

Abstract
Polymers with advanced architectures can now be readily and reproducibly synthesized using controlled living polymerization. These materials are attractive as potential drug carriers due to their tunable size, versatile methods of drug incorporation and release, and ease of functionalization with targeting ligands. In this work, we report the design and development of macrocyclic brush, or “sunflower,” polymers, synthesized by controlled radical polymerization of hydrophilic “petals” from a cyclic multimacroinitiator “core.” These nanostructures can be synthesized with low polydispersity and controlled sizes depending on polymerization time. We further demonstrate that folate-functionalized sunflower polymers facilitate receptor-mediated uptake into cancer cells. These materials therefore show potential as drug carriers for anti-cancer therapies.
Polymeric drug carriers are used to alter the biodistribution and pharmacokinetics of their drug cargo in order to improve therapeutic indices. Desirable characteristics of polymeric carriers that allow for facile adaptation to drug-specific applications include: (i) tunable size and surface charge, (ii) high drug loading capacity, (iii) controllable drug release, (iv) mechanisms for preferential accumulation at the target site based on passive or active targeting strategies, and (v) reproducible synthesis. Polymer nanostructures with advanced architectures (e.g. cyclic polymers,1-3 polymer brushes,4,5 star polymers,6,7 and hyperbranched polymers)8,9 are a promising class of materials for this application.10 Indeed, Szoka and coworkers have demonstrated that cyclic polymers and PEG-grafted cyclic polymers exhibit longer circulation times than their analogous, molecular weight-matched linear polymers.11,12 Polymer-drug conjugates prepared from polymeric nanostructures offer the additional advantage of being formulation-free; sizes of these constructs can be similar to those of polymeric micelles without the need of additional formulation and characterization steps of the self-assembled structures.
One architecture of interest is the macrocyclic brush, or “jellyfish” polymer,13 which has not to our knowledge been applied as a drug carrier. Notably, ring expansion metathesis polymerization (REMP) has been used to prepare macrocyclic brush polymers by Grubbs’ and Tew’s groups independently.14,15 REMP maximizes the formation of cyclic polymers and avoids interchain reactions at high polymerization concentrations; however, this strategy is limited by the monomer species, and suffers from the relatively complicated synthesis of various norbornene-based monomers with activated functional groups or macromonomers with polymer side-chains. In addition, “grafting from,” as compared to “grafting onto” and “grafting through,” offers much better control over the generation of uniform polymer brushes by minimizing the effect of steric hindrance and avoiding tedious purification processes. Recently, nitroxide-mediated radical polymerization (NMRP) and ring-opening polymerization (ROP) have been reported to produce macrocyclic brush polymers by the “grafting from” approach,16 but again only limited monomers can be employed by such approaches. To develop a more universal platform material based on macrocyclic brush structures for drug delivery applications, we explored herein the preparation of macrocyclic brush polymers using atom transfer radical polymerization (ATRP), which provides access to a broad range of monomers and the ability to control polymer architecture and dimensions due to its “living” characteristics.17
In this letter, we report the development of macrocyclic brush, or “sunflower” polymers (Scheme 1) as a universal platform for targeted drug delivery. Sunflower polymers are synthesized by controlled radical polymerization (CRP) using a cyclic macroinitiator “core” from which “petals” are polymerized, radiating from the core (Scheme 2). As a proof of concept, we further conjugate a commonly-used cancer targeting ligand, folate, to the petal termini and a model cargo, fluorescein, to the cyclic core buried within the polymer petals, and demonstrate receptor-mediated delivery to mammalian cells.
Scheme 1.
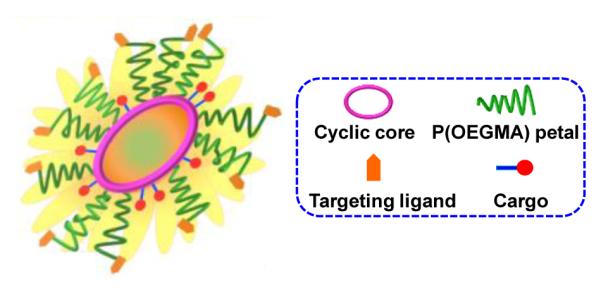
Schematic illustration of sunflower polymer containing targeting ligands and cargo.
