

Abstract

In a continued effort to improve upon the previously published 4-substituted methoxybenzoyl-arylthiazole (SMART) template, we explored chemodiverse “B” rings and “B” to “C” ring linkage. Further, to overcome the poor aqueous solubility of this series of agents, we introduced polar and ionizable hydrophilic groups to obtain water-soluble compounds. For instance, based on in vivo pharmacokinetic (PK) studies, an orally bioavailable phenyl-aminothiazole (PAT) template was designed and synthesized in which an amino linkage was inserted between “A” and “B” rings of compound 1. The PAT template maintained nanomolar (nM) range potency against cancer cell lines via inhibiting tubulin polymerization and was not susceptible to P-glycoprotein mediated multidrug resistance in vitro, and markedly improved solubility and bioavailability compared with the SMART template (45a–c (PAT) vs 1 (SMART)).
INTRODUCTION
Microtubules are cytoskeletal filaments consisting of αβ-tubulin heterodimers and are involved in a wide range of cellular functions, including shape maintenance, vesicle transport, cell motility, and division. Tubulin is the major structural component of the microtubules and a well verified target for a variety of highly successful anticancer drugs. Anticancer drugs like paclitaxel and vinblastine that are able to interfere with microtubule–tubulin equilibrium in cells are extensively used in cancer chemotherapy.1 There are two major classes of antimitotic agents: microtubule-stabilizing agents, which inhibits the microtubule depolymerization by binding to and stabilizing micro-tubule, are represented by taxanes and epothilones. Another class is microtubule-destabilizing agents, such as vinca alkaloids and colchicine, which cause microtubule disassembling and inhibit tubulin polymerization into microtubules. Vinblastine represents one of vinca alkaloids. Colchicine and colchicine-site binders are all defined as microtubule-destabilizing antimitotic agents. While no colchicine-site binders are currently approved for cancer chemotherapy, both the taxanes and vinca alkaloids are widely used to treat human cancers. However, colchicine binding agents like combretastatin A-4 (CA-4) and N-(2-(4-hydroxy phenylamino)pyridin-3-yl)-4-methoxybenzenesulfonamide (ABT-751,47) (Figure 1) are now under clinical investigation as potential new chemotherapeutic agents. 2,3,4
Figure 1.

Structures of Colchicine-Binding Site Tubulin Inhibitors.
Unfortunately, microtubule-interacting anticancer drugs in clinical use share two major problems, resistance and high lipophilicity. A common mechanism of multidrug resistance (MDR), namely ATP binding cassette (ABC) transporter protein-mediated drug efflux, limits their efficacy.5–7 P-glycoproteins (P-gp, encoded by the MDR1 gene) is an important member of the ABC superfamily.8 P-gp prevents the intracellular accumulation of many cancer drugs by increasing their efflux out of cancer cells as well as contributing to hepatic, renal, or intestinal clearance pathways. Attempts to coadminister P-gp modulators or inhibitors to increase cellular availability by blocking the actions of P-gp have met with limited success.8,9 The other major problem with taxanes, as with many biologically active natural products, is its high lipophilicity and lack of solubility in aqueous systems. This leads to the use of emulsifiers like Cremophor EL and Tween 80 in clinical preparations. A number of biologic effects related to these drug formulation vehicles have been described, including acute hypersensitivity reactions and peripheral neuropathies.10,11 Compared to compounds binding the paclitaxel- or vinca alkaloid-binding site, colchicine-binding agents usually exhibit relatively simple structures. Thus, it provides a better opportunity for oral bioavailability via structural optimization, as reported herein for compounds with improved solubility and pharmacokinetic (PK) parameters. In addition, most of these new agents appear to circumvent P-gp-mediated MDR. Therefore, these novel colchicine binding site targeted compounds hold great promise as therapeutic agents, particularly because they have improved aqueous solubility and overcome P-gp mediated MDR.
4-Substituted methoxybenzoyl-arylthiazole (SMART, 1, Figure 1) is a potential anticancer agent that was discovered recently in our laboratory which targets tubulin by binding to its colchicine binding site.12 The structure – activity-relationship (SAR) studies in the “A” and “C” rings were discussed based on synthesized analogues. Whereas, alternatives to the thiazole “B” ring and carbonyl linker were not investigated. In this article, we synthesized and evaluated the biological properties of “B” ring and carbonyl linker modified derivatives to extend the SAR studies beyond just the SMART template and found some more potent compounds. “A” ring modifications and introducing a NH linkage between “A” and “B” rings produced potent water-soluble compounds including the phenyl amino thiazole (PAT) compounds 45a–c. We demonstrated that these compounds can effectively inhibit cancer cell growth in vitro by interfering with tubulin polymerization. In experiments described herein, we have examined the antiproliferative effects of these novel compounds on a drug-sensitive ovarian cancer cell line (OVCAR-8) and its P-gp-overexpressing drug-resistant counterpart (NCI/ADR-RES). Tested compounds did not demonstrate susceptibility to P-gp mediated drug resistance. We also compared the oral bioavailability of the PAT compounds in rats. Compounds 45a and 45c showed a significant improvement in bioavailability compared with SMART compound 1. Thus, the new PAT compounds and other molecules reported herein represent new families of compounds that may be very useful in the treatment of cancer with improved PK properties.
RESULTS AND DISCUSSION
Chemistry
Design of novel chemotypes of SMART agents that target tubulin polymerization were investigated mainly by preparing three series of modifications based upon compound 1. The first series of derivatives (5,6,9a–9h) were characterized by the replacement of the thiazole in SMART molecules with a panel of different B rings as illustrated in Schemes 1–2. l-Sertine methyl ester hydrochloride was obtained from l-serine stirred in acetyl chloride/MeOH solution. Condensation of the methyl ester with ethyl benzimidate hydrochloride led to oxazoline methyl ester 2 in excellent yield (Scheme 1).13 Hydrolyzation of the methyl ester gave carboxylic acid 3 that was in turn coupled to N,O-dimethylhydroxylamine to provide Weinreb amide 4. Compound 4 was reacted with appropriate Grignard reagents in anhydrous THF to give the oxazoline 5. Oxidation of 5 with BrCCl3/DB U gave the oxazole product 6.14 Other B ring variants 9a–9f (except 9e) were obtained from different acids 7a–7f with a similar method as described above (Scheme 2). Pure compound 9e with thiophene in B ring position can not be separated from the mixture of 9e and a Grignard reagent coupling byproduct 3,4,5,3′,4′,5′-hexamethoxybiphenyl using chromatography. So we used an alternative method to prepare 9e: Weinreb amide 8e was converted into an aldehyde and then reacted with 3,4,5-trimethoxyphenylmagnesium bromide to afford the alcohol 10e, which can be easily separated from 3,4,5,3′,4′,5′ -hexamethoxybiphenyl after flash column purification. Oxidation with pyridinium dichromate (PDC) or DMSO did not afford 9e from secondary alcohol 10e with good yields, but using Dess–Martin periodinane reagent as oxidant successfully formed the desired ketone compound 9e with 81% yield.15 9g and 9h were prepared from alcohol 10g–10h using a similar method. Compound 9i was obtained via a coupling reaction from piperidine 11 and 3,4,5-trimethoxybenzoic acid.
Scheme 1a.

a (a) MeOH, CH3COCl, 83%; (b) ethyl benzimidate hydrochloride, CH2Cl2, Et3N, 96%; (c) LiOH, MeOH, H2O, 65%; (d) EDCI, HOBt, NMM, CH3OCH3NH · HCl, 61%; (e) 3,4,5-trimethoxyphenylmagnesium bromide, THF, 48–71%; (f) CBrCl3, DBU, CH2Cl2, 56%.
Scheme 2a.

a (a) EDCI, HOBt, NMM, CH3OCH3NH·HCl, CH2Cl2, 51–95%; (b) 3,4,5-trimethoxyphenyl-magnesium bromide, THF, 48–78%; (c) LAH, −78 °C, THF, 85%; (d) Dess–Martin reagent, CH2Cl2, 81%; (e) EDCI, HOBt, NMM, 3,4,5-trimethoxybenzoic acid, CH2Cl2, 58%.
In the second series of novel templates, structure modifications focused on alternatives to the carbonyl group to avoid potential metabolic problems caused by ketone reduction (Scheme 3 and Scheme 4).16 SMART compound 1 was synthesized from 2-phenyl-4,5-dihydro-thiazole-4-carboxylic acid 12a through three steps described previously.17 1 was converted to oxime isomers (13a–d) upon reaction with hydroxylamines, NH2OH or NH2OCH3. Assignments were made on the basis of chemical and spectral data as described infra. An improved Beckmann rearrangement 18 readily produced the rearranged amides 13e and 13f from the two geometric stereoisomers 13a and 13b via their reaction with tosyl chloride and subsequent basic aluminum oxide column. Hydrazide derivatives 14a and 14b were prepared by mixing 1 with hydrazine hydrate in ethanol/CH2Cl2 and refluxing for 24 h. Acrylonitriles 15a– 15b were obtained from Wittig reaction of 1 with diethyl cyanomethylphosphonate.19 Cyanoimine 16 was prepared using the procedure described by Cuccia.20 The carbonyl group in compound 1 was also reduced to a secondary alcohol (17) or converted to an alkene (18) as illustrated. We also tried to remove the carbonyl group between B and C rings in compound 1 and thus obtained compound 19 in Scheme 4. Introducing cis- and trans- double bonds into the carbonyl position formed compounds 20 and 21, which were synthesized from a Wittig reaction with 2-phenylthiazole-4-carbaldehyde. We also prepared sulfide compound 23, sulfone 24 and sulfoxide 25 using 3-aminobiphenyl as starting material through an initial Sandmeyer reaction to yield carbonodithioate 22, followed by CuI catalyzed coupling reaction and m-CPBA oxidation.21 Sulfonamide linked compound 26 was prepared from reaction of 3-biphenylsulfonyl chloride with 3,4,5-trimethoxyaniline in the presence of NEt3 in DMF.
Scheme 3a.

a (a) MeOH/pH = 6.4 phosphate buffer, RT; (b) EDCI, HOBt, NMM, HNCH3OCH3; (c) CBrCl3, DBU, CH2Cl2; (d) 3,4,5-trimethoxyphenylmagne-sium bromide, THF; (e) isopropyl triphenylphosphonium iodide, n-BuLi, THF; (f) LAH, THF; (g) for 13a and 13b, NH2OH · HCl, C2H5OH, H2O, NaOH; for 13e and 13f, NH2OMe · HCl, pyridine; (h) TsCl, NaH, basic Al2O3; (i) NH2NH2 · xH2O, CH2Cl2, C2H5OH; (j) diethyl cyanomethylphosphonate, n-BuLi, THF; (k) bis-trimethylsilylcarbodiimide, TiCl4, CH2Cl2; (l) EDCI, HOBt, Et3N, 3,4,5-trimethoxyaniline, CH2Cl2.
Scheme 4a.

a (a) Bromine, EtOH; (b) benzothioamide, EtOH, reflux; (c) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) CBrCl3, DBU, CH2Cl2; (e) LAH, THF; (f) 5-(bromomethyl)-1,2,3-trimethoxybenzene, Ph3P, THF; (g) n-BuLi, THF; (h) (1) HCl, H2O; (2) NaNO2, H2O, 0 °C; (i) ethyl potassium xanthate; (j) KOH/EtOH; (k) H2O, HCl; (l) 5-iodo-1,2,3-trimethoxybenzene, CuI, t-BuONa; (m) 2 equiv or 1 equiv m-CPBA, CH2Cl2; (n) 3,4,5-trimethoxyaniline, NEt3, DMF.
