

Abstract
This study aimed to enhance the water solubility and antitumor efficacy of (-)-gossypol. Polyethyleneimine conjugated with stearic acid (PgS) was used for loading and protecting (-)-gossypol through hydrogen bonding. Double-layered hyaluronic acid (HA)-modified PgS nanoparticles encapsulating (-)-gossypol [(-)-G-PgSHAs] were prepared through a two-step fabrication process. The nanoparticles possessed a uniform spherical shape with a dynamic size of 110.9 ± 2.4 nm, which was determined through transmission electron microscopy and dynamic light scattering analysis. The encapsulation efficiency and drug-loading capacity of (-)-G-PgSHAs were 72.6% ± 3.1% and 9.1% ± 0.42%, respectively. The IR spectra of the samples confirmed the protection effect of hydrogen bonding on the optical activity of the encapsulated (-)-gossypol. (-)-G-PgSHAs exhibited a controlled and tumor-specific release because of the high expression of HAase in the tumor region. The tumor-targeting feature of PgSHAs due to HA-receptor mediation was confirmed by in vitro cell uptake and in vivo near infrared fluorescence imaging. The in vitro test showed that the (-)-G-PgSHAs had similar cytotoxicity to free (-)-gossypol and was smaller than that of the encapsulated (±)-gossypol [(±)-G-PgSHAs]. The in vivo study of the anti-cancer effect of (-)-G-PgSHAs revealed that (-)-G-PgSHAs had a more enhanced tumor-suppression effect and reduced systemic toxicity compared with free (-)-gossypol and (±)-G-PgSHAs (P < 0.05). Therefore, PgSHA was a useful (-)-gossypol nanocarrier that exhibits high biocompatibility, tunable release of drug, and tumor-targeting characteristics for cancer treatment. In addition, this double-layered nanocarrier provided novel strategies for the encapsulation of other chiral drugs.
Introduction
Optical activity is an essential property of chiral drugs. Various chiral drugs have already been investigated and used as their racemic mixtures for clinical applications.1, 2 However, the two enantiomers of a chiral drug generally have diverse pharmacodynamic, pharmacogenetic and pharmacokinetic properties, and one of the enantiomers of the racemic drug may be pharmacologically inactive or even toxic.2, 3 Therefore, using the appropriate enantiomer of a drug is crucial in clinical therapies. Gossypol, a natural compound investigated as a promising anticancer drug, is an important chiral drug. This drug has been studied in many clinical trials in its racemic form.4-6 However, as gossypol is water-insoluble, oral administration is so far still the best treatment for cancer using this drug.
Like many other chiral drugs, notable differences were found in the metabolism between (+)- and (-)-gossypol, and only (-)-gossypol has been reported to have significant antitumor effect, because of its binding affinity to Bcl-2, Bcl-xL, and Mcl-1 proteins that are rational therapeutic targets with pathway regulation functions, leading to tumor cell apoptosis.7-9 Unfortunately, (-)-gossypol is not stable and can easily racemize in the body, which largely reduces its administrated dosage concentration, resulting in unsatisfactory drug efficacy and unwanted side-effects. Thus, its poor stability, and low water-solubility, have become the major problems in (-)-gossypol treatment. In order to solve these problems, one suitable method is the encapsulation of (-)-gossypol using nanocarriers, which increases its water-solubility for injection (e.g. intravenous injection), protects it from its environment, as well as reduces its side-effects by achieving tumor-targeting through the enhanced permeability and retention (EPR) effects and/or the incorporation of tumor-targeted moieties. Although multiple studies for gossypol antitumor effects and their mechanism have been reported, only a few studies on the gossypol nanocarriers are available. Cho et al. reported encapsulated gossypol with micelles comprising of PEG-PCl polymers that loaded paclitaxel, cyclopamine and gossypol together.10 Zhai et al. prepared gossypol liposomes to prove the low gossypol toxicity, but no in vivo study was involved.11 Furthermore, the gossypol carrier studies mentioned above only involved the racemic gossypol, which consists of both (+)-gossypol and (-)-gossypol.
To date, very few of the nanocarriers can meet the requirements of encapsulating chiral drugs. Ideally, nanocarriers for chiral drug should have adequate loading capacity, biocompatibility, and more importantly, the ability to protect the drug from racemization during the encapsulation in vitro and the delivery in vivo. Gossypol is a polyphenol with two naphthalene rings which can relatively rotate on their carbon-carbon bond (Scheme 1A), thus forming (+)- and (-)-gossypol. The delivery of (-)-gossypol is limited by the fact that the two active aldehyde groups in the (-)-gossypol structure can easily react with the surrounding materials in the solution. Once the reaction occurs, relative rotation of its two naphthalene rings may happen, resulting in the racemate product, which was observed in our previous experiments. Therefore, developing a suitable nanocarrier that encapsulates (-)-gossypol with a high drug loading efficiency and without racemization is still a challenge for the clinical application of (-)-gossypol.
Scheme 1.
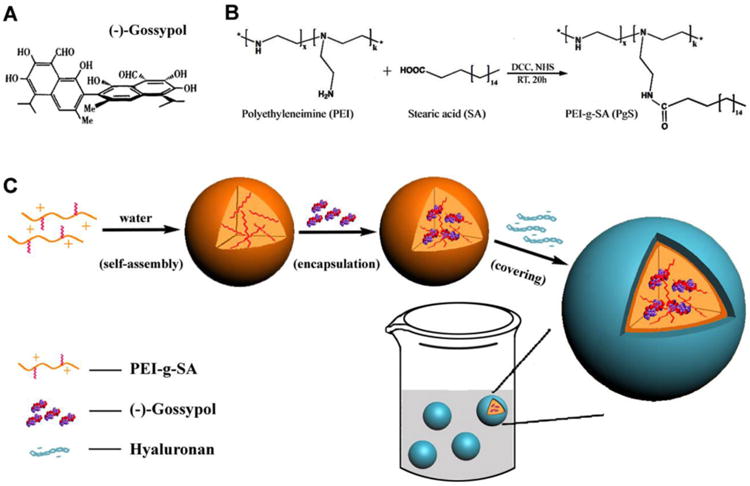
(A) Chemical structure of (-)-gossypol. (B) Chemical reaction equation of PgS synthesis; (C) Schematic representation of the fabrication process of (-)-G-PgSHAs.
As shown in Scheme 1A, (-)-gossypol has two naphthalene rings, whose relative rotation can lead to racemization of the drug. The chemical structure of (-)-gossypol indicated that, proper ‘pins’ are needed within the nanocarrier to restrain it from racemizing. Without such ‘pins’, the naphthalene rings of the (-)-gossypol can rotate freely. On the contrary, if the ‘pins’ are too strong, they may change the bioactivity of (-)-gossypol. The double-stranded DNA can maintain its secondary structure and bioactivity through hydrogen bonds, which suggests that, hydrogen bonds may function as the ‘pins’, because (-)-gossypol has eight hydroxyl groups that have the potential to form hydrogen bonds with suitable loading materials. Therefore we hypothesized that the hydrogen bonding between (-)-gossypol and its nanocarrier could avoid the racemization and keep the bioactivity of (-)-gossypol.
