

Abstract
The lager beer yeast Saccharomyces pastorianus is considered an allopolyploid hybrid species between S. cerevisiae and S. eubayanus. Many S. pastorianus strains have been isolated and classified into two groups according to geographical origin, but this classification remains controversial. Hybridization analyses and partial PCR-based sequence data have indicated a separate origin of these two groups, whereas a recent intertranslocation analysis suggested a single origin. To clarify the evolutionary history of this species, we analysed 10 S. pastorianus strains and the S. eubayanus type strain as a likely parent by Illumina next-generation sequencing. In addition to assembling the genomes of five of the strains, we obtained information on interchromosomal translocation, ploidy, and single-nucleotide variants (SNVs). Collectively, these results indicated that the two groups of strains share S. cerevisiae haploid chromosomes. We therefore conclude that both groups of S. pastorianus strains share at least one interspecific hybridization event and originated from a common parental species and that differences in ploidy and SNVs between the groups can be explained by chromosomal deletion or loss of heterozygosity.
Keywords: lager beer yeast, interspecies hybrid, Saccharomyces pastorianus, loss of heterozygosity, allopolyploid
1. Introduction
Bottom-fermenting strains of brewing yeast—also known as lager beer yeast—represent a large portion of the beer market and have therefore been the focus of mycological research for centuries. Lager beer yeasts have been taxonomically classified as Saccharomyces pastorianus since the 19th century and are classified into two groups according to geographical origin: the Saaz type (Group 1) originally used in Bohemia and the Frohberg type (Group 2) in Germany.1,2
Many pure strains have since been identified, including the type strains S. monacensis (CBS1503; Group 1), S. carlsbergensis (CBS1513; Group 1), and S. pastorianus (CBS1538; Group 1) as well as Weihenstephan 34/70 (Group 2), a widely used industrial strain. Saccharomyces pastorianus is an allopolyploid hybrid of two Saccharomyces species: the ale beer yeast S. cerevisiae and a hypothesized novel species similar to S. bayanus. In 2011, S. eubayanus3 was discovered on southern beech trees in Patagonia, South America, and identified as the absent parental species of lager beer yeast. Saccharomyces eubayanus has also recently been identified on trees in Tibet, Far East Asia, spurring a debate about the precise origin of this species.4
Genomic analysis techniques, such as array comparative genome hybridization (array-CGH), have identified a difference in ploidy between the two groups of S. pastorianus strains. Group 1 (Saaz type) strains are approximately haploid for the S. cerevisiae genome, whereas Group 2 (Frohberg type) strains are diploid for the S. cerevisiae genome.5 A Sanger sequencing-based genome sequence analysis of S. pastorianus Weihenstephan 34/706 conducted by our group revealed various genetic features, such as whole-genome structure and genomic divergence, at the single-nucleotide level. The genomes of many interspecies hybrid strains have also been sequenced, including the osmotolerant yeast species Pichia sorbitolphila,7 the Zygosaccharomyces bailii hybrid strain ISA1307,8 and the wine yeast strain VIN7.9 As in our previous study of S. pastorianus,6 most analyses of allopolyploid species genomes have been performed without the genome sequence of one or both parental species. For example, in the case of P. sorbitolphila,7 neither parental species has been isolated; its sequence data are divided into two types based on the GC content of the expected parental species. The lack of information on one or both parental genomes has complicated downstream genomic analyses. Complete (or near-complete) genomes of parental species can provide more information on genomic variation in interspecies hybrids and facilitate the identification of translocation breakpoints between interparental homologous chromosomes at the single-nucleotide level, genome rearrangements, and differences in ploidy.
In S. pastorianus, ploidy differences, copy number variations, and single-nucleotide variants (SNVs) identified by array-CGH10,11 or other approaches have suggested that the Saaz (Group 1) and Frohberg (Group 2) types originated from independent hybridization events,11–13 a theory that has gained broad acceptance. In contrast, the recent confirmation of two breakpoints associated with interchromosomal translocation of HSP82 and KEM1 genes implies a common origin.5 Therefore, the evolutionary history of S. pastorianus remains controversial, primarily because of the lack of reference genome information for S. eubayanus. For example, array-CGH analysis was performed based on the S. cerevisiae S288C and S. uvarum CBS7001 genomes,11 which are similar to non-Sc type S. pastorianus, but it was not possible to estimate differences in ploidy or sequence variations between S. pastorianus strains from the results.