Scheme 2.

Synthesis of folate-sunflower polymer.
The two main steps in sunflower polymer synthesis are (i) synthesis of the cyclic multimacroinitiator and (ii) polymerization of “petals” from the cyclized core (Scheme 2). The cyclic multimacroinitiator was first synthesized by (i) atom transfer radical polymerization (ATRP) copolymerization of ethyl glycinate methacrylamide (EGMA) and hydroxyethylmethacrylate (HEMA) using an alkyne-terminated initiator, (ii) conversion of the terminal bromine to an azide and intrachain click cyclization of the α-alkyne-ω-azide linear polymers, and (iii) conversion of HEMA monomers to alkyl halide ATRP initiators.
The ATRP conditions for the synthesis of P(HEMA-st-EGMA) copolymer were adapted from reported procedures18 with some modifications considering that ATRP of acrylamide-type monomers remains challenging,19 and more active ligands, such as 1,1,4,7,10,10-hexamethyltriethyllenetetramine (HMTETA),20 PMDETA,21 and Tris[2-(dimethylamino)ethyl]amine (Me6TREN),22 can offer improved control over the polymerization. Linear P(HEMA-st-EGMA) was first synthesized using the CuBr/PMDETA catalyst in isopropanol/N,N'-dimethylformamide (IPA/DMF) (9/1, w/w) at 65°C for 16 h. The benefits of this current polymerization condition are confirmed by a kinetics study (Figure S1). The statistical copolymer panel showed a clear shift of the GPC elution trace toward higher molecular weight with polymerization time (Figure S1a). The living characteristics were reflected by the pseudo-first order kinetics (Figure S1b) and narrow PDI (Figure S1c) during the whole polymerization process. A high conversion (~82%) of monomers was achieved after polymerization for 16 h, affording polymer of DP ~41, close to the target value of 50. Moreover, there was no obvious high molecular weight “shoulder” recorded in the GPC elution trace (Figures 1a and S1a), indicating minimal coupling termination of polymer chains during polymerization and retention of chain end functionalities for further azidonation.
Figure 1.

(a) GPC elution traces and (b) FT-IR spectra of linear and cyclic P(HEMA41-st-EGMA12).
The molar ratio of HEMA and EGMA in the resulting copolymers was determined to be 3.3:1 by 1H NMR spectroscopy (see supporting information for the detailed calculation of the copolymer composition), close to the feed molar ratio (3:1), indicating good control of the copolymer composition by ATRP (Figure S2). The resulting copolymer was thus denoted P(HEMA41-st-EGMA12). Gel permeation chromatography (GPC) shows that the copolymers have molecular weight (Mn) of 4.86 kDa and relatively low polydispersity (PDI) of 1.27, indicating controlled synthesis of copolymers (Figure 1a).
A survey of the literature showed that a large excess of sodium azide (10-40 fold molar excess compared to bromo-termini)23-26 is usually used for chain end azidonation to achieve complete conversion of the chain termini. Therefore, the linear precursor, P(HEMA-st-EGMA)-N3, was prepared by substitution with a 20-fold molar excess of sodium azide. Cyclic polymers were then prepared by intra-chain Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAc) of linear precursors following reported procedures.27,28 Successful cyclization was confirmed by FT-IR and GPC analyses, which show absence of the azide group (~2100 cm−1) after polymer cyclization and a clear shift toward longer retention times for the cyclic polymer compared to the linear precursor (Figure 1).
Cyclic poly(2-(2-bromoisobutyrnyloxy)ethyl methacrylate-st-EGMA) (p((HEMA-iBuBr)-st-EGMA)) multimacroinitiator was then prepared by esterification of cyclic p(HEMA-st-EGMA) with 2-bromoisobutyryl bromide. The disappearance of the characteristic signal at 4.8 ppm, assigned to the hydroxyl group of HEMA units, in the 1H NMR spectrum of cyclic multimacroinitiator compared to the parent cyclic polymer (Figures S3 and S4a) confirms complete conjugation of ATRP initiating sites to the pendant hydroxyl groups of the cyclic polymer.
