

Abstract
Overexpression or mutation of α-synuclein (α-Syn), a protein associated with presynaptic vesicles, causes familial forms of Parkinson’s disease in humans and is also associated with sporadic forms of the disease. We used in vivo microdialysis, tissue content analysis, behavioral assessment, and whole-cell patch clamp recordings from striatal medium-sized spiny neurons (MSSNs) in slices to examine dopamine transmission and dopaminergic modulation of corticostriatal synaptic function in mice overexpressing human wild-type α-Syn under the Thy1 promoter (α-Syn mice). Tonic striatal extracellular dopamine and 3-methoxytyramine levels were elevated in α-Syn mice at 6 months of age, prior to any reduction in total striatal tissue content, and were accompanied by an increase in open-field activity. Dopamine clearance and amphetamine-induced dopamine efflux were unchanged. The frequency of MSSN spontaneous excitatory postsynaptic currents (sEPSCs) was lower in α-Syn mice. Amphetamine reduced sEPSC frequency in wild types (WTs) but produced no effect in α-Syn mice. Furthermore, whereas quinpirole reduced and sulpiride increased sEPSC frequency in WT mice, they produced the opposite effects in α-Syn mice. These observations indicate that overexpression of α-Syn alters dopamine efflux and D2 receptor modulation of corticostriatal glutamate release at a young age. At 14 months of age, the α-Syn mice presented with significantly lower striatal tissue dopamine and tyrosine hydroxylase content relative to WT littermates, accompanied by an L-DOPA-reversible sensory motor deficit. Together, these data further validate this transgenic mouse line as a slowly progressing model of Parkinson’s disease and provide evidence for early dopamine synaptic dysfunction prior to loss of striatal dopamine.
Keywords: α-synuclein, dopamine, D2 dopamine receptors, amphetamine, microdialysis, spontaneous excitatory postsynaptic currents, behavior
α-Synuclein (α-Syn) is a protein associated with presynaptic nerve terminals throughout the central nervous system (Iwai et al., 1995; Totterdell and Meredith, 2005) and is a major constituent of Lewy bodies, a hallmark of idiopathic and familial Parkinson’s disease (PD; Spillantini et al., 1997; Trojanowski and Lee, 1998). Although rare cases of familial PD have been linked to point mutations in α-Syn (Polymeropoulos et al., 1997), the more common idiopathic form of the disease is associated with abnormal accumulation of wild-type (WT), not mutant, α-Syn. Moreover, WT α-Syn gene multiplication is pathogenic (Singleton et al., 2003), and genome-wide association studies have revealed a close relationship between α-Syn and sporadic PD (Satake et al., 2009). Therefore, animal models based on human WT α-Syn overexpression offer construct validity for modeling idiopathic PD.
Among the several different lines of mice overexpressing WT human α-Syn that have been generated (Magen and Chesselet, 2010), mice overexpressing WT human α-Syn under the Thy1 promoter (Rockenstein et al., 2002) have been extensively characterized. The Thy1 promoter confers widespread, high levels of WT human α-Syn overexpression in neurons of the brain but no loss of motor neurons (Rockenstein et al., 2002). Proteinase K-resistant α-Syn inclusions also have been identified in many brain regions of these mice (Fernagut et al., 2007; Fleming et al., 2008), and the mice show progressive sensorimotor alterations, beginning as early as 2 months of age (Fleming et al., 2004, 2006). Because the mice have a full complement of nigral dopamine (DA) cell bodies and striatal DA terminals at the times when they exhibit such deficits (Fernagut et al., 2007), it has been proposed that, at these early ages, they represent a model of premanifest PD (Chesselet, 2008), a contention supported by the demonstration of early nonmotor olfactory deficits and autonomic changes in these mice characteristic of early stages of the disease (Fleming et al., 2008; Wang et al., 2008; Magen and Chesselet, 2010).
Little is known about dopamine transmission and synaptic function at this early, premanifest stage of PD. A mouse model of α-Syn overexpression, because of the established association between excess α-Syn and the development of the disease, can provide information on these little-understood early stages of pathology. Accumulating evidence indicates that α-Syn plays a role in synaptic transmission (Abeliovich et al., 2000; Bamford et al., 2004; Yavich et al., 2004, 2005; Gureviciene et al., 2007; Burré et al., 2010) and synaptic plasticity (George et al., 1995; Liu et al., 2004; Watson et al., 2009). Previous observations by our group point to a possible α-Syn-induced disruption of nigrostriatal and/or corticostriatal synaptic transmission (Wu et al., 2009), reminiscent of our previous findings in the parkin knockout (KO) mouse model of PD (Goldberg et al., 2003). With that model, we observed an elevation in striatal extracellular DA and decreases in MSSN excitability to corticostriatal synaptic activation as well as changes in plasticity (Watson et al., 2009). We therefore embarked upon a neurochemical and electrophysiological analysis of striatal DA and DA–glutamate interactions in the Thy1-α-Syn mouse at early ages. In addition, to validate this mouse line further as a slowly progressing model of PD, we assessed aged animals (approximately 14 months old) in a sensory motor task and for postmortem striatal levels of DA and tyrosine hydroxylase (TH).
MATERIALS AND METHODS
Animals
All experimental procedures were carried out in accordance with the National Institutes of Health Guide for care and use of laboratory animals and the principles presented in the Society for Neuroscience Guidelines for the use of animals in neuroscience research under protocols approved by the UCLA Institutional Animal Care and Use Committee. Transgenic α-Syn mice initially were obtained from Eliezar Masliah at the University of California San Diego (Rockenstein et al., 2002), and a colony was maintained in our mouse breeding facilities at UCLA. Animals were maintained on the hybrid C57BL/6-DBA/2 background by mating N7 female hemizygous for the transgene (which is inserted on the X chromosome) with male WT mice on the hybrid background obtained from Charles River Laboratories (Wilmington, MA; Fleming et al., 2004, 2008; Fernagut et al., 2007). Male and female mice from the same litters were never bred together. Litter sizes ranged from 2 to 11 mice per litter. The genotype of all Thy1-aSyn and WT mice was verified by polymerase chain reaction (PCR) amplification analysis of tail DNA at the end of the experiment. Animals for the open-field experiments were maintained on a reverse light/dark cycle with lights off at 10:00 AM, and all testing was performed between 12:00 and 4:00 PM during the dark cycle under low light. Food and water were available ad libitum.