A third aim was to improve aqueous solubility and oral bioavailability of these colchicine site targeted agents. As illustrated in Scheme 5, we introduced hydroxyl and aminomethyl at the para-position of the phenyl A-ring, as well as replacing phenyl with 5-indolyl and 2-indolyl rings. Weinreb amides 27, 31, 35, and 37 were prepared by the procedure described before using aryl nitriles as starting materials.12 2-Cyano-indole 30 was prepared with reported method.22 Protections of hydroxyl (TBDMSCl), indolyl (PhSO2Cl), and amino (Boc2O) groups were used in preparations. Deprotection of TBDMS and oxidation from thiazoline (28) to thiazole (29) were finished in onestep using TBAF/THF solution. We reported this thiazoline–thiazole oxidation can happen spontaneously in the reaction of thiazoline Weinreb amide and Grignard reagent.12 We also observed the same phenomena during preparing indole compounds 32 and 36. Compound 32 was separated as a pure thiazole compound after reaction with 3,4,5-trimethoxphenyllithium and needed no further oxidation. Compound 36 was obtained after removing phenylsulfonyl protecting groups in hot NaOH ethanol solution. para-OH and NH2 on the A ring of 29 and 39 were obtained from similar Grignard reactions from Weinreb amides 27 and 37. Compound 38 was further converted to HCl salt of monomethyl amine 40 via NaH/MeI conditions, and compound 39 was converted to HCl salt of dimethylamine 42 under HCHO/NaBH3CN conditions. To improve bioavailability, we introduced an NH linker between A phenyl and B thiazole rings. We synthesized this new series of compounds as shown in Scheme 6. Reaction of 3-bromo-2-oxopropanoic acid ethyl ester and arylthiourea in ethanol under 65 °C produced 2-(arylamino)-thiazole-4-carboxylic acids 43a–c with high yields. Then these acids were converted to Weinreb amides 44a–c, followed by reactions with 3,4,5-trimethoxphenyllithium yielded aniline linked free bases 45a–c, which can be converted into HCl salts 46a–c.
Scheme 5a.

a (a) l-Cysteine, EtOH, 65 °C; (b) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (c) TBDMSCl, imidazole, THF; (d) 3,4,5-trimethoxyphenylbro-mide, BuLi, THF; (e) TBAF, THF; (f) SOCl2, Et2O; (g) NH3, MeOH; (h) POCl3; (i) PhSO2Cl, Bu4NHSO4, toluene, 50% NaOH; (j) 1N NaOH, EtOH, reflux; (k) Boc2O, 1N NaOH, 1,4-dioxane; (l) CBrCl3, DBU, CH2Cl2; (m) 2N HCl in 1,4-dioxane; (n) NaH, DMF, MeI; (o) HCHO, NaBH3- CN, Et3N.
Scheme 6a.

a (a) EtOH, 65 °C; (b) NaOH, C2H5OH, refluxing; (c) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) 3,4,5-trimethoxyphenylbromide, BuLi, THF; (e) 2 N HCl in 1, 4-dioxane.
Structural Identification of syn-/anti- Isomers
Oximes 13a–13b, hydrazides 14a–14b, and acrylonitriles 15a–15b were obtained from flash column as separated isomer pairs. Their configurations were identified using a chemistry method, NMR NOE spectrum and quantum chemical shift calculations. Improved Beckmann rearrangement was used to distinguish oxime isomers 13a and 13b as the group anti- position to the departing OTs that migrates to nitrogen (Figure 2). The syn-isomer 13a rearranged by a 1,2- shift of 3,4,5-trimethoxyphenyl (TMP) to yield amide 13e, whereas the anti-isomer 13b underwent a 1, 2-thiazolyl shift to yield 13f. An alternative coupling method was also used to prepare compound 13e via intermediate 12c (see Scheme 3),12 which was characterized and compared with two rearrangement products 13e and 13f. The NMR spectra showed that alternatively prepared 13e was identical with Beckmann rearrangement product of syn- isomer 13a. Quantum chemical calculations based on 1H NMR also corroborated the conformations of these two isomers.
Figure 2.

1H NMR of Beckmann rearrangement product from 13a isomer (A) is the same with previous prepared 4-carbonyl amide 13e (C) while rearrangement product from isomer 13b showed reversed 4-amino amide NMR signals (B).
2D NOESY NMR and 1D NOE NMR were used to identify syn/anti- isomers of hydrazides 14a–14b, and acrylonitriles 15a–15b, respectively. The 1H NMR, 2D NOESY, and 1D NOE NMR spectra of 14a–14b, 15a–15b are shown in Supporting Information. From chemical structures, we can see the distance between amino in syn- isomer (14a) hydrazine is far away from C ring protons while in anti- isomer (14b) is short, the 2D NOESY spectrum of compound 14b showed strong NOE correlations between NH2 and C ring protons and demonstrated 14b is anti- confirmation. Compound 14a which has a syn-conformation did not show any NOE on 2D NOESY. Acrylonitriles 15a and 15b were identified with the similar 1D NOE spectrum. There are no NOE correlations observed among protons from B ring (Hb)/C ring (Hc) and acrylonitrile linker (Ha), thus compound 15a is an anti- isomer. Compound 15b showed NOE between Ha and Hc protons, which corresponding to a syn- confirmation (Supporting Information).
Biological Evaluation: In Vitro Cell Growth Inhibitions
All of the reported compounds were first evaluated for cytotoxicity in a mouse melanoma cell line B16-F1, human melanoma cell lines (A375 and WM-164), and prostate cancer cell lines (DU145, PC-3, LNCaP, and PPC-1). Compounds 1 (Tables 1, 2, and 4) and 47 (E7010, Abbott Laboratories/Eisai Co Ltd., Table 4), which has entered phase II clinical studies in treating patients with different cancers, were included in the assays as examples of colchicine-site binding agents. IC50 values for cell growth inhibition are shown in Tables 1–4.
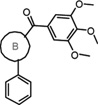
Table 1.
SAR of Alternate B ring Compounds
 | |||||||
|---|---|---|---|---|---|---|---|
| IC50 ± SEM (µM) |
|||||||
| B ring | B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |
| 1 | 2,4-thiazole | 0.055 ± 0.005 | 0.028 ± 0.005 | 0.071 ± 0.004 | 0.021 ± 0.001 | 0.028 ± 0.004 | 0.043 ± 0.005 |
| 5 | 2,4-oxazoline | 6.5 ± 0.8 | 0.5 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 |
| 6 | 2,4-oxazole | 0.6 ± 0.2 | 0.3 ± 0.1 | 0.292 ± 0.01 | 0.292 ± 0.02 | 0.331 ± 0.06 | 0.343 ± 0.04 |
| 9a | 1,3-phenyl | 0.5 ± 0.2 | 0.087 ± 0.015 | 0.171 ± 0.01 | 0.124 ± 0.02 | 0.052 ± 0.01 | 0.080 ± 0.01 |
| 9b | 4,6-pyrimidine | >30 | >30 | 6.9 ± 5.2 | 8.3 ± 4.7 | 7.0 ± 2.9 | 3.7 ± 0.2 |
| 9c | 2,6-pyridine | 0.039 ± 0.012 | 0.030 ± 0.014 | 0.033 ± 0.003 | 0.032 ± 0.002 | 0.027 ± 0.002 | 0.025 ± 0.001 |
| 9d | 2,5-furan | 0.151 ± 0.024 | 0.027 ± 0.008 | 0.035 ± 0.004 | 0.021 ± 0.002 | 0.023 ± 0.001 | 0.020 ± 0.001 |
| 9e | 2,4-thiophene | 0.072 ± 0.015 | 0.015 ± 0.006 | 0.026 ± 0.005 | 0.012 ± 0.001 | 0.017 ± 0.001 | 0.015 ± 0.001 |
| 9f | 3,5-pyrazol | 0.245 ± 0.032 | 0.100 ± 0.018 | 0.145 ± 0.01 | 0.101 ± 0.02 | 0.101 ± 0.01 | 0.084 ± 0.01 |
| 9g | 2,5-thiazole | 12.5 ± 5.2 | 13.6 ± 3.8 | >10 | >10 | >10 | >10 |
| 9h | 3,5-isoxazole | >30 | >30 | >10 | >10 | >10 | >10 |
| 9i | 1,4-piperidine | >30 | >30 | >20 | >20 | >20 | >20 |
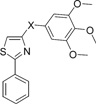
Table 2.
SAR of Alternative to the Carbonyl Linker
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 ± SEM (µM) |
||||||||
| X linker | B16-F1 | A375 | WM-164 | DU 145 | PC-3 | LNCaP | PPC-1 | |
| 1 | C═O | 0.055 ± 0.005 | 0.028 ± 0.005 | 0.064 ± 0.004 | 0.071 ± 0.004 | 0.021 ± 0.001 | 0.028 ± 0.004 | 0.043 ± 0.005 |
| 13a | syn-C═N-OH | 0.3 ± 0.1 | 0.2 ± 0.1 | NDa | 0.103 ± 0.04 | 0.120 ± 0.05 | 0.169 ± 0.06 | 0.144 ± 0.01 |
| 13b | anti-C═N-OH | 11.4 ± 2.1 | 7.8 ± 1.2 | ND | >10 | >10 | >10 | >10 |
| 13c | syn-C═N-OMe | 3.8 ± 1.6 | 2.9 ± 1.2 | 3.4 ± 1.8 | >10 | >10 | >10 | >10 |
| 13d | anti-C═N-OMe | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 13e | CONH | >30 | >30 | ND | >10 | >10 | >10 | >10 |
| 13f | NHCO | >30 | >30 | ND | >10 | >10 | >10 | >10 |
| 14a | syn-C═N-NH2 | 2.0 ± 0.8 | 0.9 ± 0.3 | ND | 1.210 | 1.120 | 1.800 | 0.872 |
| 14b | anti-C═N-NH2 | 1.8 ± 0.7 | 0.6 ± 0.2 | ND | 1.210 | 1.040 | 1.300 | 0.966 |
| 15a | syn-C═C-CN | 5.4 ± 2.1 | 4.6 ± 1.5 | 4.9 ± 1.3 | 2.28 | 0.89 ± 0.34 | 0.58 ± 0.12 | 0.90 ± 0.1 |
| 15b | anti-C═C-CN | 1.2 ± 0.3 | 1.2 ± 0.4 | 1.0 ± 0.2 | ~10 | ~10 | 1.99 | ~10 |
| 16 | C═N-CN | 0.060 ± 0.021 | 0.028 ± 0.012 | 0.027 ± 0.013 | 0.042 ± 0.002 | 0.027 ± 0.001 | 0.023 ± 0.002 | 0.020 ± 0.001 |
| 17 | CHOH | >30 | >30 | ND | >10 | >10 | >10 | >10 |
| 18 | C═CMe2 | 3.8 ± 1.3 | 1.9 ± 0.8 | 3.7 ± 1.2 | 2.65 | 2.47 | 1.39 ± 0.39 | 2.04 |
| 19 | none | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 20 | cis-C═C | 11.0 ± 2.8 | 46.5 ± 23.3 | 10.6 ± 5.8 | >10 | >10 | >10 | >10 |
| 21 | trans-C═C | 32.8 ± 13 | >100 | 30.8 ± 12 | >10 | >10 | >10 | >10 |
| 23 | S | 2.4 ± 0.9 | 1.6 ± 0.4 | 2.0 ± 1.2 | >10 | >10 | 2.3 ± 0.2 | 2.3 ± 0.1 |
| 24 | SO2 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 25 | SO | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| 26 | SONH2 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
ND = not determined.
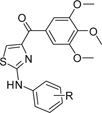
Table 4.