After many investigations, we finally found that PEI could be a good candidate for (-)-gossypol delivery. PEI is a water-soluble polymer that contains numerous primary, secondary, and tertiary amino groups, thus it can form hydrogen bonds with the hydroxyl groups of (-)-gossypol. PEI with a high molecular weight has been used to deliver various bioactive molecules, such as DNA plasmid, siRNA, and anti-sense oligodeoxynucleotide (ODN).12-14 Water-insoluble drugs can be loaded by the micelles, which were formed using modulated PEI amphiphilic materials. For example, Yim et al. synthesized amphiphilic PEI-g-all-trans-retinoic acid (PRA), which could self-assemble into micelles that encapsulate the hydrophobic anticancer drug paclitaxel. Furthermore, the cytotoxicity of PRA graft conjugates was lower than that of raw PEI, and gradually decreased with increasing PRA graft ratios.15
In addition, as a natural polysaccharide found in the extracellular matrix and synovial fluids of the body, hyaluronic acid (HA)-based nanoparticles (HA-NPs) as potential carriers of anticancer drugs have drawn substantial attention because they can specifically bind to various cancer cells that overexpress CD44 (an HA receptor) with high affinity for HA.16, 17 Some normal cells can also express CD44 at low levels. But CD44 expressed by normal cells are usually in a low affinity state that does not have the capacity to bind HA.17 Thus HA was widely used as the carrier for the targeting therapy of cancer.18-20
Based on the investigations mentioned above, we designed a novel injectable double-layered HA/PEI nanoparticles (NPs) for the delivery of (-)-gossypol. First, a PEI amphiphile, which consisted of a low-molecular weight (10 kDa) branched PEI grafted with hydrophobic stearic acid (PEI-g-SA, annotated as PgS), was synthesized (Scheme 1B). This amphiphilic PEI conjugate could reduce PEI toxicity and easily form micelles that encapsulate (-)-gossypol during the self-assembly process (Scheme 1C), and it could maintain the optical activity of (-)- gossypol. Afterwards, positively charged PgS micelles were covered with a shell of negatively charged HA via electrostatic interaction, forming double-layered HA/PEI nanoparticles [(-)-G-PgSHAs]. This kind of nanoparticle had the ability to maintain the optical activity of (-)-gossypol, low toxicity, and high antitumor therapeutic effect in vitro and in vivo, thereby serving as a useful antitumor drug carrier for cancer treatment.
Experimental section
Reagents
Cell culture medium [Dulbecco's modified Eagle medium (DMEM)], trypsin (for cell culture use, 0.25% w/v), and six-well dishes were purchased from Thermo Fisher Scientific Co., Ltd. (Shanghai, China). Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biological Technology Co., Ltd. The 10 cm cell culture dishes, 96-well plates, and other biological experimental consumables were purchased from Corning Co., Ltd. Hyaluronidase from bovine testes (HAase, 683.6 units mg-1) and (-)-gossypol with [α]D -350° to -365° (c 0.3 in CDCl3) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Branched PEI (MW 10 kDa), stearic acid (SA), N-hydroxysuccinimde (NHS), N, N-dicyclohexyl-carbodiimide (DCC), HA (MW > 100 kDa, sodium salt), penicillin/streptomycin, and rhodamine B isothiocyanate (RITC) were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Thiazolyl blue tetrazolium bromide (MTT) was purchased from Amresco, Inc. (Solon, USA). 1,1-dioctadecyl-3,3,3,3-tetramethylindotri-carbocyanine iodide (DiR) was purchased from Invitrogen (NY, USA). Other reagents were of analytical grade. All chemicals and reagents were used without further purification.
Synthesis of PgS conjugates
We grafted SA to PEI through condensation reaction between the carboxylic acid of SA and the amino groups of PEI (Scheme 1B). Three kinds of PgS conjugates with different degrees of SA substitution were prepared. SA (0.7 mmol), NHS (0.72 mmol), and DCC (0.72 mmol) were dissolved in 30 mL of absolute ethanol and the solution was stirred thoroughly for 30 min. Then, 10 mL absolute ethanol, containing 0.056, 0.092, and 0.144 mmol PEI was added into the above solution respectively, and were mixed and stirred at room temperature to start the condensation reaction. After 20 h, the reaction solution was transferred into a dialysis membrane of 3500 Da MWCO, and was dialyzed against absolute ethanol for 3 days during which the medium was routinely replaced with fresh absolute ethanol every 12 h. Finally, the solvent was evaporated to obtain the PgS conjugates. The degree of SA substitution in the three different PgS conjugates (annotated as PgS1, PgS2 and PgS3) was then measured using high-performance liquid chromatography (HPLC) analysis system (Agilent 1100, U.S.A) equipped with Agilent G1316A capillary pump, Agilent G1316A thermostatted column compartment, and Agilent G1315B diode detector. The flow rate of mobile phase [methanol/water (92:8, v/v)] was 0.8 mL min-1 at 25 °C. The UV absorbance of SA was measured at 225 nm.
The chemical structure of PgS conjugates was verified using 1H nuclear magnetic resonance (NMR) and infrared (IR) analysis. 1H NMR spectra were recorded using an Avance NMR spectrometer (Bruker Co., Switzerland) operated at 400 MHz. SA was dissolved in chloroform-d, and the PgS2 and PEI samples were prepared in methanol-d4. Tetramethylsilane was used as the internal standard. The concentration of the three samples for 1H NMR analysis was approximately 20 mg mL-1. For the IR analysis, SA, PgS, and PEI were co-pressed with potassium bromide (ca. 1.5%, w/w), and then analyzed using a Fourier-transform infrared (FTIR) spectrometer (Shimadzu Co., Japan). The transmission absorption spectra were obtained in the range of 400 cm-1 to 4000 cm-1.
Preparation of (-)-G-PgSHAs
Three kinds of amphiphilic PgS conjugates were prepared using hydrophilic PEI and hydrophobic SA at different ratios. After our investigation, the PgS2 conjugate (containing an average of 6.57 SA molecules per PEI chain) was used for the final preparation of (-)-G-PgSHAs. As shown in Scheme 1C, the (-)-G-PgSHAs were prepared through a two-step process. In the first step, 8 mg PgS2 was dispersed in 80 mL distilled water. Afterwards, 5 mL ethanol containing 15 mg (-)-gossypol was slowly added into the solution. Stirring was continued for 90 min. The (-)-gossypol-loaded PgS micelles were obtained after the mixture solution was continuously filtered using a glass funnel. In the second step, 20 mL 0.5% HA solution was added dropwise to the (-)-gossypol PgS2 micelle solution, followed by stirring for 3 h. This step allowed the binding of HA molecules onto the surface of PgS micelles through electrostatic interaction. Excess gossypol molecules were removed through extensive dialysis using a buffered solution [phosphate buffer/ethanol (7:3), pH 7.4]. For every 4 h, the buffered solution was replaced with an equal volume of fresh medium, and the optical absorbance of each solution was measured at 385 nm to determine the free (-)-gossypol concentration. This process was stopped after 28 h when free (-)-gossypol molecules were almost undetectable. The final solution of NPs was freeze-dried to obtain a yellow powder of (-)-G-PgSHAs.
Confirmation of hydrogen bonding between PgS and (-)-gossypol
IR analysis was used to study the interactions between the drug molecules and loading materials. Free (-)-gossypol, PgS, HA, drug-loaded PgS micelles, and (-)-G-PgSHAs were respectively co-pressed with potassium bromide (ca. 1.5%, w/w), and were then analyzed using an FTIR Spectrometer (Shimadzu Co., Japan). The transmission absorption spectra were obtained in the range of 400 cm-1 to 4000 cm-1.