The cyclic multimacroinitiator and its linear analogue were used for ATRP of oligoethyleneglycol methacrylate (OEGMA) to prepare sunflower and comb polymers, respectively. ATRP was performed using the catalyst CuBr/2,2′-bipyridine (bpy) in anisole/DMF (1/2, v/v) at 60°C for different lengths of time.
Three important parameters were optimized to minimize the generation of cross-linked structures and free polymer chains in the ATRP process due to the use of multimacroinitiators: (i) the feed concentration of OEGMA monomer was kept at 0.5 M; (ii) the monomer conversion was controlled to a low level (below 25%); and (iii) a small amount of CuBr2 ([CuBr2] = 0.1[CuBr]) was added to slow down the polymerization kinetics. A kinetic study was carried out using cyclic multimacroinitiator with a target DP of 100 for each radiating petal. The sunflower polymer panel showed a clear shift of the GPC elution trace toward higher MW with polymerization time. The living characteristics were reflected by the pseudo-first order kinetics and narrow PDI (around 1.3) during the whole polymerization process (Figure 2). Furthermore, the difference in topology of sunflower versus comb-like polymers was confirmed by GPC analyses. Sunflower polymers eluted at later times than comb polymers, indicating more compact structure and smaller effective molecular weight (Figure S5).
Figure 2.

ATRP results of sunflower polymer prepared from cyclic multimacroinitiator, with a target DP of 100 for each petal.
The hydrodynamic Z-average diameters of sunflower polymers were determined by dynamic light scattering (DLS). Polymer size increased with increasing p(OEGMA) petal polymerization time, ranging from 13.5 to 16.8 nm for the panel (Figure 3).
Figure 3.
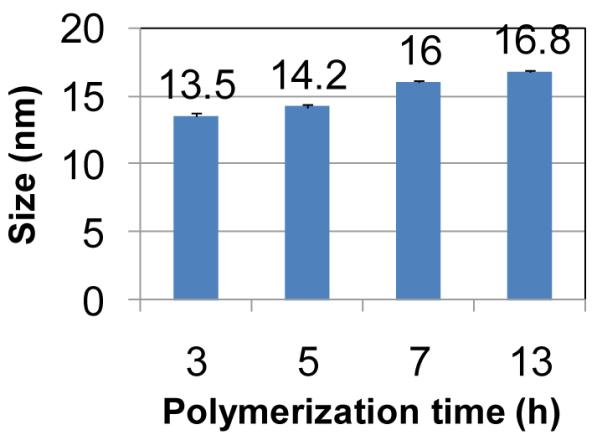
Summary of Z-average sizes of sunflower polymers with petals polymerized for different lengths of time.
The sunflower polymer with petals synthesized by 3 h ATRP of OEGMA (Mn = 101 kDa, PDI = 1.26, Rh = 14 nm) was selected for further biological evaluation. Targeted polymer constructs were prepared by conjugation of alkyne-functionalized folate (FA) to the p(OEGMA) petal termini via CuAAC “click” coupling. Folic acid was selected for targeting due to significant literature precedence establishing drug carrier targeting to cancer cells via the folic acid receptor.29-32 Folic acid conjugation was confirmed by 1H NMR analysis, which shows the appearance of characteristic signals at 6.7-8.7 ppm (assigned to the aromatic protons of folate) in the spectrum of folate-sunflower polymer (Figure S6), and by an increase in UV absorbance on GPC (Figure S7).