Microdialysis was conducted on eight male α-Syn mice and eight WT littermate controls with average ages of 182 ± 2 (mean ± SEM) and 188 ± 3 days, respectively (approximately ± months). A second round of microdialysis sampling was conducted on a subset of these animals approximately 3.5 months later (5 α-Syn and 5 WT at 286 ± 14 and 286 ± 7 days of age, respectively; approximately 9.5 months). Electrophysiological experiments were conducted on 12 α-Syn mice (5 males and 7 females) and 11 WT littermates (7 males and 4 females) with ages averaging 92.2 ± 2.1 and 92.7 ± 2.5 days, respectively (approximately 3 months). Open-field tests were conducted on a separate group of 9 male α-Syn mice and 10 male WT littermates with average ages of 219 ± 1 and 218 ± 1 days, respectively (approximately 7 months). A final group of ± male α-Syn mice and 7 male WT littermates with average ages of 452 ± 11 and 457 ± 11 days, respectively (approximately 15 months) were tested for L-DOPA reversibility of sensory motor deficits. Post-mortem striatal tissue analysis was carried out on two sets of animals as follows: the 10 male α-Syn mice and 12 male WT littermate mice (the animals used in the open-field tests mentioned above plus an additional 2 α-Syn and one WT naïve male mice) with average ages of 234 ± 1 and 233 ± 1 days, respectively (approximately 7 months) and a separate group of naive 8 male α-Syn mice and 8 WT littermate mice with average ages of 432 ± 9 and 427 ± 6 days, respectively (approximately 14 months). Group ages are presented as approximate months below for clarity.
Microdialysis
A microdialysis probe (CMA7; 2-mm cuprophan membrane), continuously perfused at a rate of 0.5 μl/min with artificial cerebrospinal fluid (ACSF, 125 mM NaCl, 2.5 mM KCl, 0.9 mM NaH2PO4, 5 mM Na2HPO4, 1.2 mM CaCl2, 1 mM MgCl2, pH 7.4), was stereotaxically implanted in the right striatum, and a microdialysis guide cannula was implanted above the left striatum with the mice under isoflurane anesthesia using the following coordinates in millimeters from bregma and the skull surface according to the atlas of Franklin and Paxinos (1997): R 0.6, L 1.8, V 4.5. After a 15–18-hr recovery period, sampling began from the right striatum at 30-min intervals. A no-net-flux experiment for DA was conducted: three baseline samples were collected into 1.5 ll perchloric acid/EDTA (12.5 mM/250 μM), and subsequently DA was added to the perfusion fluid at concentrations of 10, 20, and 40 μM in random order, each for the duration of three samples. Dialysates were stored at −80°C until analysis. On the following day, d-amphetamine (Sigma-Aldrich, St. Louis, MO) was administered by reverse dialysis at increasing concentrations (1, 10, and 100 nM) each for three 20-min samples with three intervening sampling periods of d-amphet-amine-free ACSF. Approximately 100 days later, a second microdialysis probe was lowered through the guide cannula into the left striatum, and, after a 15–18-hr recovery period, four basal 20-min samples were collected.
Extraction of Striatal Tissue for DA Tissue Content Measurement
Brains were rapidly removed post-mortem and frozen in liquid nitrogen, stored at −80°C, and subsequently sectioned on a freezing microtome at a thickness of 900 lm. Punches (2 mm diameter) of striatal tissue were weighed, homogenized in 250 μl of 0.1 M PCA/0.1% EDTA containing isoproterenol (1 μM) and dihydroxybutyrate (0.1 μM) as internal standards, and centrifuged at 10,000 rpm for 20 min at 4°C. The supernatant was filtered through a 0.22-μm filter by centrifugation at 10,000 rpm in a microfuge for 5 min at 4°C and frozen at −80°C.
HPLC Analysis of Striatal Microdialysate and Tissue Dopamine and Metabolite Content
Microdialysis samples were analyzed for DA using high-performance liquid chromatography with electrochemical detection (Antec Leyden, Leiden, The Netherlands) using a mobile phase consisting of sodium acetate (50 mM), sodium dodecane sulfonate (0.50 mM), EDTA (10 μM), acetonitrile (12%), and methanol (9%), pH 5.5, pumped at a rate of 200 μl/min (model LC-10AD; Shimadzu, Columbia, MD) through a 100 × 2 mm column (2 μm; Super-ODS C18Tosoh Bioscience, Montgomeryville, PA). The system was calibrated at regular intervals and provided a limit of detection of 0.5 fmol for a 5-μl injection of sample. DA metabolites were analyzed separately under different chromatography conditions comprising a 100 × 2 mm 3 μm Hypersil C18 Keystone Scientific column, 50 mM sodium phosphate buffer, pH 2.85, 6% methanol, 1.4 mM heptane sulfonic acid, 10 μM EDTA. The data were collected andanalyzed using ChromPerfect software (Justice Innovations, Mountain View, CA).
SDS-PAGE and Western Blot Analysis of Striatal Tissue TH Levels
Striatal tissue punches were collected as described above for DA tissue analysis but were homogenized in a lysis buffer (50 mM Tris-HCl, 50 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 50 mM NaF, 5 mM Na4P2O7, 1% Triton, 0.5 μM okadaic acid, 0.5% sodium deoxycholate, 0.1% SDS) and protein content was determined (BCA; Pierce, Thermo-Scientific, Rockford, IL). Protein samples (10 μg/ sample) were separated by SDS-PAGE (10% NuPAGE gel; Invitrogen, Carlsbad, CA) and transferred to a nitrocellulose membrane (Pierce). After several washes and a 90-min blocking step (casein/0.1% Tween-20), the membrane was incubated in anti-TH (Chemicon, Temecula, CA; 1:4,000) and antitubulin (Chemicon; 1:3,000) antibodies overnight at 4°C. The membrane was then washed, incubated in the secondary antibodies (stabilized peroxidase-conjugated anti-rabbit and anti-mouse; each at 1:10,000; Pierce) for 120 min at room temperature. The labeled protein bands (62 and 55 kDa for TH and tubulin, respectively) were visualized by chemiluminescence (Super Signal Chemiluminescent Substrate; Pierce), and signal intensity (pixels/mm2) was quantified (ImageJ; NIH). The signal intensity of TH was normalized to that of tubulin to account for any differences in protein loading, and the mean of three separate measurements of TH and tubulin protein levels in each striatal punch was used for final data analysis.
Brain Slice Preparation and Electrophysiological Recordings
Mice were deeply anesthetized with halothane prior to euthanasia, and the brains were removed and immediately placed in oxygenated ice-cold, low-Ca2+ ACSF containing (in mM) NaCl 130, NaH2PO4 1.25, NaHCO3 26, MgCl2 5, CaCl2 1, and glucose 10. The hemispheres were separated, and 350-μm coronal slices were cut and transferred to an incubating chamber containing ACSF (with 2 mM CaCl2 and 2 mM MgCl2) oxygenated with 95% O2–5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25°C ± 2°C). After 1 hr, slices were placed on the stage of an upright fixed-stage microscope (BX51; Olympus, Center Valley, PA), submerged in continuously flowing ACSF (4 ml/min).
Whole-cell patch clamp recordings in voltage-clamp mode were obtained using a MultiClamp 700A amplifier (Axon Instruments, Foster City, CA). Medium-sized spiny neurons (MSSNs) in the dorsolateral striatum were visualized with the aid of infrared videomicroscopy (Cepeda et al., 2003) and identified by somatic size and basic membrane properties (input resistance, membrane capacitance, and time constant). Series resistance (<25 MΩ) was compensated 70–80% and checked periodically. If the series resistance changed by >10% at the end of the experiment, data from the cell were discarded. The patch pipette (3–5 MΩ) contained the following solution (in mM): Cs-methanesulfonate 130, CsCl 10, NaCl 4, MgCl2 1, MgATP 5, EGTA 5, HEPES 10, GTP 0.5, phosphocreatine 10, leupeptin 0.1 (pH 7.25–7.3, osmolarity 280–290 mOsm). The use of Cs+ as the main charge carrier also blocked several K+ conductances. Passive membrane properties of MSSNs were determined by applying a depolarizing step voltage command (10 mV) and using the membrane test function integrated in pClamp8 software (Axon Instruments). This function reports membrane capacitance (in pF), input resistance (in MΩ), and time constant (in msec).