Characterizations of (-)-G-PgSHAs
The morphology of (-)-G-PgSHAs was observed via transmission electron microscopy (TEM) (H-800, Hitachi, Japan). The samples were placed on an ultra-thin carbon support film on a copper grid, and observed at different magnifications. In addition, TEM photographs with a higher resolution were also taken through another round of TEM (JEM-2100F, JEOL, Japan) to observe their core-shell structure. The zeta potential and size distribution of (-)-G-PgSHAs were measured by Dynamic Light Scattering (DLS) using a Malvern instrument (Nano-ZS90, Malvern, UK).
The encapsulation efficiency (EE) and drug loading capacity (DLC) were calculated as follows:
| (1) |
| (2) |
where Mgossypol input is the amount of (-)-gossypol input before encapsulation and Mgossypol free is the amount (mg) of (-)-gossypol in the filter residue (un-encapsulated). MG-PgSHAs is the amount of the prepared (-)-G-PgSHAs (based on total weight of the nanoparticles).
Drug-release profiles and stability of (-)-G-PgSHAs
The in vitro drug release profiles were obtained through dynamic dialysis, and the test was conducted using a dialysis membrane (MWCO 3500) in 20 mL PBS (pH 7.4) stirred at 25 rpm. All experiments were performed under sink conditions at 37° ± 0.3 °C using lyophilized (-)-G-PgSHAs containing 3 mg (-)-gossypol. The amount of released (-)-gossypol was determined using a UV-vis spectrophotometer and (-)-gossypol calibration curve obtained at 386 nm wavelength. The same amount of (-)-G-PgSHAs with 10 U mL-1 HAase were dispersed in 20 mL PBS (pH 7.4) and used as the control. The in vitro drug-release profiles were determined under the same conditions. To investigate the stability of (-)-G-PgSHAs, their samples in PBS with a (-)-gossypol concentration of 0.15 mg mL-1 were restored in a centrifuge tube inside a 4 °C refrigerator. Afterwards, the size distribution of the samples was examined at determined intervals.
Cell culture
Prostate cancer cell line PC-3 and CL-1, and myocardium cell line H9C2 were provided by the Department of Molecular Biosciences, University of Kansas and by the School of Life Science and Technology, Xi'an Jiaotong University. All cell lines were maintained in complete DMEM supplemented with 10% (w/w) FBS and penicillin/streptomycin (76 and 36 U mL-1, respectively). The cells were incubated under 5% CO2 at 37 °C in a cell incubator (MCO-18AIC, Sanyo, Japan) before they were used for in vitro tests or mice inoculation.
In vitro cytotoxicity and cellular uptake
MTT method was adopted to test the viability of PC-3 and H9C2 (myocardium) cells against PgS1 (containing an average of 3.75 SA molecules per PEI chain), PgS2 conjugates (containing an average of 6.57 SA molecules per PEI chain), and raw PEI as the control. The cells were seeded onto 96-well flat-bottomed plates with 1×105 cells/well density, and were incubated under 5% CO2 atmosphere at 37 °C. After 24 h of incubation, the cells were washed with PBS. The complete medium containing various concentration of either PgS conjugates or PEI was added to each well. After 72 h, the medium was replaced with a fresh batch, and then 20 μL of MTT solution (5 mg mL-1) was added to each well. The cells were then incubated for an additional 4 h under the same conditions. The medium with MTT was removed, and 100 μL dimethyl sulfoxide (DMSO) was added to each well. The absorbance was measured at 590 nm using an ELISA plate reader (Infinite® 200 Pro, Tecan, Switzerland).
To study the cytotoxicity of (-)-G-PgSHAs as a tumor-targeted nanomedicine and compare it with the racemic gossypol-loaded PgSHAs [(±)-G-PgSHAs], the in vitro cytotoxicity of (-)-G-PgSHAs (with a DLC of 9.6%), (±)-G-PgSHAs, free (-)-gossypol, and racemic gossypol [(±)-gossypol] against PC-3 and CL-1 cells, was measured using the MTT assay explained above. The maximal concentration of (-)-G-PgSHAs used was 0.865 mg mL-1 [containing 0.083 mg mL-1 (-)-gossypol and about 0.06 mg mL-1 PgS]. PgS samples with 0.06 mg mL-1 PgS were used to treat cells for comparison.
To confirm the CD44-mediated endocytosis of the HA-shielded nanocarrier, the cellular uptake behavior of PgSHAs was observed using a fluorescent microscope and a confocal laser scanning microscope (CLSM). Drug-free PgSHAs were labeled with RITC. PC-3 cells attached to 6-well plates were treated with RITC-labeled PgSHAs (containing 50 μg mL-1 PgS) with or without 1% HA. After incubation in the dark for a predetermined time, the cells were rinsed twice with PBS (pH 7.4), and were then visualized using an inverted fluorescence microscope (Eclipse Ti-S, Nikon, Japan). For confocal microscopy, PC-3 cells attached to 6-well plates covered with coverglass were treated with the samples as described above. After incubation in the dark for a predetermined time, the cells were rinsed with PBS and fixed using 4% paraformaldehyde solution. The nuclei of the cells were stained using 1μg mL-1 DAPI. The coverglass was then transferred onto the slide glass, followed by the observation using a CLSM (TCS SP5 II, Leica, Germany).
Animal tumor models
All animal experiments were done in compliance with the Guidelines for Use and Care of Animals in the University of Kansas. NOD.CB17-Prkdcscid (SCID) mice were from our own mice colony and NCI athymic NCr-nude mice were purchased from Harlan Laboratories. The mice were fed in a standard housing condition until 6 weeks of age and with 20 g to 25 g body weight.
To establish tumor models, 200 μL DMEM containing 1 × 106 cells (PC-3 or CL-1) were s.q. injected in the SCID or nude mice, using a sterile 23-gauge needle. Tumors were grown to predetermined sizes before the mice were used for the in vivo fluorescence imaging or drug efficacy studies.
In vivo distribution of PgSHA nanoparticles
The in vivo tumor-targeting of PgSHAs was confirmed using DiR, which is a near infrared (NIR) fluorescent dye. Four SCID mice bearing bilateral CL-1 tumors were used. Two mice were iv. injected with 200 μL DiR-loaded PgSHA nanoparticles, and another two mice were iv. injected with 200 μL DiR ethanol/water (1:4 v/v) solution as the control. The DiR concentration was 50 μmol L-1 in both groups. At different time points, the biodistributions of free DiR or DiR-loaded PgSHA nanoparticles in the four mice were imaged using a Carestream Molecular Imaging System (Carestream Health, Inc), with excitation at 750 nm and emission at 790 nm using exposure time of 60 s. All mice were euthanized by overdosing with CO2 at 96 h after the injections. The organs and tumors of mice were extracted for ex vivo imaging. In addition, the distributions of the free DiR or DiR-loaded PgSHA nanoparticles were quantified by measuring the signal intensity at the region of interest.