We next investigated folate receptor (FR)-mediated uptake of the sunflower polymers to KB (FR+) and A549 (FR−) cells. Uptake studies were performed using FA-SF-fluor (Figure S8, prepared by conjugation of the N-hydroxysuccinimide ester of fluorescein with polymer hydrazines) incubated with cells at various concentrations, followed by flow cytometry analysis for binding. Fluorescein was selected as a model drug due to its fluorescent and cell compatibility properties; in future work, aldehyde or ketone-containing drugs could be similarly conjugated to sunflowers for acid-triggered release at tumor sites. Although FA-SF-fluor uptake is dose-dependent in both cell lines, greater uptake was observed in the FR+ KB cells than in the A549 cells, as indicated by higher median fluorescence intensities of these cells at all polymer concentrations (Figure 4). Minimal uptake of untargeted SF-fluor was observed in either cell line (Figure S10a). In competition studies, cells were treated with FA-SF-fluor in the presence of 2 mM free folate. Competition with free folate significantly reduced polymer uptake in KB cells but not in A549 cells. These results further support folate receptor-mediated endocytosis as a major uptake mechanism for FA-targeted sunflower polymers by FR+ KB cells. Interestingly, when uptake of the comb-like linear analogue, FA-comb-fluor, was investigated, competition with free folate produced only a small decrease in polymer uptake in KB cells even at a low polymer concentration (Figure S10b). This may suggest higher levels of non-specific uptake of comb polymers relative to sunflower polymers. The effect of polymer architecture on internalization mechanism will therefore be an interesting topic for future investigation.
Figure 4.
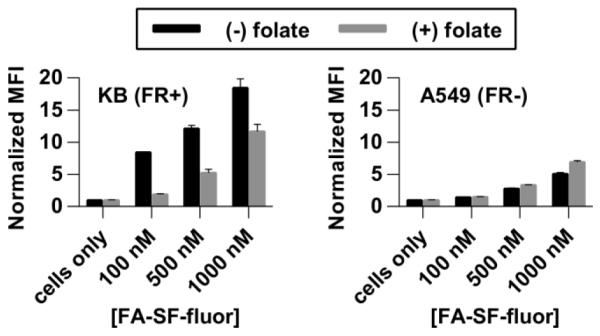
Uptake of FA-SF-fluor in FR+ KB cells and FR− A549 cells in the absence (black bars) and presence (gray bars) of 2 mM competing free folate.
Finally, because these materials demonstrate potential as drug carriers, we investigated the cytotoxicity of the sunflower polymers using KB and A549 cells. An MTS metabolic activity assay was performed to determine cell viability. The base sunflower and FA-modified SF (FA-SF) polymers were minimally toxic in either cell line, with IC50, or concentration for 50% cell killing, greater than 3.5 mg/mL in all cases.
In summary, this work introduces the sunflower polymer platform as a potential drug carrier. The ability to tailor molecular size within the relevant range for tumor delivery (5-50 nm), combined with the demonstrated features of targeted cellular uptake, support further investigation of these materials for tumor-targeted drug delivery and molecular imaging.
Supplementary Material
Figure S1. ATRP kinetics study for the synthesis of P(HEMA-st-EGMA). (a) GPC elution traces of ATRP-synthesized P(HEMA-st-EGMA) at various polymerization times; (b) pseudo-first order kinetics; and (c) plots of Mn and PDI with conversion.
The molecular weights and molecular weight distributions were determined by gel permeation chroma tography (GPC) equipped with a Waters 1515 pump and a Waters 2414 differential refractive index de tector. It used a series of three linear Styragel columns HT2, HT4, and HT5 at an oven temperature of 60°C. DMF was used as the eluent at a flow rate of 1.0 mL/min. PMMA standards were employed for the GPC calibration.
Figure S2. 1H NMR spectra of (a) EGMA monomer, and P(HEMA-st-EGMA)-Br copolymer.
Figure S3. 1H NMR spectra of (a) linear precursor P(HEMA-st-EGMA)-N3 and (b) cyclic P(HEMA-st-EGMA).
Figure S4. 1H NMR spectra of (a) cyclic multimacroinitiator and (b) sunflower polymer with azide-terminated P(OEGMA) petals.
Figure S5. GPC elution traces of sunflower polymer and comb-like polymer prepared with the same polymerization time using cyclic and linear multimacroinitiators.
Figure S6. 1H NMR spectra of (a) sunflower polymer and (b) folate-sunflower polymer.