Antiproliferative Activity of Phenyl Amino Thiazole Compounds
 | |||||||
|---|---|---|---|---|---|---|---|
| IC50 ± SEM (µM) |
|||||||
| R | B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |
| 45a | H | 0.065 ± 0.012 | 0.045 ± 0.008 | 0.070 ± 0.004 | 0.057 ± 0.003 | 0.051 ± 0.001 | 0.054 ± 0.001 |
| 46b | 4-CH3 | NDa | ND | 0.035 ± 0.001 | 0.038 ± 0.002 | 0.035 ± 0.001 | 0.036 ± 0.001 |
| 45c | 4-F | ND | ND | 0.063 ±0.001 | 0.043 ± 0.001 | 0.041 ± 0.001 | 0.037 ± 0.001 |
| 1 | 0.055 ± 0.005 | 0.028 ± 0.005 | 0.071 ± 0.004 | 0.021 ± 0.001 | 0.028 ± 0.004 | 0.043 ± 0.005 | |
| 47 | 2.127 ± 0.351 | 1.111 ± 0.108 | 0.839 ±0.719 | 0.786 ± 0.089 | 0.658 ± 0.117 | 0.701 ± 0.307 | |
ND = not determined.
SAR of Alternative “B” Ring Molecules
The first series was targeted to alternatives to the thiazole “B” ring. Accordingly, a series of heterocyclic “B” rings was examined. As shown in Table 1, the successful replacements of the thiazole were pyridine 9c, furan 9d, and thiophene 9e. The IC50s (12–35 nM against prostate cancer cells) are close to the thiazole compound 1. Introducing oxazoline (5), oxazole (6), phenyl (9a), and pyrazole (9f) maintained activity in the hundreds of nanomolar range. But introduction of pyrimidine (9b, IC50 3.7–8.3µM), a reversed 2,5-thiazole or 3,5-isoxazole (9g and 9h, IC50 > 10 µM) caused obvious losses of potency. Modification of “B” ring to the saturated ring of piperidine (9i) also totally abolished activity (9c, IC50 >20 µM).
SAR of Alternative Linkers between “B” and “C” Rings
In vitro hepatic metabolic stability studies revealed that the carbonyl linker between “B” and “C” rings in SMART compounds caused short half-lives (5–17 min) primarily due to carbonyl reduction.16 For the sake of blocking this carbonyl reduction to the inactive hydroxyl linker compound 17, we modified the carbonyl linker in the second series of compounds (Table 2). The carbonyl linker was replaced with double bonds (18, 20, 21), amides (13e, 13f), oximes (13a–13d), hydrazide (14a, 14b), acrylonitriles (15a, 15b), cyanoimine (16), sulfonyl amide (26), sulfur ether (23), and sulfonyl and sulfinyl compounds (24, 25). A direct link compound 19 without any linker between “B” and “C” rings was also prepared. Among these linker modifications, only cyanoimine linkage (16) showed promising activity (20–60 nM) compared with carbonyl compound 1, but an in vitro metabolism study showed that the half-life of 16 in human liver microsome was less than 5 min (data not shown). This result suggested that although we blocked the ketone reduction, it might introduce another new metabolic liability in compound 16. We separated the isomer pairs of compounds containing double bonds, oximes, and hydrazides. Compound 20 was designed to mimic the structure of CA-4, which contain a cis-C═C between two aryl rings, unfortunately 20(Z) and other isomer 21(E) lost activity after replacing the C═O linker. One interesting phenomenon is syn-isomer of 13a (0.1–0.3 µM) showed 10-fold more activity than its anti isomer 13b (>10 µM). The half-life of 13a in human liver microsomes is extended to 35 min, while the half-lives of compounds 14a–b were prolonged to 55 min, however, activities were much lower (~1 µM) than compound 1.
Introducing Polar and Ionizable Groups into the SMART Agents
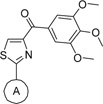
One major limitation of the SMART agents was low aqueous solubility. We used surfactant formulation strategies like Captex200/Tween80 (1/4, IP) to study in vivo SMART behavior and obtained favorable results.23 But these surfactants are biologically active and are responsible for many side effects.10 In addition, it was thought24 that low aqueous solubility of 1 resulted in low oral bioavailability (3.3%, in Table 5). In our third series of compounds, we successfully increased aqueous solubility without impacting the potency by introducing polar groups like hydroxyl and indolyls. In addition, we also designed ionizable groups like amino and alkylamino groups into “A” ring para-position. As shown in Scheme 5 and Table 3, introducing indolyl groups to the “A” ring, especially 5-indolyl (36, 7– 25 nM), increased the potency compared with the 4-OH compound 29 (76–116 nM). Aminomethyl at the “A” ring para position also maintained potency (39, 13–80 nM), but p-NHMe (40) or p-NMe2 (42) abrogated activity. As shown in Figure 3, analytical measurement to estimate aqueous solubility showed that indolyl compound 36 increased solubility in PBS from 1.1 µg/mL (compound 1) to 3.8 µg/mL. Aminomethyl compound 39 was converted to the HCl salt, which increased solubility over 35-fold (>35 µg/mL). Although compound 39 showed satisfactory aqueous solubility, the pharmacokinetic studies showed this compound still had very poor bioavailability (F = 0.2%, Table 5).
Table 5.
Pharmacokinetic Parameters for Compounds Tested in Vivo
| 1 |
39 |
46a |
46c |
|||||
|---|---|---|---|---|---|---|---|---|
| route | IV | PO | IV | PO | IV | PO | IV | PO |
| Na | 4 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| dose (mg/kg) | 2.5 | 10 | 2.5 | 4 | 5 | 10 | 5 | 10 |
| CLb (mL/min/kg) | 7.7 ± 1.0 | 22 ± 13 | 17 ± 3 | 13 ± 2 | ||||
| Vssc (L/kg) | 4.9 ± 1.9 | 0.33 ± 0.25 | 1.4 ± 0.2 | 1.4 ± 0.2 | ||||
| AUCd (min µg/mL) | 279 ± 53 | 37 ± 20 | 139 ± 77 | 0.4 | 296 ± 46 | 65 ± 20 | 381 ± 65 | 160 ± 13 |
| Cmaxe (ng/mL) | 3816 ± 509 | 212 ± 65 | 3794 ± 1580 | 3.2 ± 1.6 | 4198 ± 438 | 814 ± 255 | 3349 ± 686 | 1262 ± 362 |
| Ff (%) | 3.3 | 0.2 | 11 | 21 | ||||
Numbers of rats.
Systemic clearance.
Volume of distribution following intravenous dosing.
Area under the curve following intravenous dosing, integrated drug concentration with respect to time and integrated drug concentration with respect to time following oral dosing.
Maximum plasma concentration following intravenous dosing.
Percent oral bioavailability.
Table 3.
Antiproliferative Activity of Modified Compounds with Improved Aqueous Solubility
 | |||||||
|---|---|---|---|---|---|---|---|
| IC50 ± SEM (µM) |
|||||||
| apart | B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |
| 28 | 4-OTBDMSPh | 0.5 ± 0.2 | 0.7 ± 0.3 | 0.434 ± 0.030 | 0.183 ± 0.024 | 0.549 | 0.246 ± 0.008 |
| 29 | 4-OHPh | 0.11 ± 0.02 | 0.10 ± 0.01 | 0.116 ± 0.014 | 0.087 ± 0.005 | 0.103 ± 0.009 | 0.076 ± 0.002 |
| 32 | 2-indolyl | 0.043 ± 0.021 | 0.019 ± 0.009 | 0.032 ± 0.001 | 0.024 ± 0.004 | 0.028 ± 0.003 | 0.028 ± 0.002 |
| 36 | 5-indolyl | 0.025 ± 0.013 | 0.008 ± 0.001 | 0.013 ± 0.001 | 0.007 ± 0.001 | 0.010 ± 0.001 | 0.008 ± 0.001 |
| 38 | 4-BocNHCH2Ph | 2.9 ± 0.4 | 7.9 ± 0.5 | 4.3 ± 3.7 | 3.1 ± 1.7 | 2.6 ± 0.9 | 2.7 ± 1.5 |
| 39 | 4-NH2CH2Ph | 0.038 ± 0.011 | 0.041 ± 0.013 | 0.025 ± 0.001 | 0.080 ± 0.007 | 0.013 ± 0.001 | 0.034 ± 0.001 |
| 40 | 4-NHMeCH2Ph | >10 | >10 | ~10 | >10 | 1.14 ± 0.08 | ~10 |
| 42 | 4-NMe2CH2Ph | >10 | >10 | >10 | >10 | 1.025 ± 0.2 | >10 |
Figure 3.

Aqueous solubility of novel antitubulin compounds. Compounds 45a and 45c showed improved solubility compared with compound 1. Compound 39 is soluble at 35 µg/mL concentration.
Modifications to Improve Oral Bioavailability
Many established tubulin targeting anticancer drugs like taxanes and vinblastine require intravenous administration because of low oral bioavailability. Oral bioavailability is a complex parameter involving many chemical and physiological processes, such as solubility, permeability and metabolic stability. Our initial thought was that the low bioavailability of 1 might be caused by poor solubility. However, the water-soluble compound 39 failed to improve oral bioavailability. We further improved the solubility of these tubulin inhibitors by inserting an amino linker between the “A” and “B” rings (phenyl amino thiazole, PAT) as in 45a–c (Scheme 6), which is similar to the reported orally active colchicine binding anticancer agent 47 (Figure 1, clinical phase II trial).3,25 IC50 values (Table 4) demonstrate that these compounds (45a, 46b, and 45c) had similar potency (35–65 nM) as 1 with increased solubility (15 and 19 µg/mL for 45a and 45c, respectively (Figure 3), and they are over 20-fold more active than 47.
Rat pharmacokinetic studies were performed to study whether these new compounds exhibited improved bioavailability compared to compound 1 (Table 5). The data clearly showed that 46c (HCl salt of 45c) exhibited more than 4.3-fold increased exposure (AUC, 160 vs 37 min·µg/mL) by the oral route as compared to 1, suggesting that improved aqueous solubility by the amino linker successfully improved oral bioavailability. In addition, the maximal concentration (Cmax) of 46a and 46c by oral administration were 814 and 1262 ng/mL, respectively, while Cmax of 1 was only 212 ng/mL. Overall, the bioavailability of 46a and 46c were increased from 3.3% of 1 to 11% and 21%, respectively (Table 5). Compound 46c exhibited moderate clearance, moderate volume of distribution, and acceptable oral bioavailability. This data suggested that these new synthesized amino linked compounds have appropriate potency and PK profiles to be developed as a new class of orally bioavailable anticancer agents.
Compounds Inhibit in Vitro Tubulin Polymerization
We investigated the inhibition of tubulin polymerization of selected potent compounds 9c, 16, 36, and 45a from all three design strategies (alternative B-rings, novel linkers, and solubilizing moieties) and compared them with 1. Bovine brain tubulin (>97% pure) was incubated with the individual compounds (5 µM) to test their effect on tubulin polymerization (Figure 4). After 20 min incubation, tubulin polymerization was inhibited 45% by 1, as compared to vehicle. Compound 16 inhibited 33% of polymerization at 20 min with different inhibition patterns. Compounds 9c and 45a provided similar inhibitions of 56% and 58%, respectively. Compound 36, which showed low average IC50 of 10.1 nM, inhibited 69% of tubulin polymerization. These data suggest that these compounds exhibit strong antitubulin polymerization activity that corresponds well with their antiproliferative potency.
Figure 4.

Compounds inhibit tubulin polymerization in vitro.
Compounds Overcome P-Glycoprotein Mediated Multidrug Resistance
The P-glycoprotein (P-gp) system appears to be a primary physiological mechanism of multidrug resistance (MDR) which acts as an ATP-dependent drug efflux pump, actively removing a variety of structurally diverse cytotoxic compounds. 8,26 Enhanced efflux of these compounds reduces their intracellular accumulation and so reduces their cytotoxicity. Therefore, novel compounds which are not susceptible to drug resistance could be of high therapeutic and economic value. In addition to P-gp, clinically used antitubulin agents have other resistance mechanisms such as changes in microtubule dynamics 27 and mutations in β-tubulin which are known to limit sensitivity to the taxanes. 28 We tested our selected compounds against an ovarian cancer cell line OVCAR-8 (parent) and P-gp overexpressing NCI/ADR-RES cell line (Table 6). Notably, tested compounds demonstrated equipotent antiproliferative effects against OVCAR-8 and NCI/ADR-RES cell lines, suggesting that they are not P-gp substrates and that they function in a P-gp-independent manner. This feature is distinct from that of paclitaxel, vinblastine, and colchicine in NCI/ADR-RES cells which demonstrate 1333-, 149-, and 65-fold resistance.