In vivo tumor suppression effect of (-)-G-PgSHAs
Prostate cancer (PC-3) model was used to investigate the tumor suppression effect of (-)-G-PgSHAs. PC-3 cells were s.q. inoculated on the back of nude mice. When the tumors reached an average size of 50 mm3, the mice were randomly divided into five groups (n = 8) given as follows: (1) delivering PBS (blank control); (2) blank PgSHAs (vehicle control); (3) (-)-G-PgSHAs at 10 mg (-)-gossypol kg-1; (4) free (-)-gossypol at 10 mg kg-1, for which a 100 mg mL-1 (-)-gossypol in DMSO was diluted with PBS on the day of treatment; and (5) (±)-G-PgSHAs at 10 mg (±)-gossypol kg-1. Each group was iv. injected twice a week for three weeks. The tumor sizes and body weight of each mouse were measured twice a week, and tumor volumes were calculated as a×b2/2, where a and b represented the longest and shortest diameters of the tumor, respectively. After 30 d, all animals were sacrificed. The tumor tissues and other tissue samples, such as the heart, kidney, liver and lung, were removed. The sections of tumors and organs were stained with hematoxylin and eosin, and their histology was then studied and compared.
Statistical analysis
The data on tumor growth were reported as mean ± SEM, and were analyzed using two-way ANOVA. The data of in vitro drug release, cytotoxicity, and ex vivo imaging were reported as mean ± SD, and were analyzed using one-way ANOVA or Student's t-test to determine the difference among the samples. The data analysis was done using Prism 5.0 software (GraphPad Prism, CA, USA). P values < 0.05 were considered as statistically significant.
Results and discussion
Synthesis and characterization of PgS conjugates
Gossypol is a promising anticancer drug, which has been studied in many clinical trials in its racemic form.4-6 To date, almost no nanocarrier has been reported for (-)-gossypol, which is unstable and easily racemizes during the encapsulation in vitro and/or the delivery in vivo. Based on the chemical structure of (-)-gossypol, we hypothesized that, the formation of hydrogen bonds between (-)-gossypol and its loading material could prevent the racemization and maintain the bioactivity of (-)-gossypol. After our investigations, we found that PEI adequately protects (-)-gossypol through hydrogen bonding because of its intensive amino groups. PEI has long been investigated as an effective gene carrier and has been widely used to condense antisense ODN, pDNA, and siRNA.12-14 As gene carrier, the commonly used PEI with high molecular weight (25 kDa) possesses evident cytotoxicity, whereas PEI with low molecular weight has lower toxicity. However, the transfection efficiency of the latter is relatively weaker.21, 22 Therefore, PEI with low molecular weight is a potential material for drug delivery. However, as (-)-gossypol is hydrophobic, PEI needs to be modulated into an amphiphilic material for the encapsulation of (-)-gossypol. By contrast, stearic acid (SA) is a hydrophobic saturated fatty acid and has been used as a safe biodegradable dietary additive. SA can be grafted to low molecular weight PEI because of its good biosafety and suitable molecular weight. As a result, a new amphiphilic PEI-g-SA conjugate (PgS) was prepared through condensation reaction between the carboxyl acid of SA and the amino groups of PEI. Three kinds of PgS conjugates with different SA substitution degrees were prepared by changing the amount of PEI. The mole ratios of SA to PEI in three PgS conjugates were 11.81, 6.57, and 3.75 as determined using HPLC and 1H NMR, which means 11.81, 6.57, and 3.75 SA molecules on average were grafted to the PEI chain for each kind of PgS conjugate. These conjugates ranged from colorless viscous liquid to yellowish sticky solid with increasing SA contents.
The IR and 1H NMR analysis confirmed the successful synthesis of PgS. Fig. 1A shows the IR spectra of SA, PgS, and PEI. The IR spectra of PgS, compared with that of PEI, showed additional peaks at 1636 and 1410 cm-1. This finding indicated newly formed amide bonds (–NHCO-). With increased SA content in the PgS conjugates, the peak intensity of the amide bonds increased significantly, whereas the intensity of amine peaks from 3300 cm-1 to 3350 cm-1 became relatively weaker. Moreover, SA peaks at 1703 and 943 cm-1 that characterize carboxylic acid (–COOH) did not appear in the spectra of PgS conjugates. This result demonstrated the condensation reaction between the carboxyl acid of SA and the primary amines on PEI molecule chains. Moreover, 1H NMR was used to confirm that SA was introduced into the PEI backbone, as shown in Fig. 1B. Except for the reference and ethanol residue, all peaks were annotated. Detailed analysis of 1H NMR spectra is as follows: PEI (δ = 2.6 ppm to 2.8 ppm (A) for H in –NCH2CH2N–), PgS (δ = 2.6 ppm to 2.8 ppm (a) for H in –NCH2CH2N–;δ = 2.15 ppm (b) for H in –COCH2–; δ = 1.6 ppm (c) for H in –CH2–; δ = 1.3 ppm (d) for H in –(CH2)14–; δ = 0.9 ppm (e) for H in –CH3), SA (δ = 2.30 ppm to 2.35 ppm (B) for H in –COCH2–; δ = 1.61 ppm to 1.65 ppm (C) for H in –CH2–; δ = 1.26 ppm to 1.30 ppm (D) for H in –(CH2)14–; δ = 0.9 ppm (E) for H in –CH3). In summary, PgS had the distinctive peaks of both SA and PEI. The slight differences between the chemical shifts of peak b and B, as well as peak c and C, were mainly caused by the change of chemical environment of hydrogen atom. Thus, all the facts mentioned above demonstrated that the PgS conjugates were produced through condensation reaction between the carboxyl acid of SA and the primary amines on PEI chains.
Fig. 1.
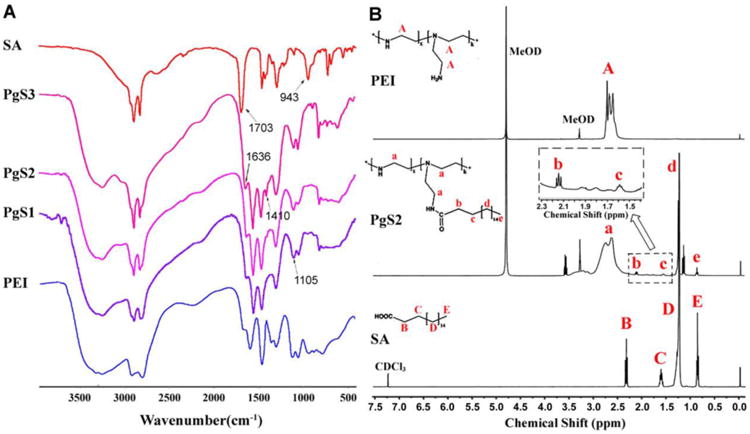
Confirmation of PgS synthesis. (A) IR spectra of SA, PgS and PEI. Samples were co-pressed with potassium bromide respectively, followed by measurement using FTIR Spectrometer; (B) 1H NMR spectra of SA, PgS2 and PEI. SA was dissolved in chloroform-d, while PgS2 and PEI were prepared in methanol-d4. Tetramethylsilane was used as the internal standard.
Cytotoxicity of PgS conjugates
Positively charged PEI can cause cytotoxicity because of its interaction with cell membranes.23 As a PEI derivative, PgS conjugates could also cause cytotoxicity when used as drug carriers. Therefore, we evaluated the cytotoxicity of PgS conjugates against PC-3 (prostate cancer cell line) and H9C2 (myocardium cell line) cells using MTT assay. As shown in Fig. 2A and 2B, the cytotoxicity of PgS conjugates was lower than that of PEI. In addition, the cytotoxicity became much lower with increasing SA contents in PgS. Under the experimental conditions mentioned above, the IC50 values of PEI against PC-3 and H9C2 cells were determined to be approximately 30 and 42 μg mL-1, respectively. The apparent IC50 values against PC-3 and H9C2 were about 79 and 72 μg mL-1, respectively, for PgS1, and about 94 and 93 μg mL-1, respectively, for PgS2. The results of MTT assay indicated that the cytotoxicity of PgS conjugates decreased with increasing SA contents in PgS, which is ascribed to the nontoxic SA portion in PgS. However, PgS also became stickier and had poorer water solubility when the SA content in PgS was very high. Therefore, for the subsequent drug encapsulation, we used PgS2 with a moderate SA substitution degree, which had a relatively low cytotoxicity but also possessed good amphiphilicity and water solubility.