In the present study, we obtained the whole-genome sequences of 10 S. pastorianus strains by Illumina next-generation sequencing (NGS) to gain insight into the evolutionary history of this species. The strains included five strains each from Groups 1 and 2, including three type strains [S. monacensis (CBS1503), S. carlsbergensis (CBS1513), and S. pastorianus (CBS1538)] and S. pastorianus Weihenstephan 34/70 (W34/70), which we sequenced previously by the Sanger method.6 NGS technology enables more accurate sequencing and produces longer continuous genome sequences. In addition to paired-end sequences, long insert mate-pair libraries were also obtained for the three type strains and W34/70 and were applied to de novo assemblies to obtain overall genomic structures, which were then compared. However, as in the case of previous studies of interspecies hybrid strains,7–9 a more detailed analysis of ploidy and evolutionary relationships based on sequence differences from the assembled sequences was difficult. To precisely determine the sequence differences between strains, we adopted mapping-based analysis using the two parent genomes as reference sequences, including the available S. cerevisiae (Sc) (S288C) genome. Because a reference sequence was not available for S. eubayanus (Se), we assembled the draft genome sequence of S. eubayanus CBS12357 using NGS data. These two reference genomes were used for mapping-based genome comparisons among the 10 S. pastorianus strains that considered the relatively high heterozygosity of S. pastorianus. Here, heterozygosity refers to the sequence difference between intrahomologous (Sc/Sc or Se/Se) chromosomes; loss of heterozygosity (LOH) refers to the loss of heterogeneity between intrahomologous chromosomes. The results provide information on ploidy, novel chromosomal translocations, mitochondrial (mt)DNA sequences, and the phylogenetic relationships among Sc and Se types that provides new insights into the origin and evolutionary history of S. pastorianus.
2. Materials and methods
2.1. Strain and sequence information
The following strains were used in this study: S. pastorianus CBS1503 (S. monacensis type strain), CBS1513 (S. carlsbergensis type strain), CBS1538 (S. pastorianus type strain), CBS1174, CBS2440, Weihenstephan 34/70 (W34/70), CBS1483, CBS1484, CBS2156, and CBS5832; S. eubayanus CBS12357 (type strain) and BaiFY1; S. bayanus NBRC1948 and CBS380; and S. cerevisiae S288C. Saccharomyces pastorianus Weihenstephan 34/70 was provided by Fachhochschule Weihenstephan (Freising, Germany); the other strains, except S. eubayanus BaiFY1, were obtained from the CBS-KNAW Culture Collection Center (Utrecht, The Netherland). Sequence data for S. eubayanus BaiFY1 were downloaded from the Sequence Read Archive, NCBI.
2.2. Genome sequencing and assembly
We prepared paired-end sequencing data with the Illumina platform (Hayward, CA, USA) for 14 strains of three species: S. pastorianus Group 1 (CBS1503, CBS1513, CBS1538, CBS1174, and CBS2440) and Group 2 (W34/70, CBS1483, CBS1484, CBS2156, and CBS5832); S. eubayanus (CBS12357 and BaiFY1); and S. bayanus (NBRC1948 and CBS380). With the exception of S. eubayanus BaiFY1, all sequence data were determined in this study by Illumina Miseq. Saccharomyces bayanus sequences were used only for mtDNA analysis. Libraries (insert size = 600 bp) were prepared using the TruSeq PCR-free DNA Sample Prep kit (Illumina) with a read length of 300 bp. Other sequence data for S. eubayanus BaiFY1 obtained from the Hiseq2000 platform with 151-bp paired-end libraries were available from the Sequence Read Archive (accession no. SRX646335). For de novo assembly, mate-pair libraries for CBS1503, CBS1513, CBS1538, W34/70, and CBS12357 were prepared using the Nextera Mate Pair Sample Prep kit (Illumina) and 3,000- to 15,000-bp fragments extracted from an agarose gel for each sample. All libraries were also sequenced using Illumina Miseq. Low-quality regions and adaptor sequences in the reads were trimmed with Platanus_trim v.1.0.7 for subsequent analyses.
