DOI] [PubMed] [Google Scholar]
- 27.Groenke S1, Larson ED, Alber S, Zhang R, Lamp ST, Ren X, Nakano H, Jordan MC, Karagueuzian HS, Roos KP, Nakano A, Proenza C, Philipson KD, Goldhaber JI. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS One. 2013;8:e81633. doi: 10.1371/journal.pone.0081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joung B, Tang L, Maruyama M, Han S, Chen Z, Stucky M, Jones LR, Fishbein MC, Weiss JN, Chen PS, Lin SF. Intracellular calcium dynamics and acceleration of sinus rhythm by beta- adrenergic stimulation. Circulation. 2009;119:788–796. doi: 10.1161/CIRCULATIONAHA.108.817379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic β-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacDonnell SM, García-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res. 2008;102:e65–72. doi: 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–855. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.