Fig. 2.
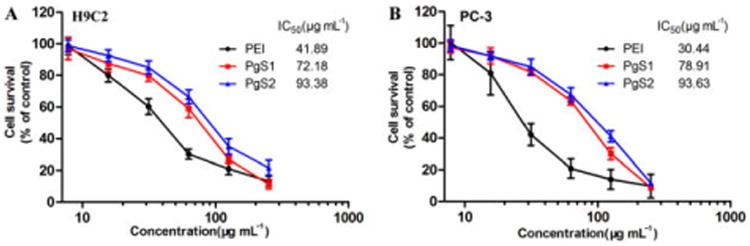
The viability of (A) H9C2 and (B) PC-3 cells treated with PEI, PgS1 and PgS2 indicated that, with increasing content of non-toxic SA portion, PgS conjugate showed less cytotoxicity. Data are presented as mean ± SD (n=3).
Preparation of (-)-G-PgSHAs
The micelles formed by PgS2 conjugate were used to encapsulate (-)-gossypol. Fig. 3 shows that, the IR absorption peak of (-)-gossypol at 1283 cm-1, which characterizes the phenolic hydroxyl groups, redshifted to 1247 cm-1 when the drug was encapsulated in PgS micelles, which indicated the formation of hydrogen bonds between the phenolic hydroxyl groups of (-)-gossypol and the amino groups of PgS. On the other hand, the peak of (-)-gossypol at 1616 cm-1, which characterizes the aldehyde groups, exhibited a slight shift after the encapsulation, suggesting that the aldehyde groups in (-)-gossypol were protected by PgS micelles and were kept intact.
Fig. 3.
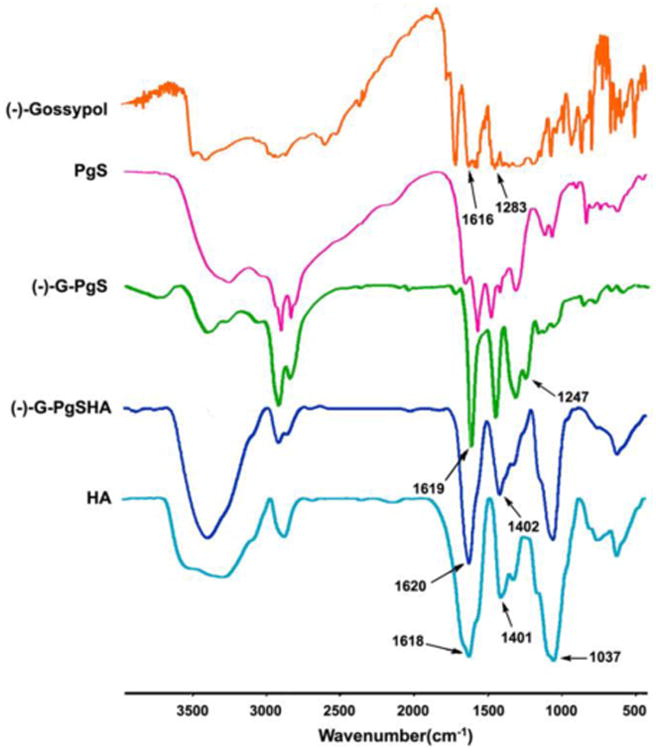
IR spectra of free drug, PgS, drug-loaded PgS micelles [(-)-G-PgS], (-)-G-PgSHA, and HA. Samples were co-pressed with potassium bromide respectively, followed by measurement using FTIR Spectrometer.
Although a relatively lower cytotoxicity was obtained when SA was grafted to PEI, tumor targeting was necessary to further reduce the toxicity and enhance the drug efficacy of chemotherapy. As a result, tumor-targeting material HA was adapted to shield the (-)-gossypol-loaded PgS micelles via electrostatic interaction. As tumor-targeting materials, HA and its derivatives have been used in the delivery of many pharmaceutics, including nucleotide therapeutics, protein and peptide therapeutics, and some small molecular drugs.24 HA have many excellent biochemical characteristics, such as biocompatibility, biodegradability and non-cytotoxicity.25 HA can also helps pharmaceutics into the intracellular matrix by forming complexes with HA receptors, which are commonly over-expressed in many tumor cells, followed by HA receptor-mediated endocytosis. Therefore, we prepared (-)-gossypol-loaded nanoparticles with intact HA outer layer [(-)-G-PgSHAs]. In Fig. 3, the IR absorption peak of HA at 1037, 1401, and 1618 cm-1 characterizing the hydroxyl groups, carboxylates, and amide bonds, respectively, also appeared in the spectrum of (-)-G-PgSHAs, indicating that the (-)-gossypol-loaded PgS micelles were shielded by HA.
Characterization of (-)-G-PgSHAs
Fig. 4A and 4B are the TEM images of (-)-G-PgSHAs at different magnifications. The nanoparticles exhibited uniform particle size and spherical shape with a clear core-shell structure. The particle size looked slightly smaller compared with the results obtained by DLS (Fig. 4C), which was probably due to the elimination of the solvent (i.e. water) around the nanoparticles when the sample was observed using TEM. The zeta potential of (-)-G-PgSHAs was approximately -30 mV (Table 1). The size distribution and zeta potential of drug-free PgS micelles and PgSHAs were also measured for comparison. The size of PgS micelles increased after being shielded with HA. The zeta potential of PgS micelles was changed from +22.8 ± 3.5 mV to –27.4 ± 2.7 mV after the covering of negatively charged HA. When loading (-)-gossypol, (-)-G-PgSHAs had a much larger particle size, and their negative charge was more notable, which may be attributed to the slightly negative charge of (-)-gossypol. In addition, the size and zeta potential of (-)-G-PgSHAs were not significantly different from those of (±)-G-PgSHAs. After storage at 4 °C and at room temperature for 6 months, only negligible differences in the (-)-G-PgSHAs were observed in terms of morphology and particle size, and the encapsulated (-)-gossypol was still maintained in its levorotatory form (data not shown), indicating a good stability of (-)-G-PgSHAs.
Fig. 4.