The genomes of five strains sequenced from the paired-end and mate-pair libraries were assembled using Platanus v.1.2.4 software.14 Contig assembly was performed using only paired-end libraries, and scaffolding and gap closing were performed using both libraries. Prior to scaffolding, mate-pair reads were mapped on contigs using the BWA program15 to remove duplicate reads generated during PCR amplification. Contig assembly for interspecies hybrid strains (CBS1503, CBS1513, CBS1538, and W34/70) was conducted with parameter −n 15 (for W34/70) or 20 (for other strains), and scaffolding was performed with parameter −u 0 to avoid removing low-coverage scaffolds derived from haploid chromosomes. The results of the assembly were evaluated by confirming the physical coverage of 6-kb mate-pair reads. Finally, contamination and mtDNA sequences were removed from scaffolds ≥500 bp by alignment with NCBI Bacteria DB, RefSeq viral DB, and BLASTN16 for mtDNA sequences (minimum identity = 90% and minimum query coverage = 50%). The assembly of mitochondrial genomes is described below. CBS12357 was assembled using Platanus v.1.2.4 with default settings.
To construct a CBS12357 draft genome for hybrid strains in subsequent analyses, scaffolds were aligned with the S. cerevisiae S288C complete genome sequence using BLASTN. Only one locus, covering the rDNA region in chromosome XII, was connected by N-runs sequences manually, and two known interchromosomal translocations6,17 (namely, II–IV and VIII–XV) that were structurally distinct from S288C were not divided and maintained. Finally, we obtained super-scaffolds that were adequate to call chromosome-level reference sequences. To estimate the genome size of the hybrid strains, paired-end reads were mapped to the parental species' genomes (S288C complete and CBS12357 draft genomes) and to the mtDNA of each strain, and the length of regions covering ≥10 reads was calculated.
2.3. Detection of interchromosomal translocations in hybrid strains
Interchromosomal translocations between Sc and Se types were detected based on mate-pair reads, which were mapped to the reference (S. cerevisiae S288C complete and S. eubayanus CBS12357 draft) genomes using BWA after removing low-identity and multihit reads. Chromosomes were divided into 10,000-bp blocks, and the number of mate-pair reads linking the blocks was tabulated. Links consisting of ≥100 pairs between Sc- and Se-type blocks were displayed in a circus plot.18 To reveal breakpoints at the nucleotide level, local assembly was performed using paired-end reads; when one of these was mapped near a breakpoint (as estimated by the procedure described above) and the opposite side was not mapped, the read corresponding to the latter was identified as a candidate covering the breakpoint region. These reads were collected and assembled.
2.4. Estimation of ploidy in hybrid strains
Sequence coverage for the 10 S. pastorianus strains was calculated based on the number of reads mapped to the two parental species [S. cerevisiae (S288C) and S. eubayanus (CBS12357)] to confirm the accuracy of the total assembly size and estimate the ploidy of each chromosome. Sequence reads were mapped to the reference genomes using BWA. Mapped reads with <90% identity or with multiple hits were removed. Following realignment using GATK realigner,19 sequence coverage at each locus was calculated using SAMtools mpileup.20 Ploidy was estimated as the ratio of the sequence coverage of the focus region to the haploid coverage obtained from the whole-genome coverage distribution. The moving average of the calculated ploidy (window size = 10,000 bp and step size = 1,000 bp) was then plotted.
2.5. SNV calling in 10 S. pastorianus strains
Mapping of paired-end reads and filtering were conducted as described above, and SNV calling was performed using SAMtools mpileup and an original Perl script that calculated the number of mapped reads, their strands, variants, and allele frequencies (AFs; i.e. the ratio of reads supporting the variant to mapped reads at the locus) from the mpileup format. SNVs were identified according to the following criteria: ≥20 reads were mapped to the locus; variants were supported by two or more mapped reads on both the forward and reverse strands; and insertion/deletion (InDel) mutations were excluded. SNVs were classified as either homo (0.8 ≤ AF) or hetero (0.2 ≤ AF < 0.8) type.