Figure S7. GPC elution traces and UV-detected signals of sunflower polymer and folate-conjugated sunflower polymer.
Figure S8. 1H NMR spectra of (a) sunflower polymer-fluorescein and (b) folate-sunflower polymer-fluorescein.
Figure S9. GPC elution trace of folate-sunflower polymer and folate-sunflower polymer-fluorescein.
Figure S10. Uptake of (a) untargeted SF-fluor and (b) FA-comb-fluor polymers in FR+ KB cells and FR− A549 cells in the absence (black bars) and presence (gray bars) of 2 mM competing free folate.
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (1R01CA177272), the National Science Foundation (DMR 1206426) and a National Science Foundation Graduate Research Fellowship to C.E.W.
Footnotes
Supporting Information. Experimental details, data and discussion on characterization of sunflower polymers, and additional results from biological studies are available in Figures S1-S10.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Laurent BA, Grayson SM. Journal of the American Chemical Society. 2011;133:13421. doi: 10.1021/ja2024355. [DOI] [PubMed] [Google Scholar]
- (2).Laurent BA, Grayson SM. Polymer Chemistry. 2012;3:1846. [Google Scholar]
- (3).Liu L, Parameswaran S, Sharma A, Grayson SM, Ashbaugh HS, Rick SW. The Journal of Physical Chemistry B. 2014;118:6491. doi: 10.1021/jp412184h. [DOI] [PubMed] [Google Scholar]
- (4).Johnson JA, Lu YY, Burts AO, Lim Y-H, Finn MG, Koberstein JT, Turro NJ, Tirrell DA, Grubbs RH. Journal of the American Chemical Society. 2011;133:559. doi: 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sheiko SS, Sumerlin BS, Matyjaszewski K. Progress in Polymer Science. 2008;33:759. [Google Scholar]
- (6).Hawker CJ, Frechet JMJ, Grubbs RB, Dao J. Journal of the American Chemical Society. 1995;117:10763. [Google Scholar]
- (7).Trollsås M, Hedrick JL. Journal of the American Chemical Society. 1998;120:4644. [Google Scholar]
- (8).Lane D, Chiu D, Su F, Srinivasan S, Kern H, Press O, Stayton P, Convertine AJ. Polymer Chemistry. 2015;6:1286. doi: 10.1039/C4PY01250J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Smet M. Hyperbranched Polymers: Synthesis, Properties, and Applications. 2011:387. [Google Scholar]
- (10).Wang C, Stayton P, Pun S, Convertine AJ. Journal of Controlled Release. 2015 doi: 10.1016/j.jconrel.2015.08.054. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen B, Jerger K, Fréchet JM, Szoka FC. Journal of Controlled Release. 2009;140:203. doi: 10.1016/j.jconrel.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nasongkla N, Chen B, Macaraeg N, Fox ME, Fréchet JM, Szoka FC. Journal of the American Chemical Society. 2009;131:3842. doi: 10.1021/ja900062u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Coulembier O, Moins S, De Winter J, Gerbaux P, Leclère P, Lazzaroni R, Dubois P. Macromolecules. 2009;43:575. [Google Scholar]
- (14).Xia Y, Boydston AJ, Grubbs RH. Angewandte Chemie. 2011;123:6004. doi: 10.1002/anie.201101860. [DOI] [PubMed] [Google Scholar]
- (15).Zhang K, Tew GN. ACS Macro Letters. 2012;1:574. doi: 10.1021/mz2001675. [DOI] [PubMed] [Google Scholar]
- (16).Jia ZF, Monteiro MJ. J. Polym. Sci. Pol. Chem. 2012;50:2085. [Google Scholar]
- (17).Matyjaszewski K, Tsarevsky NV. Journal of the American Chemical Society. 2014;136:6513. doi: 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]
- (18).Yang P, Armes SP. Macromolecular rapid communications. 2014;35:242. doi: 10.1002/marc.201300617. [DOI] [PubMed] [Google Scholar]