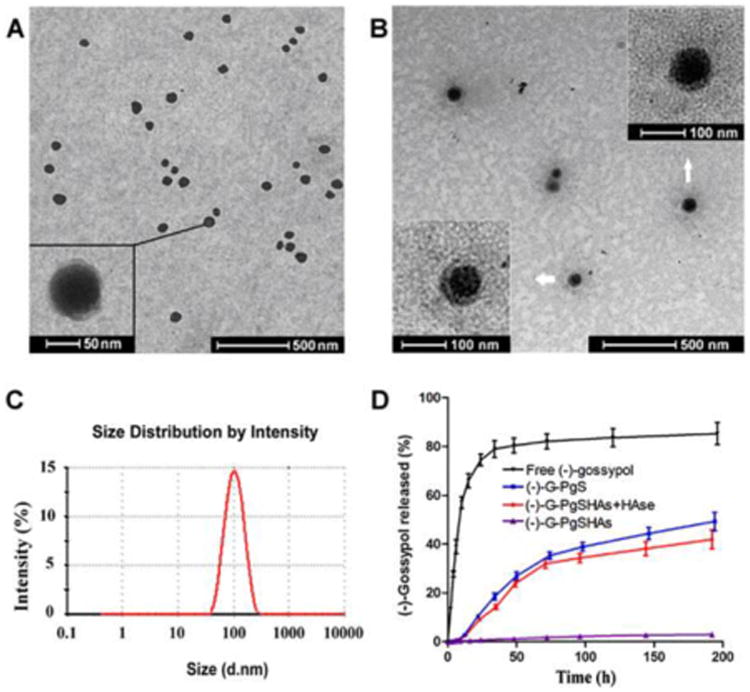
(A, B) TEM images of (-)-G-PgSHAs with different magnification; (C) In vitro release profile of gossypol from (-)-G-PgSHAs with or without HAase (10U mL-1) in PBS at 37 °C. Free (-)-gossypol and (-)-gossypol-loaded PgS micelles [(-)-G-PgS] were used for comparison; (D) Size distribution of G-PgSHAs measured using Malvern Nano-zs 90.
Table 1.
Summary of diameters and Zeta-potentials of different nanoparticles.a
| Nanoparticles | Diameters (nm) | Zeta potential (mV) |
|---|---|---|
| PgS micelles | 90.9±1.9 | +22.8±3.5 |
| PgSHAs | 99.4±2.7 | -27.4±2.7 |
| (-)-G-PgSHAs | 110.9±2.4 | -29.6±3.2 |
| (±)-G-PgSHAs | 112.3±1.6 | -28.1±3.7 |
Data are presented as mean ± SD, n=3.
The DLC and EE values of (-)-G-PgSHAs were 9.1% ± 0.42% and 72.6% ± 3.1%, respectively. The leakage or burst release of drug, which is a common phenomenon for most nanocarriers, is another problem solved by this delivery system. The in vitro release profile of (-)-gossypol from (-)-G-PgSHAs was investigated in PBS using dialysis diffusion technique. Fig. 4D shows the release profile of free drug, drug-loaded PgS micelles [(-)-G-PgS], (-)-G-PgSHAs, and (-)-G-PgSHAs with 10 U mL-1 HAase. Free (-)-gossypol molecules dispersed rapidly from dialysis membrane into the outer PBS. The dispersion of free drug was balanced within 2 days when 85% (-)-gossypol was released. In contrast, only a small amount of (-)-gossypol (about 2%) in (-)-G-PgSHAs was released into the outer phase even after a week. When HAase was added, the (-)-gossypol-loaded PgS micelles were gradually released as the HA layer was degraded. As a result, (-)-G-PgSHAs with HAase demonstrated a faster release of (-)-gossypol, similar to that of (-)-G-PgS. As many tumor tissues have much higher HAase concentration than normal tissues,26 the findings above indicated that (-)-G-PgSHAs may possess a tumor-specific release of (-)-gossypol in vivo, leading to a higher cytotoxicity of (-)-gossypol in tumor tissues than in normal tissues. However, only 43% of the total (-)-gossypol was released after 8 days, which may be ascribed not only to the hydrophobic interaction but also to the electrostatic interaction between PgS and (-)-gossypol, because (-)-gossypol molecules were slightly negatively charged at pH 7.4 based on our previous observation.
To study the potential of (-)-G-PgSHAs as an anti-cancer formulation, their cytotoxicity against CL-1 and PC-3 prostate cancer cells was evaluated using MTT assay. For both cell lines, the IC50 values of encapsulated (-)-gossypol were similar to those of free (-)-gossypol, and were almost half of those of encapsulated (±)-gossypol (Fig. 5A and 5B). These results indicated that the encapsulated (-)-gossypol was protected from racemization, and its drug efficacy was still maintained after the encapsulation. As PgS conjugates also exhibited cytotoxicity, although much less than that of PEI, we investigated the effect of drug-free PgSHAs on the same cell lines. Under the same MTT experiment conditions, the drug-free PgSHAs containing 0.06 mg mL-1 PgS (equal to the PgS content of (-)-G-PgSHAs in the maximum concentration group) was used to treat the cells. As a result, the cell viability was 86.5% ± 2.7% for PC-3 and 75.6% ± 4.1% for CL-1, which indicated that the PgSHA portion certainly contributed to (-)-G-PgSHAs cytotoxicity. However, its effect may be significantly smaller than the drug itself.
Fig. 5.
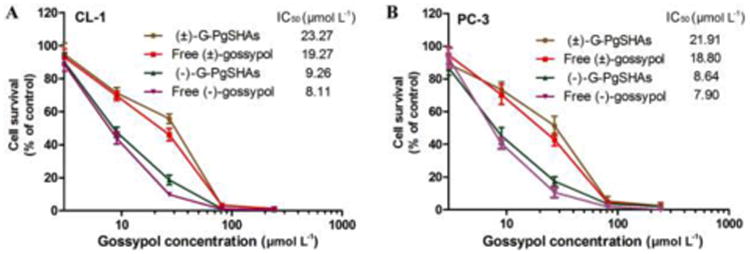
The viability of (A) CL-1 and (B) PC-3 cells treated with free (-)-gossypol, free (±)-gossypol, (-)-G-PgSHAs and (±)-G-PgSHAs indicated that, the encapsulated (-)-gossypol was protected from racemization by PgSHAs and possessed a similar activity to free (-)-gossypol. Data are presented as mean ± SD (n=3).
In vitro cellular uptake of PgSHAs
The tumor-specific delivery of PgSHAs was studied both in vitro and in vivo. Initially, cellular uptake behavior of PgSHAs was observed using fluorescent microscopy. PC-3 cells were incubated with RITC-labeled PgSHAs with or without 1% HA. Fig. 6 and S1† show the cellular uptake profiles after incubation for different time periods. Evident fluorescence was observed in cells treated with RITC-labeled PgSHAs after 50 min incubation, and became much clearer after 100 min. Most of the PgSHAs seemed to have dispersed in the cytoplasm of some cells. By contrast, very few fluorescence blots were observed even after 100 min when extra 1% HA was added, indicating that free HA molecules competed with the HA-shielded PgSHAs in the cellular uptake, which suggested that the cellular internalization of PgSHAs was dependant on HA receptor-mediated endocytosis.
Fig. 6.

Fluorescence microscopic images showing cellular uptake profile of PgSHAs. PC-3 cells attached to 6-well plates were treated with RITC-labeled PgSHAs with or without 1% HA. After incubation for different time, cells uptaking the PgSHAs were visualized with fixed excitation wavelength, light intensity and exposure time.
In vivo distribution of PgSHAs
The in vivo tumor-targeting of PgSHAs was studied using near infrared (NIR) fluorescence imaging. CL-1 tumor-bearing mice were treated with the same amount of free DiR (control group) or DiR formulated in PgSHAs (test group). Fig. 7 shows the in vivo NIR fluorescence images of mice at different time after the injection. Apparent fluorescence signal was observed 6 h after the injection in the tumors of mouse 1 and 2 (Ms#1 and Ms#2), which were given DiR-loaded PgSHAs. The signal remained strong throughout the experiment. For the free DiR group, the fluorescence intensity decreased rapidly in the control mice within 0.5 h after the injection (Fig. 7 and S2†), indicating a rapid clearance and fluorescent instability of free DiR in the body. Considerable DiR signal was also observed around the livers of test group, which may have been caused by the expression of HA receptor in the liver that also substantially binds to HA.27, 28 Some fluorescence could also be seen at the extremities and the tongue areas of the mice. This was due to their licking and touching of the urine that contained the eliminated dye, which was confirmed by the extraction of DiR from the surface of the extremities and the tongue using chloroform (data not shown).