2.6. Phylogenetic analysis
Phylogenetic analysis of the 10 S. pastorianus strains and 2 parental species was performed based on SNV sites for which ≥20 reads were mapped in all strains, excluding InDel mutations. Analyses were conducted separately for Sc and Se types to identify differences between the types. Heterozygous sites were also considered, and each allele at each SNV site was treated separately. Phylogenetic estimates were calculated by the maximum likelihood approach using phyml (−b 1,000),21 and phylogenetic trees were generated using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
2.7. Assignment of hetero-SNVs and haplotype phasing
To build two intrahomologous chromosomes from the diploid Sc-type chromosomes in Group 2 strains, the linkage relationships of adjacent hetero-SNVs were solved by pair reads mapped on a region bearing two or more hetero-SNVs. Regions in which pair reads solved the linkage relationships of consecutive adjacent hetero-SNVs were divided into two lines corresponding to chromosomal haplotypes and were defined as continuous haplotype-phased blocks. Blocks were divided by unphased (mainly caused by long no-hetero-SNVs) regions. W34/70 sequence reads were used in this analysis for the following reasons: the Sc-type genome of W34/70 has the highest heterozygosity among the five Group 2 strains; and mate-pair data (insert size 3 and 6 kb) were available. A Group 1 consensus sequence was constructed based on the major alleles in the five Group 1 strains. If the major allele was undefined at a locus, the locus was removed from comparison with W34/70 haplotypes.
2.8. Phylogenetic analysis based on haplotype-phased chromosomal SNVs
The identities between each haplotype W34/70 sequence and Group 1 consensus sequences were calculated at each block. The W34/70 haplotyped chromosomes with higher homology to Group 1 were labelled as ‘W34/70_a’, and the others were labelled as ‘W34/70_b’. Finally, the identities between Group 1 vs. W34/70_a and Group 1 vs. W34/70_b were aggregated. For comparison, W34/70 artificial intrahomologous chromosomes were virtually constructed by randomly shuffling allele nucleotides and ignoring linkage information at every locus. The obtained artificial chromosomes were divided into two groups, W34/70_a and W34/70_b, according to the same methods for real intrahomologous chromosomes. This ‘shuffling test’ was repeated 100 times, and the average was calculated.
2.9. Assembly and phylogenetic analysis of mtDNA
MtDNA assembly and phylogenetic analysis were performed for nine strains of four species, i.e. S. cerevisiae (S288C), S. pastorianus (CBS1503, CBS1513, CBS1538, and W34/70), S. eubayanus (CBS12357 and BaiFY1), and S. bayanus (NBRC1948 and CBS380). MtDNA sequences (with the exception of S288C) were assembled using only high-frequency k-mers (i.e. >2-fold higher than the average k-mer frequency value obtained from sequence reads) because the copy number of mitochondria was much higher than that of chromosomes. After gap closing, scaffolds were aligned to S. cerevisiae S288C mtDNA using BLASTN and assembled with paired-end reads that were mapped to the edges of the scaffold to generate a circle.
The mtDNA of each strain was aligned with the open reading frames (ORFs) of S. cerevisiae S288C mtDNA using BLASTX to identify the ORFs of each strain. Multiple alignment with the ORFs was performed using ClustalW2. Phylogenetic estimates using the maximum likelihood method were obtained with phyml (−b 1,000), and a phylogenetic tree was generated using FigTree.
3. Results and discussion
3.1. Genome sequencing
The sequence sizes of each strain and library are presented in Table 1 and Supplementary Table S1; 32-mer frequency distributions were calculated from the sequence reads of five strains each of S. pastorianus Groups 1 and 2 and of S. eubayanus CBS12357 (Fig. 1A–C). In haploid or low-heterozygosity diploid cases, the k-mer frequency distribution exhibited a single peak at the average sequence frequency, as observed for S. eubayanus CBS12357 (Fig. 1C). In contrast, most of the 32-mer distributions for S. pastorianus strains had two or more peaks (Fig. 1A and B), which may have resulted from the aneuploidy of hybrid strains. Based on the previous array-CGH-based analysis,11 Group 1 strains are mainly derived from haploid S. cerevisiae and diploid S. eubayanus, and the left and right peaks in the double peak correspond to sequences originating from these two species, respectively. However, the presence of three or more peaks implies that some chromosomal regions are triploid or polyploid for parental strain chromosomes. Similarly, Group 2 strains are considered to be mainly from diploid S. cerevisiae and diploid S. eubayanus, yet the 32-mer distribution did not exhibit a pure single peak and, indeed, displayed double peaks in some strains (CBS1483) or low peaks resembling shoulders occurring at approximately half the frequency of the highest peak (W34/70, CBS1484, and CBS5832). This irregular distribution may have been due to heterozygosity in addition to polyploidy. The existence of SNVs between homologous chromosomes creates different 32-mers derived from each haplotype, and highly heterozygous genomes cause significant peaks at half the frequency of diploid peaks. The 32-mer distribution graph confirmed that S. pastorianus strains exhibit complex polyploidy or heterozygosity. The left-most peaks derived from the haploid genome range from 30 to 70, suggesting that the number of sequence reads for the hybrid strains was adequate. The total sequence size of the Illumina paired-end reads for S. eubayanus CBS12357 was 1.4 Gb with an estimated genome coverage of 120-fold, whereas that of the mate-pair reads was 2.8 Gb, with insert sizes of 3–15 kb.