Table 6.
Antiproliferative Activity of Selected Compounds Against P-gp Overexpressed MDR Cell Lines
| IC50 (nM) |
|||
|---|---|---|---|
| compd | OVCAR-8 | NCI/ADR-RES | resistance factor |
| 9c | 33 ± 3 | 13 ± 0.8 | 0.4 |
| 16 | 34 ± 2 | 14 ± 1 | 0.4 |
| 36 | 10 ± 3 | 4 ± 2 | 0.4 |
| 39 | 26 ± 2 | 11 ± 2 | 0.4 |
| 45a | 46 ± 6 | 27 | 0.6 |
| 45b | 28 | 21 | 0.8 |
| 45c | 44 ± 3 | 25 ± 6 | 0.6 |
| 1 | 35 ± 2 | 13 ± 1 | 0.4 |
| paclitaxel23 | 4.7 ± 0.1 | 6263 ± 634 | 1333 |
| vinblastine | 3.9 ± 0.1 | 582 ± 57 | 149 |
| colchicine | 17 ± 1 | 1113 ± 79 | 65 |
CONCLUSION
A new series of tubulin polymerization inhibitors with acceptable oral bioavailability and equipotent activity in multidrug resistant tumor cell lines has been discovered. Medicinal chemistry efforts improved upon SMART compound 1. Chemical modifications were include alternative “B” ring and alternative linkages between “B” and “C” rings with regard to in vitro cytotoxicity against cancer cells (Figure 5) based on biological evaluation against cancer cells in vitro. SAR studies revealed that optimal “B” rings include pyridine (9c), thiophene (9e), and furan (9d), which maintain excellent in vitro potency. Replacing carbonyl linker with cyanoimine (16) between “B” and “C” ring also increased the activity. Structure modifications to increase aqueous solubility and bioavailability were performed. Introducing an amino between “A” and “B” rings gave us PAT compounds 45a–c, which showed similar in vitro antiproliferative potency against tested cancer cells as well as resistant cancer cell lines, furthermore, the solubility and in vivo bioavailability were improved greatly over those of 1. Therefore, these new compounds represent a new family of antimitotic agents that may be very useful in the treatment of cancer.
Figure 5.

Structure–activity relationship of novel anti-tubulin compounds.
EXPERIMENTAL SECTION
General
All reagents were purchased from Sigma-Aldrich Chemical Co., Fisher Scientific (Pittsburgh, PA), AK Scientific (Mountain View, CA), Oakwood Products (West Columbia, SC), etc. and were used without further purification. Moisture-sensitive reactions were carried under an argon atmosphere. 47 was prepared according methods reported by Yoshino et al.29 Routine thin layer chromatography (TLC) was performed on aluminum backed Uniplates (Analtech, Newark, DE). Melting points were measured with Fisher-Johns melting point apparatus (uncorrected). NMR spectra were obtained on a Bruker AX 300 (Billerica, MA) spectrometer or Varian Inova-500 (Vernon Hills, Illinois) spectrometer. Chemical shifts are reported as parts per million (ppm) relative to TMS in CDCl3. Mass spectral data was collected on a Bruker ESQUIRE electrospray/ion trap instrument in positive and negative ion modes. Elemental analyses were performed by Atlantic Microlab Inc. (Norcross, GA). Unless specified, all the tested compounds described in the article present >95% purity established through combustion analysis.
(2R)-(2-Phenyl-4,5-dihydro-oxazol-4-yl)-(3,4,5-trimethoxy-phenyl)-methanone (5)
To a solution of n-BuLi (1.6 M, 0.713 mL) in 8 mL of THF was added a solution of 3,4,5-trimethoxybromobenzene (1.09 mmol) in 3 mL of THF under −78 °C. The mixture was allowed to stir for 2 h, and a solution of Weinreb amide 4 (1.14 mmol) in 3 mL of THF was charged. The temperature was allowed to increase at RT and stirred overnight. The reaction mixture was quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 5 as a white solid (47.9%); mp 60–62 °C. 1H NMR (CDCl3) δ 7.97 –7.94 (m, 2 H), 7.62 (s, 2 H), 7.54–7.37 (m, 3 H), 5.61 (q, 1H, J = 7.5 Hz, 9.9 Hz), 5.12 (t, 1 H, J = 7.5 Hz), 4.57 (q, 1 H, J = 7.8 Hz, 9.9 Hz), 3.96 (s, 6 H), 3.95 (s, 3 H). MS (ESI) m/z 364.1(M + Na)+, 340.1 (M − H)−. Anal. (C19H19NO4S) C, H, N.
(2R)-(2-Phenyl-oxazol-4-yl)-(3,4,5-trimethoxy-phenyl)-methanone (6)
A mixture of 5 (1.48 mmol), CBrCl3 (2.59 mmol), and DBU (2.97 mmol) in CH2Cl2 (20 mL) was stirred overnight. The reaction mixture was absorbed on silica gel and purified by column chromatography to yield pure compound 6 as desired (61.6%); mp 138–139 C 1H NMR (CDCl3) δ 8.37 (s, 1 H), 8.14–8.12 (m, 2 H), 7.74 (s, 2 H), 7.52–7.49 (m, 3 H), 3.97 (s, 9 H). MS (ESI) m/z 362.1 (M + Na)+. Anal. (C19H17NO5) C, H, N.
Biphenyl-3-yl(3,4,5-trimethoxyphenyl)methanone (9a)
To a solution of 8a (0.174 g, 0.72 mmoL) in 5 mL of THF was added a THF solution of 3,4,5-trimethoxyphenylmagnesiumbromide (0.5 N, 1.08 mmol) at 0 °C. The mixture was allowed to stir for 30 min and quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 9a as a white solid (43.8%); mp 87–89 °C. 1HNMR (CDCl3) δ 8.02 (t, 1 H), 7.84–7.74 (m, 2 H), 7.64–7.38 (m, 6 H), 7.11 (s, 2 H), 3.95 (s, 3 H), 3.88 (s, 6 H). MS (ESI) m/z 371.1 (M + Na)+. Anal. (C22H20O4) C, H, N.
(6-Phenylpyrimidin-4-yl)(3,4,5-trimethoxyphenyl)methanone (9b)
To a solution of 8b (0.243 g, 1 mmoL) in 5 mL of THF was added a THF solution of 3,4,5-trimethoxyphenylmagnesiumbromide (0.5 N, 5.6 mL, 1.4 mmol) at 0 °C. The mixture was allowed to stir for 30 min and quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 9b (52.3%); mp 132–133 °C. 1H NMR (CDCl3) δ 9.40 (d, 1 H, J = 1.5 Hz), 8.29 (d, 1 H, J = 1.5 Hz), 8.22–8.18,7.57–7.54 (m, 5 H), 7.46 (s, 2 H), 3.96 (s, 3 H), 3.91 (s, 6 H). MS (ESI) m/z 351.1 (M + H)+. Anal. (C20H18N2O4) C, H, N.
(6-Phenylpyridin-2-yl)(3,4,5-trimethoxyphenyl)methanone (9c)
To a solution of 8c (0.210 g, 0.86 mmoL) in 5 mL of THF was added a THF solution of 3,4,5-trimethoxyphenylmagnesiumbromide (0.5 N, 3.5 mL, 1.73 mmol) at 0 °C. The mixture was allowed to stir for 30 min and quenched with water, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 9c as white needle crystals (78%); mp 116–117°C 1H NMR (CDCl3) δ 8.10 (d, br, 2 H), 8.02–8.00 (m, 1 H), 7.97–7.96 (m, 2 H), 7.66 (s, 2 H), 7.49–7.43 (m, 3 H), 3.97 (s, 3 H), 3.89 (s, 6 H). MS (ESI) m/z 372.6 (M + Na)+. Anal. (C21H19NO4) C, H, N.
(5-Phenylfuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (9d)
To a solution of 8d (0.231 g, 1 mmoL) in 5 mL of THF was added a THF solution of 3,4,5-trimethoxyphenylmagnesiumbromide (0.5 N, 4.0 mL, 2 mmol) at 0 °C. The mixture was allowed to stir for 30 min and quenched with water, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 9d as white crystals (35.5%); mp 114–116 °C. 1H NMR (CDCl3) δ 7.85–7.82 (m, 1 H), 7.48–7.36 (m, 4 H), 7.35 (s, 2 H), 7.25 (d, 1 H, J = 4.0 Hz), 6.86 (d, 1 H, J = 4.2 Hz), 3.96 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 339.1 (M + H)+. Anal. (C20H18O5) C, H.
(5-Phenylthiophen-3-yl)(3,4,5-trimethoxyphenyl)methanone (9e)
To a solution of 10e (0.260 g, 0.73 mmoL) in 20 mL of anhydrous CH2Cl2 was added Dess–Martin reagent (0.465 g, 1.36 mmol). The mixture was allowed to stir for 30 min and quenched with satd Na2S2O3 solution, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to give pure compound 9e as light-yellow crystals (81.0%); mp 140–141 °C. 1H NMR (CDCl3) δ 7.97 (d, 1 H, J = 1.5 Hz), 7.82 (d, 1 H, J = 1.5 Hz), 7.59–7.57 (m, 2 H), 7.45–7.34 (m, 3 H), 7.19 (s, 2 H), 3.95 (s, 3 H), 3.93 (s, 6 H). MS (ESI) m/z 355.1 (M + H)+. Anal. (C20H18O4S C, H.
(3-Phenyl-1H–pyrazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (9f)
Compound 9f was prepared using the same method as used of compound 9c from 3-phenyl-1H-pyrazole-5-carboxylic acid 7f via 8f as intermidiate. 1H NMR (500M, CDCl3) δ 10.97 (br, 1 H), 7.77 (s, br, 2 H), 7.48–7.38 (m, 5 H), 7.14 (s, br, 1 H), 3.96 (s, 3 H), 3.94 (s, 6 H). MS (ESI) m/z 361.1 (M + Na)+ , 337.0 (M − H)−. Anal. (C19H18 N2O4) C, H, N.
(2-Phenylthiazol-5-yl)(3,4,5-trimethoxyphenyl)methanone (9g)
To a solution of 10g (0.357 g, 1 mmoL) in 40 mL of anhydrous CH2Cl2 was added Dess–Martin reagent (0.848 g, 2 mmol). The mixture was allowed to stir for 30 min and quenched with satd Na2S2O3 solution, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to give pure compound 9 g (80.1%); mp 147–148 °C. 1H NMR (CDCl3) δ 8.33 (s, 1 H), 8.04 (m, 2 H), 7.51 (m, 3 H), 7.18 (s, 2 H), 3.96 (s, 3 H), 3.93 (s, 6 H). MS (ESI) m/z 378.1 (M + H)+. Anal. (C19H17NO4S) C, H, N.
(5-Phenylisoxazol-3-yl)(3,4,5-trimethoxyphenyl)methanone (9h)
To a solution of 10h (0.110 g, 0.73 mmoL) in 8 mL of anhydrous CH2Cl2 was added Dess–Martin reagent (0.274 g, 0.645 mmol). The mixture was allowed to stir for 30 min and quenched with satd Na2S2O3 solution, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to give pure compound 9h (70.1%); mp 143–144 °C. 1H NMR (CDCl3) δ 7.87–7.85 (m, 2 H), 7.72 (s, 2 H), 7.53–7.49 (m, 3 H), 7.05 (s, 1 H), 7.82 (d, 1 H, J = 1.5 Hz), 3.97 (s, 3 H), 3.96 (s, 6 H). MS (ESI) m/z 362.1 (M + H).+ Anal. (C19H17NO5) C, H, N.