Fig. 7.
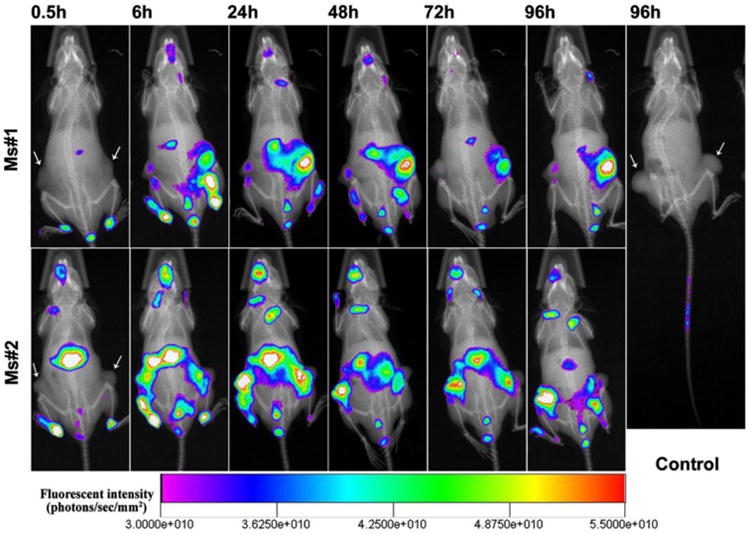
In vivo NIR fluorescence images of CL-1 tumor-bearing SCID mice at different time after i.v. injection of DiR-loaded PgSHAs. The sites of tumors are indicated by arrows. Free DiR dissolved in ethanol solution was used as the control.
To investigate the biodistribution of DiR-loaded PgSHAs, ex vivo NIRF images of tumors and organs harvested 96 h after the injection were obtained (Fig. S3†). In all four mice, DiR signals in the liver, lung, and spleen were stronger than those in other organs. In addition, as a result of faster elimination of free DiR molecules, DiR signal in the control group was weaker than that in the test group. However, compared with the control group, DiR in the test group also tended to accumulate in tumors other than the liver, lung, and spleen. In order to better compare the two groups, we quantified the tumors/liver and tumor/spleen ratios of DiR signal intensity of each mouse, and found that both ratios in the test group were significantly higher than those of control group (Fig. S4†). The results above suggested that PgSHAs could facilitate their payload into tumors and prolong its retention in the body, and therefore may enhance the efficacy of loaded anticancer drugs and reduce the systemic toxicity.
In vivo tumor suppression effect of (-)-G-PgSHAs
PC-3 tumor-bearing mice were used to investigate the tumor suppression effect of (-)-G-PgSHAs. Fig. 8A shows that, within the investigated dose schedule of each treatment, tumors in both blank control and vehicle control groups showed rapid growth as a function of time, whereas the tumors in the free (-)-gossypol, (-)-G-PgSHA, and (±)-G-PgSHAs groups showed a significantly slower growth (P < 0.001, compared with blank control group), indicating a effective tumor suppression of each treatments. The body weight of mice in free (-)-gossypol was significantly decreased, as expected (Fig. 8B). The systemic toxicity of free drug was supposed to be mainly caused by non-specific biodistribution. On the contrary, the tumor-targeted PgSHA nanoparticles weakened the systemic toxicity of the drug and improved the tumor inhibition. Moreover, the tumor growth in (-)-G-PgSHAs group was significantly slower than that of (±)-G-PgSHAs group (P < 0.05), suggesting that (-)-gossypol in the nanocarrier kept levorotatory in the body and maintained its bioactivity, thus showing a better tumor suppression effect.
Fig. 8.
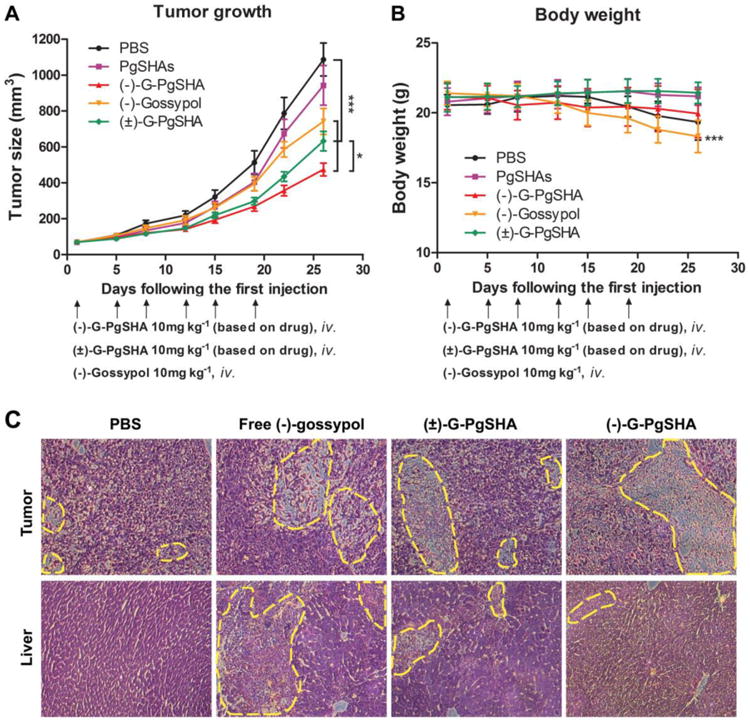
(A) In vivo tumor growth curves. Nude mice were inoculated s.q. with PC-3 cells on the back. Error bars represent SEM. *P < 0.05 [(-)-G-PgSHAs vs. (±) -G-PgSHAs] and ***P < 0.001 (each treatment vs. PBS), n=8. (B) Body weight of PC-3 tumor-bearing mice receiving various treatments. Error bars represent SD, n=8. ***P < 0.001 as compared with original body weight. (C) Pathological section images of the tumor (upper row) and liver (lower row) tissues of PC-3 tumor-bearing nude mice treated by PBS, free (-)-gossypol, (±)-G-PgSHAs and (-)-G-PgSHAs, respectively. All the samples were hematoxylin and eosin stained. Magnification was 200×.
The pathological section images of the tumor and liver tissues agreed with the in vivo results. Tumor tissues in free (-)-gossypol, (-)-G-PgSHAs, and (±)-G-PgSHAs groups showed notable necrosis, with the smallest amount of tumor cells found in the (-)-G-PgSHAs-treated group, indicating an excellent therapeutic effect of (-)-G-PgSHAs (Fig. 8C). In addition, according to the NIRF imaging, the DiR fluorescence was also observed in the liver region of the mouse, which indicated that some of the nanoparticles also accumulated in the liver. Therefore the repeated long-term treatment with gossypol may also cause damages to the liver.29, 30 In this study, the liver tissues in (-)-G-PgSHAs and (±)-G-PgSHAs groups exhibited less damage than that in the free (-)-gossypol group (Fig. 8C), which demonstrated a low toxicity of drug brought about by the tumor-targeted PgSHA nanocarrier with good biocompatibility.