Table 1.
Strains used in this study and total sequence sizes (Mb)
| Strains | Libraries (target insert size, bp) |
Remarks | ||||
|---|---|---|---|---|---|---|
| Paired end (600) | Mate pair (3k) | Mate pair (6k) | Mate pair (10k) | Mate pair (15k) | ||
| S. pastorianus | ||||||
| Group 1 | ||||||
| CBS1503 | 5,975.6 | 913.6 | 773.1 | — | — | S. monacensis type strain |
| CBS1513 | 5,231.5 | 882.9 | 923.5 | — | — | S. carlsbergensis type strain |
| CBS1538 | 5,400.5 | 915.3 | 766.3 | — | — | S. pastorianus type strain |
| CBS1174 | 3,277.6 | — | — | — | — | |
| CBS2440 | 3,126.8 | — | — | — | — | |
| Group 2 | ||||||
| W34/70 | 3,947.7 | 916.0 | 824.2 | — | — | Widely used industrial strain |
| CBS1483 | 2,388.4 | — | — | — | — | |
| CBS1484 | 2,559.7 | — | — | — | — | |
| CBS2156 | 2,493.8 | — | — | — | — | |
| CBS5832 | 3,113.3 | — | — | — | — | |
| S. eubayanus | ||||||
| CBS12357 | 1,408.3 | 670.1 | 748.0 | 719.0 | 680.5 | S. eubayanus type strain |
| BaiFY1 | 1,570.3 | — | — | — | — | |
| S. bayanus | ||||||
| NBRC1948 | 1,343.6 | — | — | — | — | |
| CBS380 | 3,778.9 | — | — | — | — | S. bayanus type strain |
Figure 1.
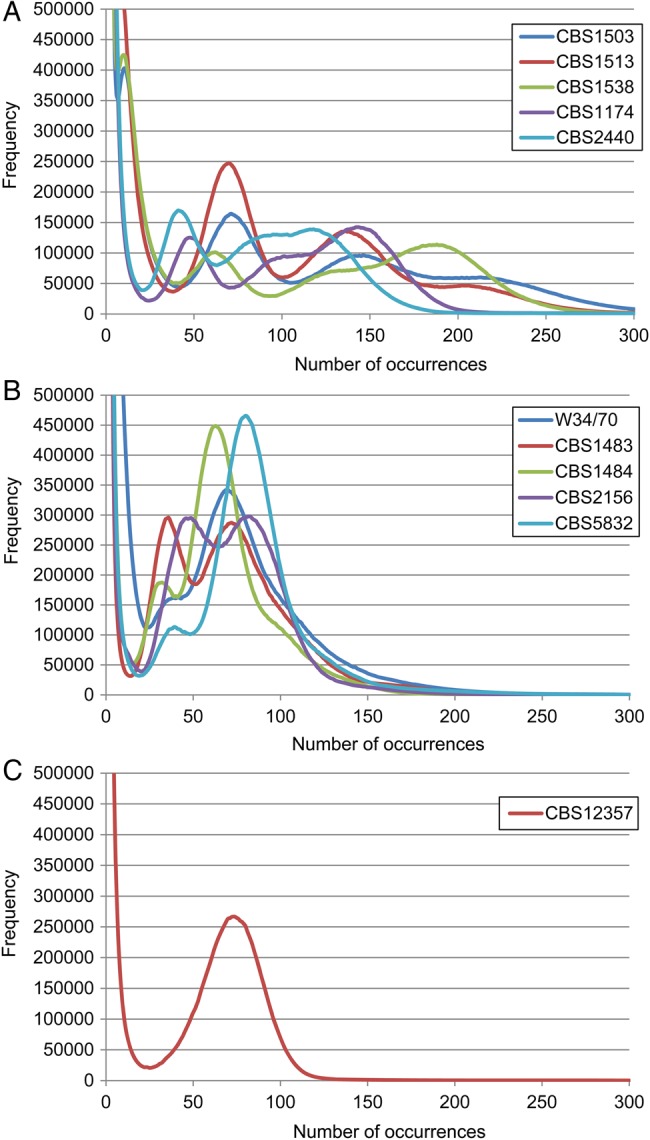
Frequency distribution of 32-mers in 10 S. pastorianus and S. eubayanus strains. Shown are the 32-mer distributions of (A) five Group 1 S. pastorianus strains; (B) five Group 2 S. pastorianus strains; and (C) the S. eubayanus strain. This figure is available in black and white in print and in colour at DNA Research online.