After characterizing the basic membrane properties of the neuron, spontaneous excitatory postsynaptic currents (EPSCs) were recorded for 3–6 min in ACSF. For EPSCs, cells were held at −70 mV to minimize the contribution of GABAA receptors and that of voltage-gated conductances. In addition, in all experiments, bicuculline (BIC; 20 μM) was added to block the contribution of spontaneous currents mediated by activation of GABAA receptors. To determine alterations in DA modulation, after a control period, d-amphetamine (25 μM), quinpirole (10 μM), or sulpiride (10 μM) was added to the bath. After a 5-min application, spontaneous EPSCs were recorded for 3–6 min. For all recordings, the membrane current was filtered at 1 kHz and digitized at 100 ls using Clampex (gap-free mode; Axon Instruments).
For electrophysiological experiments, drugs were purchased from Sigma-Aldrich, except for BIC (Tocris, Ellisville, MO). d-Amphetamine and quinpirole were prepared as stock solutions in double-distilled water and diluted to their final concentration before each experiment. Sulpiride was freshly made for each experiment.
Locomotor Activity in an Open Field
Spontaneous activity in an open field (25.5 cm × 25.5 cm) was monitored for 15 min using an automated system (Truscan system for mice; Coulbourn Instruments, Allentown, PA). Testing took place within the first 2–4 hr of the dark cycle after animals had first been habituated to the testing room for 1 hr. The open field was illuminated with an anglepoise lamp equipped with a 25-W red bulb. Distance covered in the horizontal plane and time spent in motion were analyzed in 3 × 5-min time blocks (modified from Hickey et al., 2005). Movement velocity was calculated by dividing the distance traveled by the time in motion for each of the 5-min blocks.
Adhesive Removal Sensory Motor Test
Motor response to sensory stimuli was measured as described by Fleming et al. (2004). Briefly, a small adhesive stimulus (Avery adhesive-backed labels, one-fourth inch round) was placed on the snout of the mouse. Latency to initial stimulus contact, removal time, and initial contact-to-removal time were noted. If the animal did not remove the stimulus within 30 sec, the stimulus was removed by the experimenter. Each animal was tested three times in each session, alternating between mice within a session, and an average value was calculated for each animal. Each mouse was tested in two sessions with a 2-day break between sessions. LDOPA (25 mg/kg, i.p.; Sigma) or saline vehicle (0.9%, i.p.) was administered 30 min prior to each session in a counterbalanced manner. The peripheral decarboxylase inhibitor benserazide (12.5 mg/kg, i.p.; Sigma) was administered 20 min prior to L-DOPA, whereas saline-treated animals received an additional injection of saline at this point.
Data Analysis and Statistics
The difference in basal dialysate DA and metabolite concentrations between genotypes was determined by averaging the three baseline values for each animal and comparing the mean of these values between the genotypes using Student’s t-test. Quantitative no-net-flux data were analyzed by plotting the concentration of DA in the dialysis perfusion fluid on the X-axis against the difference between that value and the measured concentration in the dialysate, on the Y-axis, and fitting a linear line-of-best-fit to the data for each animal. The point of intercept with the X-axis (the point of no-net-flux) and the slope of the line (extraction fraction) were averaged across animals and genotype differences analyzed by Student’s t-test. The effect of d-amphetamine on DA efflux was assessed by calculating the area under the curve for each of the three amphetamine concentrations and comparing this value across genotype by repeated-measures analysis of variance. Striatal tissue content of DA, its metabolites, and TH were compared across genotypes by using an unpaired Student’s t-test.
Spontaneous synaptic events were analyzed off-line in the Mini Analysis Program (Jaejin Software, Leonia, NJ). This software was used to calculate spontaneous EPSC frequency and amplitude of events. The threshold amplitude for the detection of an event was set at ± pA for spontaneous EPSCs [2–3 times above root mean square noise level (generally ~2– × pA at −70 mV)]. All events were confirmed visually by an experimenter blind to genotype. Frequencies were expressed as number of events per second (Hz). Differences among group means were assessed with appropriately designed analyses of variance, followed by multiple comparisons using Bon-ferroni t-tests. Student’s t-tests alone were used when only two group means were compared.
The difference in open-field activity between genotypes was compared across time using a repeated-measures ANOVA followed by unpaired Student’s t-test on individual time epochs. A Mann-Whitney U-test was used to compare WT and α-Syn mice in the adhesive removal task, after saline or L-DOPA treatment. Unless otherwise noted, values in the figures and text are mean ± standard error of the mean (SEM). Differences between means were considered statistically signif-icant at P < 0.05.
RESULTS
Basal Striatal Extracellular DA Is Elevated in α-Syn Mice at ± Months of Age
Dialysis probe placement was confirmed to be within the targeted region of the striatum by post-mortem observation in all cases. The striatal dialysate DA concentration was significantly higher in α-Syn mice (5.7 ± 0.6 nM, n = 8) relative to WT controls (3.0 ± 0.8 nM; n = 8, P < 0.05; Fig. 1A) at approximately ± months of age. There was a single outlier in the WT group as shown by Grubbs test (Grubbs, 1969). When this outlier was removed, the mean and SEM of that group were reduced to 2.2 ± 0.4 nM and the statistical significance of the difference was increased to P < 0.001 (Fig. 1B). The data from this outlier mouse were therefore removed from DA no-net-flux and metabolite analyses presented below.
Fig. 1.

A: Scatterplot comparison of striatal dialysate DA concentration in WT and α-Syn mice showing the single statistical outlier in the WT group, which was removed from subsequent analyses. B: Comparison of mean striatal dialysate DA, 3MT, DOPAC, and HVA concentrations between the genotypes following removal of the outlier, demonstrating a significant elevation in DA and 3MT but not DOPAC or HVA in the α-Syn mice. *P < 0.005, **P < 0.001.
Dialysate 3MT but Not DOPAC and HVA Is Elevated in α-Syn Mice at ± Months
To determine whether the observed increase in extracellular DA in the α-Syn mice was the result of a general decrease in DA metabolism or, conversely, was accompanied by an increase in DA turnover, we also measured dialysate DA metabolite concentrations. As shown in Figure 1B, DOPAC and HVA concentrations were not different between genotypes (P = 0.46 and P = 0.63, respectively). Thus, a deficit in extraor intraneuronal metabolism cannot account for the elevated extracellular DA. In contrast, extracellular 3MT concentrations were significantly higher in α-Syn mice (P < 0.005; Fig. 1B). 3MT is generally considered to be produced only from DA that has been released from the presynaptic terminal, because catechol-O-methyltransferase is not present in DA terminals (Kaakkola et al., 1987). Therefore, the selective increase in extracellular 3MT points to an increase in vesicular-mediated DA efflux in the α-Syn mice rather than a deficit in vesicular packaging accompanied by reverse transport of cytosolic DA via the DA transporter (DAT).