(4-Phenylpiperidin-1-yl)(3,4,5-trimethoxyphenyl)methanone (9i)
To a mixture of 4-phenylpiperidine 11 (5 mmol), EDCI (6 mmol), HOBt (5.5 mmol), and NMM (6 mmol) in CH2Cl2 (50 mL) was added 3,4,5-trimethoxybenzoic acid (5.3 mmol) and stirring continued at RT for overnight. The reaction mixture was diluted with CH2Cl2 (100 mL) and sequentially washed with water, satd NaHCO3, and brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 9i. (57.9%); mp 141 – 142 °C. 1H NMR (CDCl3) δ 7.35–7.21(m, 5 H), 6.66 (s, 2 H), 4.84 (br, 1 H), 3.95 (br, 1 H), 3.88 (s, 6 H), 3.86 (s, 3 H), 3.20–2.87 (br, 2 H), 2.85–2.74 (tt, 1 H, J = 3.6 Hz, J = 15.6 Hz) 1.92 (br, 2 H), 1.70 (br, 2 H). MS (ESI) m/z 378.1 (M + Na).+ Anal. (C21H25NO4) C, H, N.
(Z)-(2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methanoneOxime (13a)
To a suspension of 1 (210 mg, 0.59 mmol) in 10 mL of ethanol was added an aqueous solution (2 mL) of hydroxylamine hydrochloride (127 mg, 1.83 mmol). Then 2 mL of 1N NaOH was added dropwise to the reaction mixture and the mixture was stirred at 55 °C for 3 h. After completion of the reaction, the residue was absorbed on silica gel and purified by column chromatography to give compounds 13a (85 mg) and 13b (50 mg); mp 150–152 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.95 (s, 1 H), 8.35 (s, 1 H), 7.91–7.89 (m, 2 H), 7.50–7.44 (br, 3 H), 6.85 (s, 2 H), 3.73 (s, 6 H), 3.70 (s, 3 H). MS (ESI) m/z 393.1 (M + Na)+; 368.9 (M − H)−. Anal. (C19H18N2O4S) C, H, N.
(E)-(2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (13b)
Melting point: 176–177 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.49 (s, 1 H), 7.92–7.89 (m, 2 H), 7.64 (s, 1 H), 7.51–7.49 (m, 3 H), 7.34 (s, 1 H), 6.75 (s, 2 H), 3.75 (s, 6 H), 3.72 (s, 3 H). MS (ESI) m/z 393.1 (M + Na)+ ; 368.9 (M − H)−. Anal. (C19H18-N2O4S) C, H, N.
(Z)-(2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone O-Methyl Oxime (13c)
To a suspension of 1 (110 mg, 0.59 mmol) in 10 mL of pyridine was added O-methylhydroxylamine hydrochloride (52 mg, 0.63 mmol) and the mixture was stirred at 60 °C for overnight. The reaction was quenched with 1 N HCl solution, extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to give pure compounds 13c (41 mg) and 13d (33 mg); mp 116–117 °C. 1H NMR (500 MHz, CDCl3) δ 8.13 (s, 1 H), 7.96–7.94 (m, 2 H), 7.45–7.44 (m, 3 H), 6.94 (s, 2 H), 4.13 (s, 3 H), 3.91 (s, 6 H), 3.88 (s, 3 H). MS (ESI) m/z 407.2 (M + Na)+. Anal. (C20H20N2O4) C, H, N.
(E)-(2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone O-Methyl Oxime (13d)
Melting point: 91–92 °C. 1H NMR (500 MHz, CDCl3) δ 8.00–7.98 (m, 2 H), 7.44–7.43 (m, 3 H), 7.28 (s, 1 H), 6.70 (s, 2 H), 4.08 (s, 3 H), 3.91 (s, 6 H), 3.85 (s, 3 H). MS (ESI) m/z 407.0 (M + Na)+. Anal. (C20H20N2O4) C, H, N.
2-Phenyl-N-(3,4,5-trimethoxyphenyl)thiazole-4-carboxamide (13e)
To a solution of 13a (21 mg, 0.06 mmol) in 5 mL of CH2Cl2 was added p-toluenesulfonyl chloride (23 mg, 0.12 mmol) and NaH (5 mg, 60% in light mineral oil). Then the reaction mixture was stirred for 20 min. After completion of the reaction, the residue was absorbed on silica gel and purified by Al2O3 column chromatography to give compound 13e (15 mg); mp 157–158 °C. 1H NMR (300 MHz, CDCl3) δ 9.22 (s, 1 H), 8.19 (s, 1 H), 8.02–7.99 (m, 2 H), 7.52–7.50 (m, 3 H), 7.07 (s, 2 H), 3.92 (s, 6 H), 3.85 (s, 3 H). MS (ESI) m/z 371.1 (M + H)+. Anal. (C19H18N2O4S) C, H, N.
3,4,5-Trimethoxy-N-(2-phenylthiazol-4-yl)benzamide (13f)
To a solution of 13b (26 mg, 0.07 mmol) in 5 mL of CH2Cl2 was added p-toluenesulfonyl chloride (27 mg, 0.14 mmol) and NaH (5 mg, 60% in light mineral oil). Then the reaction mixture was stirred for 20 min. After completion of the reaction, the residue was absorbed on silica gel and purified by Al2O3 column chromatography to give compound 13f (15 mg); mp 154–156 °C. 1H NMR (300 MHz, CDCl3) δ 8.88 (s, 1 H), 7.94–7.91 (m, 2 H), 7.83 (s, 1 H), 7.48–7.46 (m, 3 H), 7.18 (s, 2 H), 3.97 (s, 6 H), 3.94 (s, 3 H). MS (ESI) m/z 393.1 (M + Na)+. Anal. (C19H18N2O4S) C, H, N.
(Z)-4-(Hydrazono(3,4,5-trimethoxyphenyl)methyl)-2-phenylthiazole (14a)
To a mixture of 1 (230 mg, 0.65 mmol) in 3 mL of CH2Cl2 and 3 mL of ethanol was added hydrazine hydrate (2 mL). Then the mixture was refluxed for overnight. After completion of the reaction, the residue was absorbed on silica gel and purified by column chromatography to give compounds 14a (80 mg) and 14b (56 mg); mp 117–119°C 1H NMR (300 MHz, CDCl3) δ 8.01–7.98 (m, 2 H), 7.49–7.46 (m, 5 H), 7.33 (s, 1 H), 6.82 (s, 2 H), 3.87 (s, 3 H), 3.85 (s, 6 H). MS (ESI) m/z 370.1 (M + H)+. Anal. (C19H19N3O3S) C, H, N.
(E)-4-(Hydrazono(3,4,5-trimethoxyphenyl)methyl)-2-phenylthiazole (14b)
Melting point: 65–66 °C. 1H NMR (300 MHz, CDCl3) δ 8.04–8.01 (m, 2 H), 7.44–7.40 (m, 3 H), 6.95 (s, 1 H), 6.65 (s, 2 H), 5.62 (s, 2 H), 3.93 (s, 3 H), 3.87 (s, 6 H). MS (ESI) m/z 370.1 (M + H)+. Anal. (C19H19N3O3S) C, H, N.
(Z)-3-(2-Phenylthiazol-4-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile (15a)
To a solution of 0.4 mL of 2.5 N n-BuLi in hexane and 10 mL of THF was added dropwise a solution of 177 mg (1 mmol) of diethyl cyanomethylphosphonate in 5 mL of THF at 0 °C under Ar2. The ice bath was removed, and the mixture was stirred at 25 °C for 40 min. A solution of 200 mg (0.56 mmol) of 1 in 10 mL of THF was added dropwise at 0 °C, and the mixture was stirred for 1 h at RT. The reaction mixture was treated with saturated NH4C1 solution. After a conventional workup, column chromatography (silica gel, petroleum ether/ ethyl acetate) gave compounds 15a (83 mg) and 15b (76 mg); mp:192–193 °C. 1H NMR (300 MHz, CDCl3) δ 8.01–7.99 (m, 2 H), 7.44–7.40 (m, 3 H), 7.21 (s, 1 H), 6.74 (s, 2 H), 6.67 (s, 1 H), 3.93 (s, 3 H), 3.89 (s, 6 H). MS (ESI) m/z 401.1 (M + Na)+. Anal. (C21H18N2O3S) C, H, N.
(E)-3-(2-Phenylthiazol-4-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile (15b)
Melting point: 111 – 114 °C. 1H NMR (300 MHz, CDCl3) δ 8.07–8.05 (m, 2 H), 7.49–7.46 (m, 4 H), 6.66 (s, 2 H), 5.64 (s, 1 H), 3.91 (s, 3 H), 3.86 (s, 6 H). MS (ESI) m/z 401.1 (M + Na)+. Anal. (C21H18N2O3S) C, H, N.
N-((2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methylene)cyanamide (16)
First, 100 mg of 1 (0.28 mmol, 1 equiv) was dissolved in 10 mL of methylene chloride. Then titanium tetrachloride in methylene chloride (1.0 N, 0.7 mL, 2.5 equiv) was added dropwise at 0 °C and stirred for 30 min. Bis-trimethylsilylcarbodiimide (2.4 equiv) in 2 mL of methylene chloride was added and the reaction stirred overnight protected from air and moisture. The reaction was treated with ice–water mixture followed by extraction with methylene chloride. The organic phase was dried over magnesium sulfate, filtered through Celite, and concentrated to give the crude acetophenone cyanoimines, which were purified by flash column as isomers (35 mg) with a ratio of 3:7. 1H NMR (300 MHz, CDCl3) δ 8.72 (br, 0.3 H), 8.63 (s, 0.7 H), 8.09–8.07 (m, 1.4 H), 7.99 (br, 0.6 H), 7.58–7.56 (br, 3 H), 7.26 (s, 1.4 H), 7.18 (s, 0.6 H), 3.84, 3.83 (s, s, 6 H), 3.82 (s, 3 H). MS (ESI) m/z 402.1 (M + Na)+. Anal. (C20H17N3O3S) C, H, N.
(2-Phenylthiazol-4-yl)(3,4,5-trimethoxyphenyl)methanol (17)
At 0 °C, to a solution of 104 mg of 12b (0.55 mmol, 1 equiv) in 6 mL of THF was added 3,4,5-trimethoxyphenylmagnesium bromide (0.5 N in THF, 2.9 mL). The mixtures were stirred for 30 min until aldehyde disappeared on TLC plates. The reaction mixture was quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 17; mp 49–51 °C. 1H NMR (300 MHz, CDCl3) δ 7.95–7.92 (m, 2 H), 7.44–7.43 (m, 4 H), 6.97 (s, 1 H), 6.76 (s, 2 H), 5.93 (d, 1 H, J = 3.6 Hz), 3.86 (s, 9 H). MS (ESI) m/z 402.1 (M + Na)+. Anal. (C19H19NO4S) C, H, N.
4-(2-Methyl-1-(3,4,5-trimethoxyphenyl)prop-1-enyl)-2-phenylthia-zole (18)
At −78 °C, to a solution of 223 mg of isopropyl triphenylphosphonium iodide (0.52 mmol) in 5 mL of THF was added dropwise 0.4 mL of 1.6 N n-BuLi in hexane under Ar2 protection. And the mixture was stirred at 0 °C for 40 min. A solution of 140 mg (0.39 mmol) of 1 in 5 mL of THF was added dropwise at 0 °C. The mixture was stirred for 1 h at RT. The reaction mixture was treated with saturated NH4C1 solution. After a conventional workup, column chromatography (silica gel, petroleum ether/ethyl acetate) gave compound 18 (86 mg, 57.3%). 1H NMR (300 MHz, CDCl3) δ 7.98–7.97 (m, 2 H), 7.45–7.40 (m, 3 H), 6.77 (s, 1 H), 6.48 (s, 2 H), 3.86 (s, 3 H), 3.82 (s, 6 H), 2.15 (s, 3 H), 1.81 (s, 3 H). MS (ESI) m/z 404.1 (M + Na)+. Anal. (C22H23NO3S) C, H, N.