- (19).Teodorescu M, Matyjaszewski K. Macromolecules. 1999;32:4826. [Google Scholar]
- (20).Xu F, Zheng SZ, Luo YL. Journal of Polymer Science Part A: Polymer Chemistry. 2013;51:4429. [Google Scholar]
- (21).Wang J, Gao P, Ye L, Zhang A.-y., Feng Z.-g. Polymer Chemistry. 2011;2:931. [Google Scholar]
- (22).Xia Y, Yin X, Burke NA, Stöver HD. Macromolecules. 2005;38:5937. [Google Scholar]
- (23).Cai T, Yang WJ, Neoh K-G, Kang E-T. Polymer Chemistry. 2012;3:1061. [Google Scholar]
- (24).Chen F, Liu G, Zhang G. Journal of Polymer Science Part A: Polymer Chemistry. 2012;50:831. [Google Scholar]
- (25).Jiang X, Shi Y, Zhu W, Chen Y, Xi F. Journal of Polymer Science Part A: Polymer Chemistry. 2012;50:4239. [Google Scholar]
- (26).Ren JM, Satoh K, Goh TK, Blencowe A, Nagai K, Ishitake K, Christofferson AJ, Yiapanis G, Yarovsky I, Kamigaito M. Angewandte Chemie International Edition. 2014;53:459. doi: 10.1002/anie.201308366. [DOI] [PubMed] [Google Scholar]
- (27).Laurent BA, Grayson SM. Journal of the American Chemical Society. 2006;128:4238. doi: 10.1021/ja0585836. [DOI] [PubMed] [Google Scholar]
- (28).Wei H, Chu DS, Zhao J, Pahang JA, Pun SH. ACS macro letters. 2013;2:1047. doi: 10.1021/mz400560y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lee S-M, Chen H, O’Halloran TV, Nguyen ST. Journal of the American Chemical Society. 2009;131:9311. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Low PS, Henne WA, Doorneweerd DD. Accounts of Chemical Research. 2008;41:120. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- (31).Lu Y, Low PS. Advanced drug delivery reviews. 2012;64:342. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- (32).Yang X, Grailer JJ, Pilla S, Steeber DA, Gong S. Bioconjugate chemistry. 2010;21:496. doi: 10.1021/bc900422j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ATRP kinetics study for the synthesis of P(HEMA-st-EGMA). (a) GPC elution traces of ATRP-synthesized P(HEMA-st-EGMA) at various polymerization times; (b) pseudo-first order kinetics; and (c) plots of Mn and PDI with conversion.
The molecular weights and molecular weight distributions were determined by gel permeation chroma tography (GPC) equipped with a Waters 1515 pump and a Waters 2414 differential refractive index de tector. It used a series of three linear Styragel columns HT2, HT4, and HT5 at an oven temperature of 60°C. DMF was used as the eluent at a flow rate of 1.0 mL/min. PMMA standards were employed for the GPC calibration.
Figure S2. 1H NMR spectra of (a) EGMA monomer, and P(HEMA-st-EGMA)-Br copolymer.
Figure S3. 1H NMR spectra of (a) linear precursor P(HEMA-st-EGMA)-N3 and (b) cyclic P(HEMA-st-EGMA).
Figure S4. 1H NMR spectra of (a) cyclic multimacroinitiator and (b) sunflower polymer with azide-terminated P(OEGMA) petals.
Figure S5. GPC elution traces of sunflower polymer and comb-like polymer prepared with the same polymerization time using cyclic and linear multimacroinitiators.
Figure S6. 1H NMR spectra of (a) sunflower polymer and (b) folate-sunflower polymer.
Figure S7. GPC elution traces and UV-detected signals of sunflower polymer and folate-conjugated sunflower polymer.
Figure S8. 1H NMR spectra of (a) sunflower polymer-fluorescein and (b) folate-sunflower polymer-fluorescein.
Figure S9. GPC elution trace of folate-sunflower polymer and folate-sunflower polymer-fluorescein.
Figure S10. Uptake of (a) untargeted SF-fluor and (b) FA-comb-fluor polymers in FR+ KB cells and FR− A549 cells in the absence (black bars) and presence (gray bars) of 2 mM competing free folate.