DA Reuptake Is Unchanged in α-Syn Mice at ± Months
no-net-flux analysis provided no indication of compromised DA reuptake, insofar as the extraction fraction, indicated by the slopes of the lines in Figure 2, were not significantly different between genotypes (0.49 ± 0.04 vs. 0.41 ± 0.04 in α-Syn and WT, respectively; P = 0.18). This analysis also established that the estimated extracellular DA concentration was higher in the α-Syn mice relative to WT (11.3 ± 1.3 vs. 6.3 ± 1.5 nM, respectively; P < 0.05).
Fig. 2.
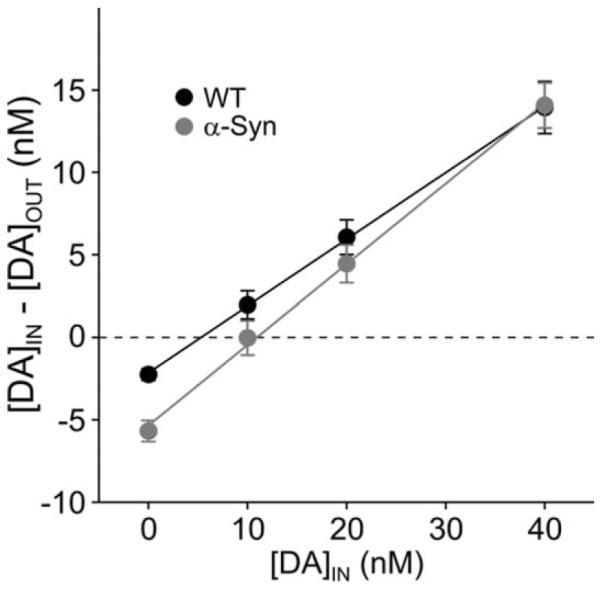
Mean extraction fraction plots showing the different points of no-net-flux (intercept on the X-axis) between the genotypes but no significant difference in the extraction fraction (line slope), an indirect measure of DA reuptake.
Amphetamine-Induced DA efflux Is Unchanged in α-Syn Mice at ± Months
Because the stereotypic behavioral effect of amphetamine is conspicuously absent in α-Syn mice (Fleming et al., 2006), we examined whether amphetamine-evoked DA efflux, induced by reverse dialysis of the drug, was similarly perturbed on a second day of microdialysis measurements. This proved not to be the case (Fig. 3); amphetamine dose dependently elevated dialysate DA to levels that were not significantly different between genotypes (F2,24 5 36.42 P < 0.05 for effect of dose; F1,24 5 1.01 P > 0.05 for effect of genotype, n = 7 per genotype) despite the fact that basal levels remained twice as high in the α-Syn mice (4.5 ± 0.9 vs. 2.2 ± 0.6; this difference narrowly failed to attain statistical significance; P = 0.06). This suggests that the previously reported compromised behavioral response to amphetamine (Fleming et al., 2006) probably was due to alterations postsynaptic to DA terminals, as shown by our electrophysiological results.
Fig. 3.
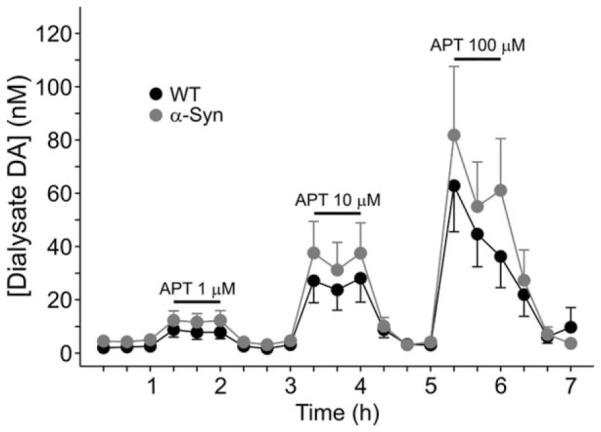
Incorporation of amphetamine (APT) into the dialysis perfusion fluid at increasing concentrations dose dependently elevated dialysate DA to a similar degree in WT and α-Syn mice.
Basal Striatal Extracellular DA Is Normalized in α-Syn Mice at 9.5 Months of Age
Analysis of basal dialysate sampled from the contralateral striatum of a subset (n = 5 per genotype) of previously sampled animals approximately 3.5 months later (at approximately 9.5 months of age) revealed normalization of DA concentration, although the data were much more variable (2.9 ± 1.2 nM and 3.5 ± 1.3 nM for α-Syn and WT, respectively).
Open-Field Activity Is Increased in α-Syn Mice at 7 Months
Increased extracellular DA is known to induce hyperactivity in mice (Zhuang et al., 2001). In line with the microdialysis data showing elevated extracellular DA at ± months, we observed an increase in open-field activity of α-Syn mice at approximately 7 months of age in a separate cohort of animals. Data include measurements in mice that were naïve to the open field (n = 5 in the WT group, n = 4 in the α-Syn group) and mice that were tested for the second time (n = 5 per group) having been previously tested at 5 months (data not shown). However, because t-tests did not reveal any significant differences between mice tested for the first time and those tested for the second time and also between different time points in the same mice (P > 0.4), the results of naïve and nonnaïve mice were pooled. Time spent in motion was significantly higher in α-Syn mice compared with WT mice (Fig. 4A; main effect of genotype, F1,17 5 11.45, P < 0.005), an effect that was apparent at each of the three 5-min recording epochs (P < 0.05 to P < 0.01). Total distance traveled was also significantly greater in α-Syn mice (Fig. 4B; main effect of genotype, F1,17 5 9.36, P < 0.01), which, again, was significant throughout the recording period (P < 0.05 to P < 0.01). Movement velocity was significantly higher in α-Syn mice (Fig. 4C; main genotype effect, F1,17 5 4.96, P = 0.04), an effect that attained significance in the second 5-min recording epoch only (P < 0.05).apparent at each of the three 5-min recording epochs (P < 0.05 to P < 0.01). Total distance traveled was also significantly greater in α-Syn mice (Fig. 4B; main effect of genotype, F1,17 5 9.36, P < 0.01), which, again, was significant throughout the recording period (P < 0.05 to P < 0.01). Movement velocity was significantly higher in α-Syn mice (Fig. 4C; main genotype effect, F1,17 5 4.96, P = 0.04), an effect that attained significance in the second 5-min recording epoch only (P < 0.05).apparent at each of the three 5-min recording epochs (P < 0.05 to P < 0.01). Total distance traveled was also significantly greater in α-Syn mice (Fig. 4B; main effect of genotype, F1,17 5 9.36, P < 0.01), which, again, was significant throughout the recording period (P < 0.05 to P < 0.01). Movement velocity was significantly higher in α-Syn mice (Fig. 4C; main genotype effect, F1,17 5 4.96, P = 0.04), an effect that attained significance in the second 5-min recording epoch only (P < 0.05).apparent at each of the three 5-min recording epochs (P < 0.05 to P < 0.01). Total distance traveled was also significantly greater in α-Syn mice (Fig. 4B; main effect of genotype, F1,17 5 9.36, P < 0.01), which, again, was significant throughout the recording period (P < 0.05 to P < 0.01). Movement velocity was significantly higher in α-Syn mice (Fig. 4C; main genotype effect, F1,17 5 4.96, P = 0.04), an effect that attained significance in the second 5-min recording epoch only (P < 0.05).