3.2. Genome assembly
The assembly statistics for S. pastorianus CBS1503, CBS1513, CBS1538, and W34/70 and S. eubayanus CBS12357 are presented in Table 2 and were, in general, very robust. For example, all N50 lengths exceeded 400 kb, and except in CBS1538, the longest scaffold lengths exceeded 1 Mb, comparable to the length of the longest chromosome of S. cerevisiae S288C (chromosome IV, 1.5 Mb). We previously reported an N50 length of 108 kb for W34/70,6 only approximately one-seventh of the value observed here. In particular, the assembly for S. eubayanus CBS12357 yielded 17 scaffolds >5,000 bp, one more than the chromosome number of 16. The S. eubayanus CBS12357 chromosome corresponding to chromosome XII of S. cerevisiae S288C was divided into two scaffolds that overlapped the rDNA region and were connected by ‘N run 5000 bp’. We ultimately obtained 16 super-scaffolds with a total genome size of 11.7 Mb—slightly smaller than that of S288C—with 30 gaps totalling 22,137 bp. Based on the total size of the scaffolds, the number of scaffolds, and the small number of gaps, the S. eubayanus CBS12357 draft genome was deemed adequate for use as a reference genome.
Table 2.
Assembly statistics for five strains of two species
| Strains | Total length (bp) | Number of sequences | N50 length (bp) | N50 # | Minimum length (bp) | Maximum length (bp) |
|---|---|---|---|---|---|---|
| S. pastorianus | ||||||
| Group 1 | ||||||
| CBS1503 | 17,195,167 | 631 | 484,478 | 12 | 500 | 1,060,739 |
| CBS1513 | 19,248,212 | 178 | 644,406 | 12 | 502 | 1,050,489 |
| CBS1538 | 14,404,124 | 277 | 428,791 | 13 | 501 | 760,567 |
| Group 2 | ||||||
| W34/70 | 22,500,926 | 495 | 723,289 | 13 | 500 | 1,455,873 |
| S. eubayanus | ||||||
| CBS12357 (super-scaffolds) | 11,666,993 | 17 | 833,488 | 6 | 196,426 | 1,269,399 |
| (11,671,993) | 16 | 903,844 | 6 | 196,426 | 1,269,399 | |
The assembly statistics revealed some interesting features. The total sequence size of the Group 2 W34/70 strain (22.5 Mb) was nearly equal to the sum of the S. cerevisiae S288C and S. eubayanus CBS12357 genome sizes (12.1 and 11.7 Mb, respectively). In contrast, the genome sizes of Group 1 strains—which ranged from 14.4 to 19.2 Mb—were much smaller than this value, suggesting partial or complete chromosomal deletion. Additionally, although the four S. pastorianus strains exhibited robust statistics (e.g. N50 and maximum scaffold length), the scaffold numbers were much higher than those of S. eubayanus CBS12357. Large N50 values and the considerable number of scaffolds suggest the existence of many short scaffolds, potentially due to the complexity of chromosome structure. During interchromosomal translocation, a homologous diploid or triploid chromosome may translocate to another interparental chromosome, leaving behind the remaining one or two homologous chromosomes. A junctional structure would then be present in the chromosome, and assembly results would be divided. Interchromosomal translocations have been reported,5,6,2,2 and this scenario is also supported by the unusual 32-mer distribution (Fig. 1). These complex chromosome structures may not be resolved by the assembly method; in the following sections, we describe our investigation of translocation structure performed by mapping mate-pair reads and the elucidation of ploidy distribution by mapping paired-end reads.