2-Phenyl-4-(3,4,5-trimethoxyphenyl)thiazole (19)
Bromine (160 mg, 1 mmol) was added dropwise to a stirred solution of 1-(3,4,5-trimethoxyphenyl)ethanone (210 mg, 1 mmol) in ethanol (30 mL), and the solution was stirred at 0 °C for 1 h and then poured into water to form a precipitate. This was recrystallized from ethanol to give bromoacetophenone (70%) and used directly for the next step. A mixture of bromoacetophenone (288 mg, 1 mmol) and benzothioamide (137 mg, 1 mmol) in ethanol was refluxed for 1 h. The reaction mixture was concentrated in vacuo and purified with a flash column to give 19 (167 mg, 51.1%); mp: 95–96 °C. 1H NMR (500 MHz, CDCl3) δ 8.05–8.03 (m, 2 H), 7.48–7.44 (m, 3 H), 7.41 (s, 1 H), 7.22 (s, 2 H), 3.97 (s, 6 H), 3.89 (s, 3 H). MS (ESI) m/z 350.1 (M + Na)+. Anal. (C18H17NO3S) C, H, N.
(Z)-2-Phenyl-4-(3,4,5-trimethoxystyryl)thiazole (20)
Triphenylphosphine (3.41 g, 13 mmol) was added to a solution of 5-(bromomethyl)-1,2,3-trimethoxybenzene (2.61 g, 10 mmol) in dry THF (30 mL). The mixture was refluxed with stirring for 6 h. The resulting white solid was filtered and washed with ether/hexane to afford the product 3,4,5-trimethoxybenzyltriphenylphosphonium bromide in 96.4% yield. 1H NMR (500MHz, CDCl3) δ 7.77–7.73, 7.65–7.61 (m, 15 H), 6.44 (d, 2 H, J = 1.5 Hz), 5.37 (d, 2 H, J = 14 Hz), 3.76 (s, 3 H), 3.51 (d, 6 H). MS (ES m/z 443.1 (M − Br]+. At −78°C, n-BuLi (0.42 mL, 2.5 N in hexane) was added to a solution of 3,4,5-trimethoxybenzyltriphenyl- phosphonium bromide (500 mg, 0.96 mmol) in 10 mL of THF. After stirring at RT for 2 h, aldehyde 12b (109 mg, 0.58 mmol) in 3 mL of THF was charged and stirred for 30 min. The reaction mixture was treated with saturated NH4C1 solution. After a conventional workup, column chromatography (silica gel, petroleum ether/ethyl acetate) gave compounds 20(Z) (57 mg and 21(E) (99 mg. 1H NMR (500 MHz, CDCl3) δ 7.90–7.89 (m, 2 H), 7.42–7.40 (m, 3 H), 7.07 (s, 1 H), 6.71 (s, 2 H), 6.66 (s, 1 H), 3.87 (s, 6 H), 3.75 (s, 3 H). MS (ESI) m/z 376.1 (M + Na)+. Anal. (C20H19NO3S C, H, N.
(E)-2-Phenyl-4-(3,4,5-trimethoxystyryl)thiazole (21)
1H NMR (500 MHz, CDCl3) δ 8.03–8.01 (m, 2 H), 7.52 (d, 1 H, J = 16 Hz), 7.47–7.44 (m, 3 H), 7.16 (s, 1 H), 7.05 (d, 1 H, J = 16 Hz), 6.79 (s, 2 H), 3.92 (s, 6 H), 3.88 (s, 3 H). MS (ESI) m/z 354.1 (M + H)+. Anal. (C20H19NO3S) C, H, N.
S-Biphenyl-3-yl-O-Ethyl-Carbonodithioate (22)
To a solution of 1 equiv of biphenyl-3-amine (1 g, 5.92 mmol) in water (7.3 mL) at 0 °C was added concentrated hydrochloric acid (1 mL). A cold solution of 1.1 equiv of sodium nitrite (450 mg, 6.5 mmol) in water (3 mL) was added slowly and stirred for 15 min. The cold diazonium solution was added slowly to a solution of 1.3 equiv of potassium ethyl xanthate (1.16 g, 1.3 mmol) in water (1.3 mL) at 45 °C. The reaction mixture was stirred for an additional 30 min at 45 °C and then cooled to RT. The reaction mixture was extracted with diethyl ether (3 × 50 mL). The combined organic extracts were washed with 1 N NaOH solution (100 mL), water (3 × 50 mL), brine (50 mL), dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting crude xanthate 22 was used directly in the next step without further purification. MS (ESI) m/z 275.0 (M + H)+.
Biphenyl-3-yl(3,4,5-trimethoxyphenyl)sulfane (23)
To a solution of 22 (1.1 g, crude compound) in ethanol (8 mL) was added potassium hydroxide (2.1 g, 12 mL) and heated to reflux for overnight. The solution was cooled to RT, and the ethanol was evaporated under reduced pressure. The residue was dissolved in water and washed with diethyl ether (10 mL). The aqueous layer was acidified with 2 N HCl and extracted with diethyl ether (3 × 50 mL). The organic extracts were washed with water (50 mL), brine (50 mL), dried over MgSO4, filtered, and evaporated under reduced pressure to afford 0.85 g (77.3%) of crude biphenyl-3-thiol product (overall, 3 steps). Into a round-bottomed flask, stirred magnetically, were placed 0.1 g (1.04 mmol) of sodium tert-butoxide and 83 mg of copper iodide (0.43 mmol). After the reaction vessel was sealed, 0.13 g (0.71 mmol) of 4-methoxybenzenethiol and 0.19 g (0.65 mmol) of 5-iodo-1,2,3-trimethoxybenzene in 3.0 mL of toluene were injected through the septum. The reaction mixture was heated for overnight at 110 °C. Purification was performed by flash chromatography and colorless oil 23 was obtained (40% yield). 1H NMR (500 MHz, CDCl3) δ 7.54–7.52 (m, 3 H), 7.44–7.41 (m, 3 H), 7.37–7.33 (m, 2 H), 7.23 (s, br, 1 H), 6.69 (s, 2 H), 3.86 (s, 3 H), 3.80 (s, 6 H). MS (ESI) m/z 353.2 (M + H)+. Anal. (C21H20O3S) C, H.
3-(3,4,5-Trimethoxyphenylsulfonyl)biphenyl (24)
To a solution of 60 mg (0.17 mmol) of compound 23 and 5 mL of dichloromethane was added very slowly 2 equiv of m-CPBA over 3 h. Sulfoxide formation was monitored by thin-layer chromatography. Purification was performed with a flash chromatographic column, and an amorphous powder of 24 was obtained (73% yield); mp 99–101 °C. 1H NMR (500 MHz, CDCl3) δ 8.14 (br, 1 H), 7.89 (d, 1 H), 7.78 (d, 1 H), 7.59–7.56 (m, 3 H), 7.49–7.39 (m, 3 H), 7.19 (s, 2 H), 3.89 (s, 6 H), 3.87 (s, 3 H). MS (ESI) m/z 385.0 (M + Na)+. Anal. (C21H20O5S) C, H.
3-(3,4,5-Trimethoxyphenylsulfinyl)biphenyl (25)
At 0 °C, to a solution of 500 mg (1.42 mmol) of compound 23 and 5 mL of dichloromethane was added very slowly 1 equiv of m-CPBA over 3 h. Sulfoxide formation was monitored by thin-layer chromatography. Purification was performed with a flash chromatographic column, and an amorphous powder of 25 was obtained (87% yield); mp 108–109 °C. 1H NMR (500 MHz, CDCl3) δ 7.92 (br, 1 H), 7.71 (d, 2 H), 7.62–7.60 (m, 3 H), 7.58–7.40 (m, 4 H), 6.94 (s, 2 H), 3.79 (s, 3 H), 3.74 (s, 6 H). MS (ESI) m/z 369.1 (M + H)+. Anal. (C21H20O4S) C, H.
N-(3,4,5-Trimethoxyphenyl)biphenyl-3-sulfinamide (26)
A mixture of 65 mg of biphenyl-3-sulfonyl chloride (0.25 mmol), 44 mg of 3,4,5-trimethoxyaniline (0.24 mmol), and 0.3 mmol of triethylamine in 5 mL of DMF was stirred overnight. The reaction mixture was treated with water and extracted with ethyl acetate. After a conventional workup, column chromatography (silica gel, petroleum ether/ethyl acetate) gave 88 mg of 26 (91.7%); mp 48–50 °C. 1H NMR (500 MHz, CDCl3) δ 7.96 (t, 1 H, J = 1.8 Hz), 7.81–7.74 (m, 2 H), 7.57–7.40 (m, 6 H), 6.33 (s, 2 H), 3.86 (s, 3 H), 3.80 (s, 6 H). MS (ESI) m/z 422.1 (M + Na)+. Anal. (C21H21NO5S) C, H, N.
(2-(4-Hydroxyphenyl)thiazol-4-yl)(3,4,5-trimethoxyphenyl)metha-none (29)
At 0 °C, to a solution of 28 (0.2 mmol) in 5 mL of CH2Cl2 was added a solution of tetrabutylammonium fluoride in THF (1 N, 0.6 mmol) and stirred at RT for around 14 h until reaction was finished by TLC monitor; 67.0% yield. 1H NMR (500 MHz, DMSO-d6) δ 10.1 (s, 1 H), 8.51 (s, 1 H), 7.85 (d, 2 H, J = 8.50 Hz), 7.62 (s, 2 H), 6.91 (d, 2 H, J = 8.5 Hz), 3.86 (s, 6 H), 3.79 (s, 3 H). MS (ESI) m/z 394.1 (M + Na)+ , 369.9 (M − H)−. Anal. (C19H17FNO5S) C, H, N.
(2-(1H–Indol-2-yl)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (32) Was Synthesized from 31 Using the Same Method As Used for 5
Yield 45.8%. 1H NMR (500 MHz, DMSO-d6) δ 9.26 (s, 1 H), 8.11 (s, 1 H), 7.66 (d, 1 H, J = 8.0 Hz), 7.46 (s, 2 H), 7.42 (d, 1 H, J = 8.0 Hz), 7.29 (t, 1 H, J = 7.5 Hz), 7.16 (t, 1 H, J = 7.5 Hz), 7.10 (s, 1 H), 3.97 (s, 3 H), 3.93 (s, 6 H). MS (ESI) m/z417.1 (M+ Na)+ , 392.9 (M − H)−. Anal. (C21H18N2O4S) C, H, N.
(2-(1H–Indol-5-yl)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (36)
To a solution of n-BuLi (1.6M, 1.7 mL) in 8 mL of THF was added a solution of 3,4,5-trimethoxybromobenzene (2.47 mmol) in 3 mL of THF under −78 °C. The mixture was allowed to stir for 2 h, and a solution of Weinreb amide 35(1.24 mmol) in 3 mL of THF was charged. The temperature was allowed to increase at RT and stirred overnight. The reaction mixture was quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was refluxed in 1 N NaOH in 5 mL of ethanol solution to obtain the deprotected compound 36 and purified by column chromatography to obtain pure compound as a light-yellow solid (36.3%); mp 162–164 °C 1H NMR (300M, CDCl3) δ 8.36 (br, s, 1 H), 8.31 (s, 1 H), 8.21 (s, 1 H), 7.92, 7.89 (dd, 1 H, J = 1.8, 2.7 Hz), 7.46 (d, 1 H), 7.62 (s, 2 H, J = 8.7 Hz), 7.29 (t, 1 H, J = 2.7 Hz), 6.64 (br, 1 H), 3.97 (s, 6 H), 3.97 (s, 3 H). MS (ESI) m/z 417.1 (M + Na)+, 392.9 (M − H)−. Anal. (C21H18-N2O4S) C, H, N.
tert-Butyl4-(4-(3,4,5-Trimethoxybenzoyl)thiazol-2-yl)benzylcarbamate (38)
A mixture of 37 (2.5 mmol), CBrCl3 (3.2 mmol), and DBU (5.0 mmol) in CH2Cl2 (20 mL) was stirred overnight. The reaction mixture was absorbed on silica gel and purified by column chromatography to yield an intermediate thiazole Weinreb amide. To a solution of (3,4,5-trimethoxyphenyl)magnesium bromide (0.5 M, 5.5 mL) in THF was added a solution of the intermediate thiazole Weinreb amide (1.83 mmol) in 10 mL of THF under 0 °C and stirred for 30 min. The reaction mixture was quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound as a light-yellow solid (32.3%); mp 123–124 °C. 1H NMR (300M, CDCl3) δ 8.27 (s, 1 H), 7.98 (d, 2 H, J = 8.1 Hz), 7.78 (s, 2 H), 7.39 (d, 2 H, J = 8.1 Hz), 4.93 (br, 1 H), 4.37 (br, d, 2 H), 3.96 (s, 3 H), 3.95 (s, 6 H), 1.47 (s, 9 H). MS (ESI) m/z 507.1 (M + Na)+. Anal. (C25H28N2O6S) C, H, N.