Fig. 4.

Open-field activity in 7-month-old WT and α-Syn mice. Time in motion (A), distance traveled (B), and movement velocity (C) were all significantly lower in α-Syn mice. *P < 0.05, **P < 0.01.
α-Syn Mice Exhibit Abnormal DA Modulation of Spontaneous EPSCs
Our previously published data demonstrated that the frequency of spontaneous EPSCs of MSSNs is significantly reduced in α-Syn mice (Wu et al., 2009). In the present experiment, we first examined spontaneous EPSCs in the presence of BIC and replicated our previously published data (Fig. 5A,B). The mean frequency of sEPSCs was significantly reduced in MSSNs from α-Syn mice in the present experiments [1.51 ± 0.14 Hz (n = 38) vs. 0.73 ± 0.12 Hz (n = 32), P < 0.001]. We have shown that the frequency of spontaneous inhibitory postsynaptic currents is also altered in α-Syn mice at × months of age (Wu et al., 2009).
Fig. 5.
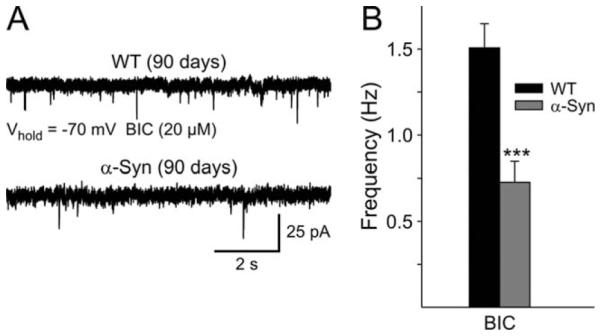
A: Representative traces showing spontaneous EPSCs from WT and α-Syn mice in BIC (Vhold = −70 mV). B: Mean frequency of sEPSCs from MSSNs for WT and α-Syn mice in BIC. ***P < 0.001.
We, and others, have shown that DA and amphetamine reduce the frequency of MSSN spontaneous EPSCs by activating presynaptic D2 receptors on corticostriatal terminals (Flores-Hernández et al., 1997; Cepeda et al., 2001; Bamford et al., 2004; Wu et al., 2007). To examine DA modulation in α-Syn mice, amphetamine (25 μM) was bath applied. Amphetamine produced a significant decrease in spontaneous EPSC frequency in cells from WT mice [−27% ± 8% (n = 13); P < 0.001; Fig. 6], whereas it had inconsistent effects in cells from α-Syn mice (four cells showed increases and seven cells showed decreases; −1% ± 8% average change). Amphetamine application did not affect the amplitude of spontaneous synaptic currents.
Fig. 6.
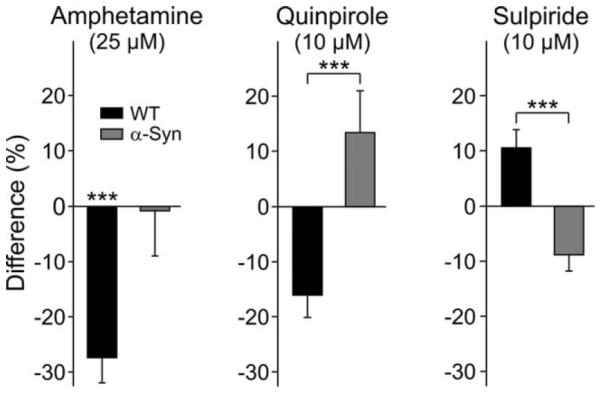
Left bar graphs show that amphetamine significantly reduced spontaneous EPSC frequency in WT mice but failed to affect spontaneous EPSC frequency in α-Syn mice. Middle bar graphs show that quinpirole decreased the frequency of spontaneous EPSCs in WT mice but produced opposite effects in MSSNs from α-Syn mice. Right bar graphs show that sulpiride increased the frequency of sEPSCs of MSSNs in WT mice but produced opposite effects in MSSNs from α-Syn mice. All recordings were made in the presence of BIC. ***P < 0.001.
Because amphetamine-induced DA efflux was unperturbed in vivo (Fig. 3), and given the recognized role of presynaptic D2 receptors in modulating corticostriatal glutamate transmission, the observation that the frequency of events was not significantly altered by amphetamine in α-Syn mice indicated that D2 receptors in these animals may be less sensitive. To test this hypothesis more directly, we applied quinpirole (10 μM), a selective D2 receptor agonist. Quinpirole produced a significantly different effect in MSSNs from α-Syn compared with those from WTs (P < 0.003). The frequency of spontaneous EPSCs in MSSNs was significantly reduced by quinpirole in WTs [−16% ± 5% (n = 13); P = 0.001; Fig. 6], whereas it was increased in MSSNs from α-Syn mice [13% ± 8% (n = 10)]. Although this increase was not statistically significant, the drug by genotype interaction was significant (P = 0.001), indicating that quinpirole produced different effects in α-Syn neurons compared with those from WTs. Sulpiride (10 μM), a selective D2 receptor antagonist, also produced a differential effect: the frequency of spontaneous EPSCs in MSSNs was significantly increased in WT cells [11% ± 3% (n = 12); P < 0.001], whereas it was decreased in α-Syn cells [−9% ± 3% (n = 11); Fig. 6]. Again, although the decrease narrowly failed to attain statistical significance (P = 0.06), the drug by genotype interaction was significant (P = 0.001), indicating that quinpirole produced different effects in α-Syn neurons compared with those from WTs. Amplitudes of spontaneous EPSCs were not significantly affected by quinpirole or sulpiride in either genotype. Taken together, these results support the hypothesis that the function of D2 receptors, probably located on corticostriatal glutamatergic terminals, is altered in α-Syn mice.
Striatal Tissue DA and Metabolite Content Is Normal at 7 Months but Reduced, Together With TH, at 14 Months in α-Syn Mice
There were no significant differences between the genotypes in striatal tissue content of DA, DOPAC, HVA, or 3MT at approximately 7 months of age (n = 8 and 11, for α-Syn and WT, respectively; data from 2 α-Syn and 1 WT were omitted as a result of a tissue weighing error; Table I). However, at approximately 14 months of age, α-Syn mice presented with 42% lower tissue levels of DA relative to WTs (P < 0.05; n = 8 and 8, respectively) accompanied by similarly significant reductions in DOPAC (35%; P < 0.05), HVA (25%; P < 0.01), and 3MT (49%; P < 0.01; Table I). These changes were accompanied by 24% lower TH levels relative to WT littermates (n = 8 per genotype) at 14 months of age as assessed by Western blot (Fig. 7).
TABLE I.