In summary, the PgSHA is an excellent nanocarrier for the chiral drug (-)-gossypol. The optical activity of (-)-gossypol was protected by PgSHAs through hydrogen bonding in vitro and in vivo, which was proven by the IR analysis, stability studies, MTT experiments, and in vivo anticancer efficacy studies.
Conclusions
We designed and prepared a novel double-layered PgSHA nanoparticle encapsulating the chiral drug (-)-gossypol through a two-step fabrication process. The nanoparticle can easily disperse in water, and has great potential for therapeutic applications because of its substantial tumor-targeting ability, sustained release profile, and evident tumor-inhibition effects. Moreover, this bilayered nanocarrier may also provide an applicable strategy for the encapsulation of other chiral drugs.
Supplementary Material
Acknowledgments
This study was supported in part by National Basic Research Program 973 of China (No. 2010CB732603 and No. 2011CB707903), the National Nature Science Foundation of China (Nos. 81271686 and 81228011), and the grants of Shaanxi province science and technology and innovation project (Nos. 2011KTCL03-07). This study was also supported in part by US National Institutes of Health grant (R01 CA121830 S1), K-INBRE (P20 GM103418), and Kansas Bioscience Authority Rising Star Award (to L. X.).
Footnotes
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/b000000x/
Contributor Information
Liang Xu, Email: xul@ku.edu.
Daocheng Wu, Email: wudaocheng@mail.xjtu.edu.cn.
References
- 1.Ali I, Gupta VK, Aboul-Enein HY, Singh P, Sharma B. Chirality. 2007;19:453–463. doi: 10.1002/chir.20397. [DOI] [PubMed] [Google Scholar]
- 2.Ali I. Combinatorial chemistry & high throughput screening. 2007;10:326–335. doi: 10.2174/138620707781662835. [DOI] [PubMed] [Google Scholar]
- 3.Lu H. Expert opinion on drug metabolism & toxicology. 2007;3:149–158. doi: 10.1517/17425255.3.2.149. [DOI] [PubMed] [Google Scholar]
- 4.Baggstrom MQ, Qi Y, Koczywas M, Argiris A, Johnson EA, Millward MJ, Murphy SC, Erlichman C, Rudin CM, Govindan R, Mayo Phase C, California C. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:1757–1760. doi: 10.1097/JTO.0b013e31822e2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, Borgen P, Gollub M, Bacotti D, Yao TJ, Bloch R, Ligueros M, Sonenberg M, Norton L, Hudis C. Breast cancer research and treatment. 2001;66:239–248. doi: 10.1023/a:1010686204736. [DOI] [PubMed] [Google Scholar]
- 6.Stein RC, Joseph AE, Matlin SA, Cunningham DC, Ford HT, Coombes RC. Cancer chemotherapy and pharmacology. 1992;30:480–482. doi: 10.1007/BF00685601. [DOI] [PubMed] [Google Scholar]
- 7.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan JY. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 8.Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu XH, Cao YY, Guo RB, Li BH, Zhu XF, Huang Y, Long YQ, Roller PP, Yang DJ, Wang SM. J Med Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 9.Yu YW. Journal of ethnopharmacology. 1987;20:65–78. doi: 10.1016/0378-8741(87)90120-6. [DOI] [PubMed] [Google Scholar]
- 10.Cho H, Lai TC, Kwon GS. Journal of controlled release: official journal of the Controlled Release Society. 2013;166:1–9. doi: 10.1016/j.jconrel.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhai GX, Wu J, Zhao XB, Yu B, Li H, Lu YH, Ye WP, Lin YC, Lee RJ. Anticancer Res. 2008;28:2801–2805. [PubMed] [Google Scholar]
- 12.Jeong JH, Kim SH, Kim SW, Park TG. Bioconjugate chemistry. 2005;16:1034–1037. doi: 10.1021/bc0497315. [DOI] [PubMed] [Google Scholar]
- 13.Boussif O, Lezoualch F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. P Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park K, Lee MY, Kim KS, Hahn SK. Biomaterials. 2010;31:5258–5265. doi: 10.1016/j.biomaterials.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Yim H, Jo EA, Na K. Macromol Res. 2010;18:913–918. [Google Scholar]
- 16.Surace C, Arpicco S, Dufay-Wojcicki A, Marsaud V, Bouclier C, Clay D, Cattel L, Renoir JM, Fattal E. Molecular pharmaceutics. 2009;6:1062–1073. doi: 10.1021/mp800215d. [DOI] [PubMed] [Google Scholar]
- 17.Cichy J, Pure E. The Journal of cell biology. 2003;161:839–843. doi: 10.1083/jcb.200302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maya S, Sarmento B, Nair A, Rejinold NS, Nair SV, Jayakumar R. Current pharmaceutical design. 2013;19:7203–7218. doi: 10.2174/138161281941131219124142. [DOI] [PubMed] [Google Scholar]
- 19.Choi KY, Saravanakumar G, Park JH, Park K. Colloids and surfaces B, Biointerfaces. 2012;99:82–94. doi: 10.1016/j.colsurfb.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murano E, Perin D, Khan R, Bergamin M. Natural product communications. 2011;6:555–572. [PubMed] [Google Scholar]
- 21.Forrest ML, Koerber JT, Pack DW. Bioconjugate chemistry. 2003;14:934–940. doi: 10.1021/bc034014g. [DOI] [PubMed] [Google Scholar]
- 22.Dong W, Jin GH, Li SF, Sun QM, Ma DY, Hua ZC. Acta biochimica et biophysica Sinica. 2006;38:780–787. doi: 10.1111/j.1745-7270.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- 23.Lecocq M, Wattiaux-De Coninck S, Laurent N, Wattiaux R, Jadot M. Biochemical and biophysical research communications. 2000;278:414–418. doi: 10.1006/bbrc.2000.3809. [DOI] [PubMed] [Google Scholar]
- 24.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, Lee MY, Hoffman AS, Hahn SK. Journal of Controlled Release. 2010;141:2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Necas J, Bartosikova L, Brauner P, Kolar J. Vet Med-Czech. 2008;53:397–411. [Google Scholar]
- 26.Liu DC, Pearlman E, Diaconu E, Guo K, Mori H, Haqqi T, Markowitz S, Willson J, Sy MS. P Natl Acad Sci USA. 1996;93:7832–7837. doi: 10.1073/pnas.93.15.7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takei Y, Maruyama A, Ferdous A, Nishimura Y, Kawano S, Ikejima K, Okumura S, Asayama S, Nogawa M, Hashimoto M, Makino Y, Kinoshita M, Watanabe S, Akaike T, Lemasters JJ, Sato N. Faseb J. 2004;18:699–+. doi: 10.1096/fj.03-0494fje. [DOI] [PubMed] [Google Scholar]
- 28.Toriyabe N, Hayashi Y, Hyodo M, Harashima H. Biol Pharm Bull. 2011;34:1084–1089. doi: 10.1248/bpb.34.1084. [DOI] [PubMed] [Google Scholar]
- 29.Ali SF, El-Sewedy SM. Toxicology letters. 1984;23:299–306. doi: 10.1016/0378-4274(84)90025-0. [DOI] [PubMed] [Google Scholar]
- 30.de Peyster A, Quintanilha A, Packer L, Smith MT. Biochemical and biophysical research communications. 1984;118:573–579. doi: 10.1016/0006-291x(84)91341-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.