(2-(4-(Aminomethyl)phenyl)thiazol-4-yl)(3,4,5-trimethoxyphenyl)-methanone Hydrochloride (39)
At 0 °C, to a solution of 38 (200 mg) in 10 mL of CH2Cl2 was added a solution of HCl in 1,4-dioxane (4 N, 2 mL) and stirred at RT for 4 h. The precipitate (39) was filtered and washed with diethyl ether. Yield 81.3%; mp 200–203 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.68 (s, 1 H), 8.38 (br, 3 H), 8.10 (d, 2 H, J = 8.4 Hz), 7.66 (d, 2 H, J = 8.4 Hz), 7.62 (s, 2 H), 4.11 (s, 2 H), 3.87 (s, 6 H), 3.80 (s, 3 H). MS (ESI) m/z 385.1 (M + H)+. Anal. (C20H20N2O4S · HCl) C, H, N, Cl.
(2-(4-((Methylamino)methyl)phenyl)thiazol-4-yl)(3,4,5-trimeth-oxyphenyl)methanone Hydrochloride (40)
At 0 °C, to a solution of 41 (60 mg) in 5 mL of CH2Cl2 was added a solution of HCl in 1,4-dioxane (4 N, 2 mL) and stirred at RT for overnight. The precipitate (40) was filtered and washed with diethyl ether. Yield 81.3%; mp 197–200°C. 1H NMR (500MHz, CDCl3) δ 10.0 (s, 1 H), 8.29 (s, 1 H), 8.05 (d, 2 H, J = 6.0 Hz), 7.74 (s, 2 H), 7.72 (d, 2 H, J = 6.0 Hz), 4.15 (s, 2 H), 3.99 (s, 3 H), 3.96 (s, 6 H), 2.61 (s, 3 H). MS (ESI) m/z 399.1 (M + H)+.; Anal. (C21H22N2O4S·HCl·H2O) C, H, N.
(2-(4-((Dimethylamino)methyl)phenyl)thiazol-4-yl)(3,4,5-trimeth-oxyphenyl)methanone Hydrochloride (42)
To a solution of 39 (53 mg, 0.14 mmol) in 5 mL of CH2Cl2 was added formaldehyde solution (37% in H2O, 340 mg, 4.2 mmol) and sodium cyanoborohydride (34 mg, 0.55 mmol), the reaction mixture was absorbed on silica gel, and free base was purified after flash column (41 mg, 70.9%). At 0 °C, to a solution of free base (41 mg) in 5mL of CH2Cl2 was added a solution of HCl in 1, 4-dioxane (4 N, 2 mL) and stirred at RT for overnight. The precipitate (42) was filtered and washed with diethyl ether. Yield 71.3%; mp 94–96 °C. 1H NMR (500 MHz, CDCl3) δ 13.0 (s, 1 H), 8.34 (s, 1 H), 8.13 (d, 2 H, J = 7.0 Hz), 7.82 (d, 2 H, J = 7.5 Hz), 7.75 (s, 2 H), 4.24 (s, 2 H), 3.99 (s, 3 H), 3.97 (s, 6 H), 2.83 (s, 6 H). MS (ESI) m/z 413.1 (M + H)+. Anal. (C22H24N2O4S·HCl) C, H, N.
General Procedure for the Synthesis of (2-(Arylamino)-thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanones (45a–c)
At −78 °C, to a solution of 5-bromo-1,2,3-trimethoxybenzene (1.235 g, 5.0 mmol) in 30 mL of THF was charged n-BuLi in hexane (2.5 N, 2.4 mL, 6 mmol) under Ar2 protection and stirred for 10 min. Weinreb amide 44a–c (1 mmol) in 10 mL of THF was added to the lithium reagent and allowed to stir at RT for 2 h. The reaction mixture was quenched with satd NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 45a–c.
(2-(Phenylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (45a)
Yield 33.3%; mp 149–151 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.4 (s, 1 H), 7.85 (s, 1 H), 7.68 (d, 2 H, J = 8.0 Hz), 7.31 (t, 2 H, J = 8.0 Hz), 6.98 (t, 1 H, J = 8.0 Hz), 3.83 (s, 6 H), 3.78 (s, 3 H). MS (ESI) m/z 393.1 (M + H)+, 368.9 (M − H)−. Anal. (C19H18N2O4S) C, H, N.
(2-(p-Tolylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (45b)
Yield 40.6%; mp 139–140 °C. 1H NMR (500 MHz, CDCl3) δ 7.48 (s, 1 H), 7.47 (s, 2 H), 7.30 (br, 1 H), 7.27 (d, 2 H, J = 8.5 Hz), 7.17 (d, 2 H, J = 8.5 Hz), 3.93 (s, 3 H). 3.90 (s, 6 H), 2.34 (s, 3 H). MS (ESI) m/z 385.1 (M + H)+ , 382.9 (M − H)−. Anal. (C20H20-N2O4S) C, H, N.
(2-(p-Fluorophenylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)-methanone (45c)
Yield 39.6%; mp 129–130 °C. 1H NMR (500 MHz, CDCl3) δ 7.52 (br, 1 H), 7.49 (s, 1 H), 7.45 (s, 2 H), 7.40–7.37 (q, 2 H, J = 4.5 Hz), 7.08–7.04 (t, 2 H, J = 8.0 Hz), 3.93 (s, 3 H), 3.89 (s, 6 H). MS (ESI) m/z 389.3 (M + H)+, 386.9 (M − H)−. Anal. (C19H17-FN2O4S) C, H, N.
General Procedure for the Synthesis of Hydrochloride Salts (46a–c)
At 0°C, to a solution of compound 45a–c (0.1 mmol) in 5 mL of CH2Cl2 was added a solution of HCl in 1,4-dioxane (4 N, 2 mL) and stirred at RT for overnight. The precipitates 46a–c were collected and washed with diethyl ether.
(2-(Phenylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone Hydrochloride Salt (46a)
Yield 91.6%; mp 94–96 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.9 (br, 1 H), 7.49–7.46 (m, 2 H), 7.42–7.40 (m, 2 H), 7.37–7.34 (m, br, 2 H), 7.11 (s, 2 H), 3.94 (s, 3 H), 3.92 (s, 6 H), 3.57 (br, H2O). MS (ESI) m/z 389.1 (M + H)+. Anal. (C19H18N2O4S • HCl · H2O) C, H, N.
(2-(p-Tolylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone Hydrochloride Salt (46b)
Yield 39.6%;mp 115– 118°C. 1H NMR (500 MHz, CDCl3) δ 7.30–7.25 (m, br, 6 H), 7.12 (s, 2 H), 3.94 (s, 3 H), 3.92 (s, 6 H), 2.38 (s, 3 H). MS (ESI) m/z 389.1 (M + H)+. Anal. (C20H20N2O4S·2 HCl) C, H, N.
(2-(p-Fluorophenylamino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)-methanone Hydrochloride Salt (46c)
Yield 89.3%; mp 102–104 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.55 (s, 1 H), 7.85 (s, 1 H), 7.72–7.69 (q, 2 H, J = 4.5 Hz), 7.50 (s, 2 H), 7.18–7.15 (t, 2 H, J = 8.5 Hz), 4.30 (br, H2O), 3.82 (s, 6 H), 3.78 (s, 3 H). MS (ESI) m/z 389.3 (M + H)+. Anal. (C19H17FN2O4S· 1.5 HCl·0.5 H2O) C, H, N.
Cell Culture and Cytotoxicity Assay of Prostate Cancer and Melanoma
All cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), while cell culture supplies were purchased from Cellgro Mediatech (Herndon, VA, USA). We examined the antiproliferative activity of our antitubulin compounds in four human prostate cancer cell lines (LNCaP, DU 145, PC-3, and PPC-1) and three melanoma cell lines (A375, B16–F1 and WM-164). Human ovarian cell line OVCAR-8 and its resistant cell line that overexpresses P-gp (NCI/ADR-RES) were used as MDR models. Both ovarian cell lines were obtained from National Cancer Institutes (NCI). All cell lines were tested and authenticated by either ATCC or NCI. All prostate cancer and ovarian cancer cell lines were cultured in RPMI 1640 and supplemented with 10% fetal bovine serum (FBS). Melanoma cells were cultured in DMEM, supplemented with 5% FBS, 1% antibiotic/ antimycotic mixture (Sigma-Aldrich, Inc., St. Louis, MO, USA), and bovine insulin (5 µg/mL; Sigma-Aldrich). The cytotoxic potential of the antitubulin compounds was evaluated using the sulforhodamine B (SRB) assay after 96 h of treatment.
Aqueous Solubility
The solubility of drugs was determined by a Multiscreen solubility filter plate (Millipore Corporate, Billerica, MA) coupled with LC-MS/MS. Briefly, 198 µL of phosphate buffered saline (PBS) buffer (pH 7.4) was loaded into 96-well plate, and 2 µL of 10 mM test compounds (in DMSO) was dispensed and mixed with gentle shaking (200–300 rpm) for 1.5 h at RT (N = 3). The plate was centrifuged at 800g for 5 min, and the filtrate was used to determine its concentration and solubility of test compound by LC-MS/MS as described below. Concentrations of tested compounds were determined with individual calibration curve with satisfied linearity (r > 0.995).
Pharmacokinetic Study
Female Sprague–Dawley rats (n = 3 or 4; 254 ± 4 g) were purchased from Harlan Inc. (Indianapolis, IN). Rat thoracic jugular vein catheters were purchased from Braintree Scientific Inc. (Braintree, MA). On arrival at the animal facility, the animals were acclimated for 3 days in a temperature-controlled room (20–22 °C) with a 12 h light/dark cycle before any treatment. Compound 1 was administered intravenously (IV) into the jugular vein catheters at a dose of 2.5 mg/kg (in DMSO/PEG300, 2/8), whereas 46a and 46c were dosed at 5 mg/kg (in DMSO/PEG300, 1/9). An equal volume of heparinized saline was injected to replace the removed blood, and blood samples (250 µL) were collected via the jugular vein catheters at 10, 20, 30 min, and 1, 2, 4, 8, 12, 24 h. Compounds 1, 46a, and 46c were given (PO) by oral gavage at 10 mg/kg (in Tween80/DMSO/H2O, 2/1/7). All blood samples (250 µL) after oral administration were collected via the jugular vein catheters at 30, 60, 90, 120, 150, 180, 210, 240 min, and 8, 12, 24 h. Heparinized syringes and vials were prepared prior to blood collection. Plasma samples were prepared by centrifuging the blood samples at 8000g for 5 min. All plasma samples were stored immediately at −80 °C until analyzed.