Striatal Tissue Content of DA and Metabolites at 7 and 14 Months of Age†
| 7 Months |
14 Months |
|||
|---|---|---|---|---|
| WT | α-Syn | WT | α-Syn | |
| DA | 39.6 ± 5.6 | 36.1 ± 2.3 ns | 23.3 ± 2.5 | 13.5 ± 2.6* |
| DOPAC | 2.9 ± 0.3 | 2.4 ± 0.3 ns | 2.0 ± 0.2 | 1.3 ± 0.2* |
| HVA | 5.0 ± 0.5 | 5.0 ± 0.3 ns | 2.4 ± 0.1 | 1.8 ± 0.1** |
| 3MT | 2.0 ± 0.3 | 1.5 ± 0.2 ns | 0.84 ± 0.3 | 0.43 ± 0.1** |
There was no statistically significant difference between the genotypes in striatal tissue content of dopamine and its three major metabolites at 7 months of age, but all four compounds were significantly lower in α-Syn mice at 14 months. Values are pmol/mg tissue ± SEM.
P < 0.05 vs. WT at 14 months.
P < 0.01 vs. WT at 14 months
Fig. 7.
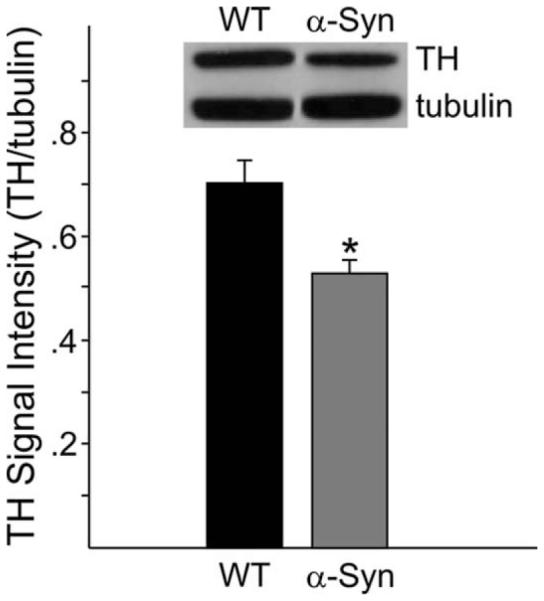
At 14 months of age, α-Syn mice presented with lower striatal TH levels as measured by Western blot. *P < 0.01.
L-DOPA-Reversible Sensory Motor deficits Are Apparent in α-Syn Mice at 15 Months
To determine whether the loss of striatal DA observed in 14-month-old mice was functionally significant, α-Syn mice at approximately 15 months of age (n = 6) were tested in a sensorimotor test known to reflect nigrostriatal DA deficits, the adhesion removal test (Schallert et al., 1982; Fleming et al., 2004). The transgenic animals took significantly longer to remove a sensory stimulus compared with WT (n = 7) mice (P < 0.05). Confirming the involvement of DA deficits in the behavioral impairment, this effect was reversed by L-DOPA, there being no significant difference between the genotypes following treatment with this drug (P > 0.05; Fig. 8).
Fig. 8.
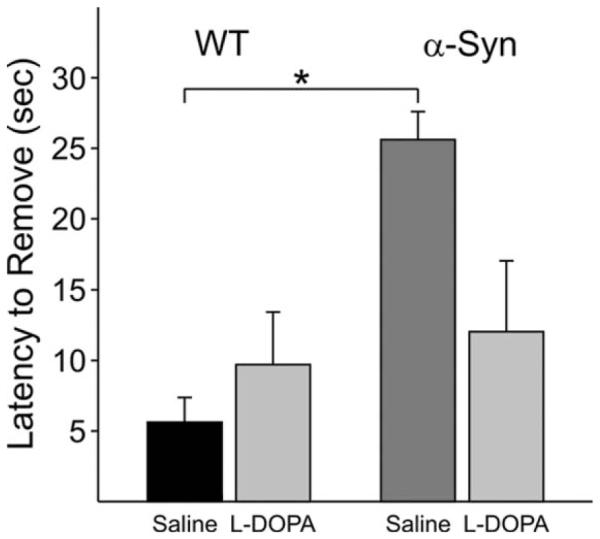
The time taken by a mouse to remove an adhesive sticker from its snout was significantly longer for α-Syn than for WT mice at 15 months. This genotype difference was erased by administration of L-DOPA. *P < 0.05.
DISCUSSION
The present study demonstrates a biphasic alteration in DA in mice overexpressing human WT α-Syn under the Thy1 promoter, with corresponding behavioral anomalies. We observed a striking elevation of striatal extracellular DA in young (6 months old) α-Syn mice relative to WTs that appears to be of vesicular origin, to be independent of changes in DA reuptake or metabolism, and to be functionally relevant as revealed by an increased activity in the open field. Whereas damphetamine-induced DA efflux was normal in these mice, the ability of this drug, and of specific D2 agonists and antagonists, to modulate striatal MSSN spontaneous EPSCs was severely compromised. All of these effects occurred at ages prior to those at which both striatal DA levels (current data) and number of nigral DA cell bodies (Fernagut et al., 2007) were shown to remain normal. Importantly, however, as these mice age (14 months), they present with significant and substantial reductions in striatal tissue content of DA and its metabolites and in striatal TH expression relative to WT mice. Furthermore, whereas previously described motor deficits at early ages are not L-DOPA-reversible (Fleming et al., 2006), by 14 months of age the sensory-motor deficits apparent in the α-Syn mice are reversed by L-DOPA, in line with the DA-depletion data. Thus, at 14–15 months of age, these mice exhibit canonical deficits associated with manifest PD. Our findings further validate use of this α-Syn-overexpressing mouse line at early ages as a model of premanifest PD and identify significant disruption of pre- and postsynaptic DA transmission prior to loss of striatal DA.
Extensive evidence supports a role for α-Syn in release mechanisms. α-Syn KO mice exhibit a subtle facilitation of DA release under paired-pulse conditions (Abeliovich et al., 2000) and an increased rate of refilling of the readily releasable pool of DA (Yavich et al., 2004). Furthermore, double KO of α-Syn and γ-synuclein results in enhanced electrical stimulation-evoked release of striatal DA under both single and burst stimulation conditions (Senior et al., 2008). These data have been taken to implicate α-Syn as a negative regulator of DA release (but see Cabin et al., 2002; Chandra et al., 2004). Accordingly, similar studies using either intact mice (Yavich et al., 2005) or cells (Larsen et al., 2006; Nemani et al., 2010) overexpressing WT or mutated human α-Syn have shown deficits in the readily releasable pool of DA. Our data, at first glance, might be considered inconsistent with these findings. However, it must be emphasized that, although such studies have measured the releasability of DA under electrically evoked conditions, they have not assessed the tonic level of extracellular DA as measured in the current study. This distinction leads us to propose that, although the releasability of striatal DA may indeed be compromised in α-Syn mice, other mechanisms, perhaps compensatory in nature, result in more DA being available extracellularly under basal conditions in vivo.
There are many potential causes of the elevated tonic extracellular DA but several were rendered unlikely by our results. The no-net-flux experiment served two functions, one of which was to confirm that the extracellular concentration of DA was indeed higher in the α-Syn mice. More importantly, the no-net-flux experiment failed to provide evidence of compromised DA reuptake: the in vivo extraction fraction, an indirect measure of DAT activity (Justice, 1993; Smith and Justice, 1994), was similar between genotypes. In fact, there was a trend toward increased brain extraction of perfused DA in α-Syn mice. This trend is consistent with models in which the normal function of α-Syn is considered to curtail translocation of DAT to the terminal membrane, a function compromised by mutation or overexpression of α-Syn (Sidhu et al., 2004). It should be noted, how-ever, that A53T-overexpressing mice have been reported to present with an impediment in DA reuptake assessed by chronoamperometry (Unger et al., 2006). The enhanced temporal resolution of the latter technique permits a more detailed kinetic analysis of reuptake than that offered by microdialysis. Therefore, it may be premature to rule out a compromised reuptake in the α-Syn mice on the basis of the no-net-flux data alone.
A simple reduction in DA metabolism is ruled out by our observation that extracellular DOPAC and HVA levels were not different from WT. Moreover, dialysate 3MT concentrations were elevated in α-Syn mice. These metabolite data are important for another reason; there is evidence in the literature supporting the idea that cytosolic DA is elevated, perhaps as a result of disrupted vesicular packaging, in mice in which human A30P α-Syn is expressed (Mosharov et al., 2006). This being the case, it is feasible that the elevated extracellular DA that we observe in the Thy-1 human WT α-Syn line results from reverse transport of DA from the cytoplasm. However, the metabolite data suggest otherwise. The only DA metabolite that was elevated, 3MT, is produced directly from DA that is first released, because COMT is not present in DA terminals but is instead found in nondopaminergic striatal neurons and glia (Kaakkola et al., 1987; Karhunen et al., 1995; Matsumoto et al., 2003). If cytosolic, rather than vesicular, DA was the source of the elevated extracellular DA in these mice, one might expect it to be accompanied by an increase in DOPAC and HVA as a result of metabolism initiated intraneuronally. Thus, it seems more likely that the elevated extracellular DA that we observe is of a vesicular origin. Given the absence of data implicating compromised reuptake, an alternative explanation is that DA neurons may be more tonically active. Such a scenario could transpire as a result of compensation for the reported α-Syn-induced deficits in the intrinsic releasability of DA cited above.
The functional significance of our observed tonic extracellular DA elevation at approximately ± months is demonstrated by the increased open-field activity at a similar age, a phenotype shared by other α-Syn-overex-pressing models (Unger et al., 2006; Graham and Sidhu, 2010). Interestingly, the elevated extracellular DA phenotype resolves by 9.5 months, perhaps reflecting a transition to the TH and DA depletion observed at 14 months.
Because α-Syn mice fail to show the normal stereotyped behavioral response to amphetamine (Fleming et al., 2006), we sought to determine whether this was due to a deficit in amphetamine-induced DA efflux, but we found this not to be the case: the levels of dialysate DA attained by amphetamine administered at concentrations over three orders of magnitude were similar. Amphetamine elevates extracellular DA by displacing DA from vesicles and promoting reverse transport (Sulzer et al., 1993), so these data further argue against a deficit in vesicular packaging as a source of the elevated extracellular DA in α-Syn mice. Moreover, they suggest that the source of the amphetamine-induced behavioral deficit lies postsynaptically to the DA terminals. In support of this, we found marked alterations in amphetamineand D2-receptor-mediated modulation of corticostriatal glutamate release based on measures of MSSN spontaneous EPSCs. Particularly striking was the reversal of the effect of D2 activation on the frequency of spontaneous EPSCs from a reduction in WTs to an increase in α-Syn mice. Why this should be the case is currently unclear, but it may be an effect secondary to chronically elevated extracellular DA, especially insofar as the present electro-physiological results resemble those found in DAT knock-down mice (Wu et al., 2007), which exhibit elevated extracellular DA levels. It should be noted, however, that the electrophysiological data were collected at an earlier time point than the microdialysis data (approximately × vs. ± months), so the sequence of events is as yet unclear. Moreover, studies in CHO cells indicate that α-Syn may modulate presynaptic D2 receptor signaling directly, independent of changes in extracellular DA (Kim et al., 2006). The observed effects may therefore be mediated directly via α-Syn overexpression in cortical glutamatergic neurons. It is also noteworthy that D2 receptor signaling appears to be affected in other genetic models of PD. For example, hyposensitivity of D2 receptors was observed in mice lacking the DJ-1 gene (Goldberg et al., 2005).
In conclusion, we have demonstrated a marked elevation in striatal extracellular DA accompanied by an alteration in DA modulation of corticostriatal glutamate transmission in the α-Syn mice at ages when DA terminals and cell bodies remain intact. Although a causal link with toxicity has not been demonstrated by the current data, they nevertheless raise the potential importance of α-Syn overexpression-induced elevation in extracellular, as opposed to cytoplasmic, DA as a factor in disease progression, because, as we show here, these mice go on to lose striatal terminal TH and DA as they age, ultimately presenting with sensorimotor deficits that are reversed by L-DOPA. It is generally considered that DA-related oxidative stress within the DA neuron is the major factor contributing to dopaminergic neurodegeneration (Sulzer, 2007; Mosharov et al., 2009), but it has long been recognized that exogenously applied DA is toxic to DA neurons (Michel and Hefti, 1990; Filloux and Townsend, 1993; Hastings et al., 1996). significantly, a recent study employing a viral model of α-Syn overexpression in rats reported an increase in striatal tissue DOPAC:DA ratio, potentially reflecting an increase in tonic DA release similar to that observed in our model and which, again, presented prior to DA neuron degeneration (Chung et al., 2009). Increased striatal extracellular DA was also apparent in another model of PD, the parkin KO mouse (Goldberg et al., 2003), suggesting that increased extracellular DA may be a common mechanism contributing to substantia nigra DA neuron stress and eventual demise. It is particularly noteworthy in this regard that recent epidemiological data indicate that increased PD risk is associated not only with compromised VMAT function (Glatt et al., 2006) but also with compromised DAT function, the latter in conjunction with pesticide exposure (Ritz et al., 2009). That these and many other α-Syn mouse models of PD have thus far proved to retain their full complement of DA cell bodies, relative to WT, presumably indicates a resistance of mice, generally, to the ultimate toxic effect of α-Syn. We are currently aging these mice still further to determine whether they ultimately present with lower numbers of nigral DA neurons relative to WT. However, given the observed loss of striatal DA and TH at 14 months, we posit that younger α-Syn mice represent an early stage of PD and can provide important information on mechanisms that initiate later pathophysiological processes.
ACKNOWLEDGMENTS
We thank Donna Crandall for help with the illustrations and J. Watson for critical evaluation of earlier versions of the manuscript.
Contract grant sponsor: UPHS; Contract grant number: P50NS38367; Contract grant number: U54ES12078; Contract grant number: NS33538.