Analytes were extracted from 100 µL of plasma with 200 µL of acetonitrile containing 200 nM of the internal standard ((3,5-dimethoxyphenyl)(2-phenyl-1H–imidazol-4-yl)methanone). The samples were thoroughly mixed, centrifuged, and the organic extract was transferred to autosampler for LC-MS/MS analysis. Multiple reaction monitoring (MRM) mode, scanning m/z 356 → 188 (compound 1), m/z 371 → 203 (compound 46a), m/z 389 → 221 (compound 46c), and m/z 309 → 171 (the internal standard), was used to obtain the most sensitive signals. The pharmacokinetic parameters were determined using noncompartmental analysis (WinNonlin, Pharsight Corporation, Mountain View, CA)
Analytical Method
Sample solution (10µL) was injected into an Agilent series HPLC system (Agilent 1100 Series Agilent 1100 Chem-station, Agilent Technology Co, Ltd.). All analytes were separated on a narrow-bore C18 column (Alltech Alltima HP, 2.1 mm × 100 mm, 3 µm, Fisher, Fair Lawn, NJ). Two gradient modes were used. Gradient mode was used to achieve the separation of analytes using mixtures of mobile phase A [ACN/H2O (5%/95%, v/v) containing 0.1% formic acid] and mobile phase B [ACN/H2O (95%/5%, v/v) containing 0.1% formic acid] at a flow rate of 300 µL/min. Mobile phase A was used at 15% from 0 to 1 min, followed by a linearly programmed gradient to 100% of mobile phase B within 6 min, 100% of mobile phase B was maintained for 0.5 min before a quick ramp to 15% mobile phase A. Mobile phase A was continued for another 12 min toward the end of analysis.
A triple-quadruple mass spectrometer, API Qtrap 4000 (Applied Biosystems/MDS SCIEX, Concord, Ontario, Canada), operating with a TurboIonSpray source was used. The spraying needle voltage was set at 5 kV for positive mode. Curtain gas was set at 10; gas 1 and gas 2 were set 50. Collision-assisted dissociation (CAD) gas at medium and the source heater probe temperature at 500 °C. Data acquisition and quantitative processing were accomplished using Analyst software, Ver. 1.4.1 (Applied Biosystems).
In Vitro Tubulin Polymerization Assay
Bovine brain tubulin (0.4 mg, >97% pure) (Cytoskeleton, Denver, CO) was mixed with 5 µm of the test compounds and incubated in 100µL of general tubulin buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP) at pH 6.9. The absorbance of wavelength at 340 nm was monitored every 1 min for 20 min by the SYNERGY 4 Microplate Reader (Bio-Tek Instruments, Winooski, VT). The spectrophotometer was set at 37 °C for tubulin polymerization.
Supplementary Material
Acknowledgments
This research was supported by funds from Department of Pharmaceutical Sciences, College of Pharmacy, and University of Tennessee Health Science Center; the Van Vleet Endowed Professorship; USPHS grants 1R15CA125623 and 1RO1CA-148706. We thank Terrence A. Costello, Katie N. Kail, and Stacey L. Barnett for providing technical support for animal studies at GTx, Inc.
ABBREVIATIONS USED
- ABC
ATP binding cassette
- Boc2O
tert-butyl dicarbonate
- CA-4
combretastatin A-4
- DMSO
dimethyl sulfoxide
- MDR
multidrug resistance
- MRM
Multiple reaction monitoring
- NMM
N-methylmorpholine
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- PDC
pyridinium dichromate
- P-gp
P-glycoprotein
- PAT
phenylamino-thiazole
- PK
pharmacokinetic
- RT
Room temperature
- SAR
Structure-activity relationship
- SMART
4-substituted methoxybenzoyl-arylthiazole
- TBAF
tetrabutylammonium fluoride
- TBDMS
tert-butyldimethylsilyl
- THF
tetrahydrofuran
- TMP
3,4,5-tri-methoxyphenyl
Footnotes
ASSOCIATED CONTENT
Supporting Information. 2D NOESY NMR and 1D NOE NMR to identify syn/anti-isomers of hydrazides 14a–14b and acrylonitriles 15a–15b, synthesis, mass, and NMR of intermediates 1–4, 8a–e, 10e–g, 12a, 12b, 27, 30, 31, 34, 35, 37, 38, 41, 43a–c, 44a–c, and quantum chemical shifts calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Kiselyov A, Balakin KV, Tkachenko SE, Savchuk NI, vachtchenko AV. Recent progress in discovery and development of antimitotic agents. Anticancer Agents Med. Chem. 2007;7(2):189–208. doi: 10.2174/187152007780058650. [DOI] [PubMed] [Google Scholar]
- 2.Luo Y, Hradil VP, Frost DJ, Rosenberg SH, Gordon GB, Morgan SJ, Gagne GD, Cox BF, Tahir SK, Fox GB. ABT-751, a novel tubulin-binding agent, decreases tumor perfusion and disrupts tumor vasculature. Anticancer Drugs. 2009;20(6):483–492. doi: 10.1097/CAD.0b013e32832c0acf. [DOI] [PubMed] [Google Scholar]
- 3.Mauer AM, Cohen EE, Ma PC, Kozloff MF, Schwartzberg L, Coates AI, Qian J, Hagey AE, Gordon GB. A phase II study of ABT-751 in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2008;3(6):631–636. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]
- 4.Rustin GJ, Shreeves G, Nathan PD, Gaya A, Ganesan TS, Wang D, Boxall J, Poupard L, Chaplin DJ, Stratford MR, Balkissoon J, Zweifel M. A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Br. J. Cancer. 2010;102(9):1355–1360. doi: 10.1038/sj.bjc.6605650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green H, Rosenberg P, Soderkvist P, Horvath G, Peterson C. beta-Tubulin mutations in ovarian cancer using single strand conformation analysis-risk of false positive results from paraffin embedded tissues. Cancer Lett. 2006;236(1):148–154. doi: 10.1016/j.canlet.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Cabral F. Paclitaxel resistance in cells with reduced beta-tubulin. Biochim. Biophys. Acta, Mol. Cell Res. 2005;1744(2):245–255. doi: 10.1016/j.bbamcr.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Leslie EM, Deeley RG, Cole SP. C Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005;204(3):216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman MM, Pastan I. The multidrug transporter, a double-edged sword. J. Biol. Chem. 1988;263(25):12163–12166. [PubMed] [Google Scholar]
- 9.Fisher GA, Sikic BI. Clinical studies with modulators of multidrug resistance. Hematol./Oncol. Clin. North Am. 1995;9(2):363–382. [PubMed] [Google Scholar]
- 10.Hennenfent KL, Govindan R. Novel formulations of taxanes: a review Old wine in a new bottle? Ann. Oncol. 2006;17(5):735–749. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 11.ten Tije AJ, Verweij J, Loos WJ, Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin. Pharmacokinet. 2003;42(7):665–685. doi: 10.2165/00003088-200342070-00005. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Zhao W, Li C-ML, Chen J, Dalton JT, Li W, Miller DD. Discovery of 4-substituted methoxybenzoyl-arylthiazole as novel anticancer agents: synthesis, biological evaluation structure-activity relationships. J. Med. Chem. 2009;52(6):1701–1711. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.At-Haddou H, Hoarau O, Cramailere D, Pezet F, Daran JC, Balavoine GG. New dihydroxy bis(oxazoline) ligands for the palladium-catalyzed asymmetric allylic alkylation: experimental investigations of the origin of the reversal of the enantioselectivity. Chemistry. 2004;10(3):699–707. doi: 10.1002/chem.200204649. [DOI] [PubMed] [Google Scholar]
- 14.Williams DR, Lowder PD, Gu Y-G, Brooks DA. Studies of mild dehydrogenations in heterocyclic systems. Tetrahedron Lett. 1997;38(3):331–334. [Google Scholar]
- 15.Dess DB, Martin JC. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983;48(22):4155–4156. [Google Scholar]
- 16.Li CM, Lu Y, Narayanan R, Miller DD, Dalton JT. Drug Metabolism and Pharmacokinetics of 4-Substituted Methoxyben-zoyl-aryl-thiazoles (SMART) Drug Metab. Dispos. 2010;38(11):2032–2039. doi: 10.1124/dmd.110.034348. [DOI] [PubMed] [Google Scholar]
- 17.Lu Y, Wang Z, Li CM, Chen J, Dalton JT, Li W, Miller DD. Synthesis, in vitro structure-activity relationship, and in vivo studies of 2-arylthiazolidine-4-carboxylic acid amides as anticancer agents. Bioorg. Med. Chem. 2010;18(2):477–495. doi: 10.1016/j.bmc.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DD. Ph.D Dissertation. Seattle, WA: University of Washington; 1969. Tricyclic norephedrine analogs. Isomeric 9-hydroxy-10-amino-1,2,3,4,4a,9,10,10a-(trans-4a,10a)octahydrophenanthrenes. [DOI] [PubMed] [Google Scholar]
- 19.Park SU, Chung SK, Newcomb MA. Acceptor, donor, and captodative stabilization in transition states of 5-hexen-1-yl radical cyclizations. J. Am. Chem. Soc. 1986;108(2):240–244. [Google Scholar]
- 20.Cuccia SJ, Fleming LB, France DJ. A novel and efficient synthesis of 4-phenyl-2-chloropyrimidines from acetophenone cyano-imines. Synth. Commun. 2002;32(19):3011–3018. [Google Scholar]
- 21.Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, Davies P, Chiosis G. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J. Med. Chem. 2005;48(8):2892–2905. doi: 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 22.Pletnev AA, Tian Q, Larock RC. Carbopalladation of nitriles: synthesis of 2,3-diarylindenones and polycyclic aromatic ketones by the Pd-catalyzed annulation of alkynes and bicyclic alkenes by 2-iodoarenenitriles. J. Org. Chem. 2002;67(26):9276–9287. doi: 10.1021/jo026178g. [DOI] [PubMed] [Google Scholar]
- 23.Li CM, Wang Z, Lu Y, Ahn S, Narayanan R, Kearbey JD, Parke DN, Li W, Miller DD, Dalton JT. Biological activity of 4-substituted methoxybenzoyl-arylthiazole: an active microtubule in-hibitor. Cancer Res. 2011;71(1):216–224. doi: 10.1158/0008-5472.CAN-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Wang Z, Li CM, Lu Y, Vaddady PK, Meibohm B, Dalton JT, Miller DD, Li W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J. Med. Chem. 2010;53(20):7414–7427. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox E, Maris JM, Widemann BC, Meek K, Goodwin A, Goodspeed W, Kromplewski M, Fouts ME, Medina D, Cho SY, Cohn SL, Krivoshik A, Hagey AE, Adamson PC, Balis FM. A phase 1 study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 7 days every 21 days in pediatric patients with solid tumors. Clin. Cancer Res. 2006;12(16):4882–4887. doi: 10.1158/1078-0432.CCR-06-0534. [DOI] [PubMed] [Google Scholar]
- 26.Mickisch GH, Licht T, Merlino GT, Gottesman MM, Pastan I. Chemotherapy and chemosensitization of transgenic mice which express the human multidrug resistance gene in bone marrow: efficacy, potency, and toxicity. Cancer Res. 1991;51(19):5417–5424. [PubMed] [Google Scholar]
- 27.Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, Wilson L, Jordan MA. Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proc. Natl. Acad. Sci. U.S.A. 2001;98(20):11737–11742. doi: 10.1073/pnas.191388598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monzo M, Rosell R, Sanchez JJ, Lee JS, O’Brate A, Gonzalez-Larriba JL, Alberola V, Lorenzo JC, Nunez L, Ro JY, Martin C. Paclitaxel resistance in non-small-cell lung cancer associated with beta-tubulin gene mutations. J. Clin. Oncol. 1999;17(6):1786–1793. doi: 10.1200/JCO.1999.17.6.1786. [DOI] [PubMed] [Google Scholar]
- 29.Yoshino H, Ueda N, Niijima J, Sugumi H, Kotake Y, Koyanagi N, Yoshimatsu K, Asada M, Watanabe T. Novel sulfon-amides as potential, systemically active antitumor agents. J. Med. Chem. 1992;35(13):2496–2497. doi: 10.1021/jm00091a018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.