REFERENCES
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-Synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. alpha-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-Synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Altemus KL, Flores-Hernández J, Calvert CR, Jokel ES, Grandy DK, Low MJ, Rubinstein M, Ariano MA, Levine MS. Facilitated glutamatergic transmission in the striatum of D2 dopamine receptor-deficient mice. J Neurophysiol. 2001;85:659–670. doi: 10.1152/jn.2001.85.2.659. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernández-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC. Doubleknockout mice for alphaand beta-Synuclein: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-Synuclein overexpression in rodents: a useful model of Parkinson’s disease? Exp Neurol. 2008;209:22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-Synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-Synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux F, Townsend JJ. Preand postsynaptic neurotoxic effects of dopamine demonstrated by intrastriatal injection. Exp Neurol. 1993;119:79–88. doi: 10.1006/exnr.1993.1008. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-Synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-Synuclein. Neuroscience. 2006;142:1245–1253. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF. Olfactory deficits in mice overexpressing human wildtype alpha-Synuclein. Eur J Neurosci. 2008;28:247–256. doi: 10.1111/j.1460-9568.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Hernández J, Galarraga E, Bargas J. Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse. 1997;25:185–195. doi: 10.1002/(SICI)1098-2396(199702)25:2<185::AID-SYN9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; New York: 1997. [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Glatt CE, Wahner AD, White DJ, Ruiz-Linares A, Ritz B. Gain-of-function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Hum Mol Genet. 2006;15:299–305. doi: 10.1093/hmg/ddi445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopa-minergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Graham DR, Sidhu A. Mice expressing the A53T mutant form of human alpha-Synuclein exhibit hyperactivity and reduced anxiety-like behavior. J Neurosci Res. 2010;88:1777–1783. doi: 10.1002/jnr.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubbs F. Procedures for detecting outlying observations in samples. Technometrics. 1969;11:1–21. [Google Scholar]
- Gureviciene I, Gurevicius K, Tanila H. Role of alpha-Synuclein in synaptic glutamate release. Neurobiol Dis. 2007;28:83–89. doi: 10.1016/j.nbd.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci U S A. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Gallant K, Gross GG, Levine MS, Chesselet MF. Early behavioral deficits in R6/2 mice suitable for use in preclinical testing. Neurobiol Dis. 2005;20:1–11. doi: 10.1016/j.nbd.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Justice JB., Jr Quantitative microdialysis of neurotransmitters. J Neurosci Methods. 1993;48:263–276. doi: 10.1016/0165-0270(93)90097-b. [DOI] [PubMed] [Google Scholar]
- Kaakkola S, Mannisto PT, Nissinen E. Striatal membrane-bound and soluble catechol O-methyl-transferase after selective neuronal lesions in the rat. J Neural Transmiss. 1987;69:221–228. doi: 10.1007/BF01244343. [DOI] [PubMed] [Google Scholar]
- Karhunen T, Tilgmann C, Ulmanen I, Panula P. Catechol O-methyltransferase (COMT) in rat brain: immunoelectron microscopic study with an antiserum against rat recombinant COMT protein. Neurosci Lett. 1995;187:57–60. doi: 10.1016/0304-3940(95)11337-v. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim SY, Na YS, Lee HJ, Chung KC, Baik JH. Alphasynuclein enhances dopamine D2 receptor signaling. Brain Res. 2006;1124:5–9. doi: 10.1016/j.brainres.2006.09.079. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. Neuroscience. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I, Chesselet M-F. Genetic mouse models of Parkinson’s disease: the state of the art. Prog Brain Res. 2010;184:53–87. doi: 10.1016/S0079-6123(10)84004-X. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Weickert CS, Akil M, Lipska BK, Hyde TM, Herman MM, Kleinman JE, Weinberger DR. Catechol O-methyltransferase mRNA expression in human rat brain: evidence for a role in cortical neuronal function. Neuroscience. 2003;116:127–137. doi: 10.1016/s0306-4522(02)00556-0. [DOI] [PubMed] [Google Scholar]
- Michel PP, Hefti F. Toxicity of 6-hydroxydopamine and dopamine for dopaminergic neurons in culture. J Neurosci Res. 1990;26:428–435. doi: 10.1002/jnr.490260405. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bové J, Prou D, Hananiya A, Markov D, Poulsen N, Larsen KE, Moore CM, Troyer MD, Edwards RH, Przedborski S, Sulzer D. alpha-Synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-Synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-Synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-Synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Ritz BR, Manthripragada AD, Costello S, Lincoln SJ, Farrer MJ, Cockburn M, Bronstein J. Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ Health Perspect. 2009;117:964–969. doi: 10.1289/ehp.0800277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-Synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Schallert T, Upchurch M, Lobaugh N, Farrar SB, Spirduso WW, Gilliam P, Vaughn D, Wilcox RE. Tactile extinction: distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol Biochem Behav. 1982;16:455–462. doi: 10.1016/0091-3057(82)90452-x. [DOI] [PubMed] [Google Scholar]
- Senior SL, Ninkina N, Deacon R, Bannerman D, Buchman VL, Cragg SJ, Wade-Martins R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-Synuclein and gamma-Synuclein. Eur J Neurosci. 2008;27:947–957. doi: 10.1111/j.1460-9568.2008.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P. alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004;565:1–5. doi: 10.1016/j.febslet.2004.03.063. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. alpha-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Par-kinson’s disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Totterdell S, Meredith GE. Localization of alpha-Synuclein to identified fibers and synapses in the normal mouse brain. Neuroscience. 2005;135:907–913. doi: 10.1016/j.neuroscience.2005.06.047. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Aggregation of neurofilament and alpha-Synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- Unger EL, Eve DJ, Perez ZA, Reichenbach DK, Xu Y, Lee MK, Andrews AM. Locomotor hyperactivity and alterations in dopamine neurotransmission are associated with overexpression of A53T mutant human alpha-Synuclein mice. Neurobiol Dis. 2006;21:431–443. doi: 10.1016/j.nbd.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Wang L, Fleming SM, Chesselet MF, Taché Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-Synuclein. Neuroreport. 2008;19:873–876. doi: 10.1097/WNR.0b013e3282ffda5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JB, Hatami A, David H, Masliah E, Roberts K, Evans CE, Levine MS. Alterations in corticostriatal synaptic plasticity in mice overexpressing human apla synuclein. Neuroscience. 2009;159:501–513. doi: 10.1016/j.neuroscience.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Cepeda C, Zhuang X, Levine MS. Altered corticostriatal neurotransmission and modulation in dopamine transporter knockdown mice. J Neurophysiol. 2007;98:423–432. doi: 10.1152/jn.00971.2006. [DOI] [PubMed] [Google Scholar]
- Wu N, Joshi PR, Cepeda C, Masliah E, Levine MS. α-Synuclein overexpresion in mice alters synaptic communication in the corticostriatal pathway. J Neurosci Res. 2009;8:1764–1776. doi: 10.1002/jnr.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of alpha-Synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, Oksman M, Tanila H, Kerokoski P, Hiltunen M, van Groen T, Puolivali J, Mannisto PT, Garcia-Horsman A, MacDonald E, Beyreuther K, Hartmann T, Jakala P. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-Synuclein. Neurobiol Dis. 2005;20:303–313. doi: 10.1016/j.nbd.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci U S A. 2001;98:1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]