3.3. Estimation of ploidy in S. pastorianus strains
The ploidy of the 10 S. pastorianus strains was estimated based on the sequence coverage of paired-end reads mapped onto the genomes of the parental species (Fig. 2). Clear differences in ploidy between Groups 1 and 2 were observed. In the former, the Sc-type genome was haploid in most cases, and partial or complete chromosomal deletions were frequently observed; chromosomes I, IX, X, and the left arm of VII were present in these strains, and deletions of the right arm of chromosome IV, left arm of XIII, and the entire chromosome XII were common. Most Sc-type chromosomes in Group 1 were haploid or missing, whereas most Se-type chromosomes were diploid or triploid. In contrast, Group 2 Sc-type chromosomes were always haploid or diploid, whereas Se-type chromosomes ranged from haploid to triploid. Each parental type appeared aneuploid in each strain, but the sum of interhomologous chromosomes (between Sc- and Se-type) was approximately identical. Specifically, five strains in Group 1 and CBS2156 were basically triploid, and those in Group 2—except for CBS2156—were tetraploid. Despite the chromosomal deletion trails that were observed, a mechanism for maintaining a constant total number of intra- and interparental homologous chromosomes appears to exist in Sc and Se types. The differences in ploidy between the two groups provide additional evidence that both groups originated independently from the same parental species.
Figure 2.

Ploidy distribution in S. pastorianus based on sequence coverage mapped onto parental genomes for (A) Group 1 and (B) Group 2 strains. The blue and orange dots indicate the ploidy of Sc- (left) and Se- (right) type chromosomes, respectively. The vertical axis represents the estimated ploidy, which was calculated as the ratio of sequence coverage at each locus to the estimated haploid coverage; the horizontal axis represents chromosomal loci. In general, Group 1 strains had triploid genomes (haploid or non-Sc type, and diploid or triploid Se type), whereas Group 2 strains were tetraploid (haploid to triploid Sc and Se types). This figure is available in black and white in print and in colour at DNA Research online.
3.4. Interchromosomal translocations between Sc- and Se-type genomes
Interchromosomal translocations were detected by mate-pair reads in CBS1503, CBS1513, CBS1538, and W34/70 (Fig. 3). Previous studies have detected 11, 9, and 8 Sc-/Se-type translocations in CBS1503,5 CBS1513,5,2,2 and W34/70,6 respectively, and these translocations were verified by PCR amplification prior to the start of our study. Our analysis confirmed these known translocations in the three strains, demonstrating that this method is capable of detecting these events. Moreover, two translocations present at the HSP825,2,2 and KEM1 loci5 were also confirmed in the other six S. pastorianus strains using paired-end sequences (Supplementary Fig. S1). The two breakpoints were completely matched at the nucleotide level among all Group 1 and 2 strains, with the exception of a chromosomal deletion (HSP82 of CBS1538 and CBS1174). Novel interchromosomal translocations were also detected by our study—one each in CBS1503, CBS1513, and CBS1538 and two in W34/70. One of the novel translocations common to the four strains was further investigated by paired-end sequence alignment, revealing that all 10 S. pastorianus strains share a breakpoint within ZUO1 (Supplementary Fig. S2). The remaining translocation present in W34/70 spans from Sc-type chromosome VIII PRP8 to Se-type chromosome XV–VIII (Supplementary Fig. S3).
Figure 3.

Interchromosomal translocations between Sc- and Se-type genomes. The circular layouts show interchromosomal translocations between Sc and Se types detected by mate-pair links in Group 1 (CBS15103, CBS1513, and CBS1538) and Group 2 (W34/70) strains. Blue and orange bars represent Sc-type (left) and Se-type (right) chromosomes, respectively. The grey histograms within the circles represent sequence coverage, and regions without bars indicate chromosomal deletions. Grey intersecting lines indicate interchromosomal translocations supported by mate-pair reads bridging the Sc and Se types. Green lines indicate translocations shared by the two groups. Three interchromosomal translocations were common to both groups. The line thickness is proportional to the number of links supported by mate-pair reads. This figure is available in black and white in print and in colour at DNA Research online.
The identification of both known and novel interchromosomal translocations with identical breakpoints at the nucleotide level in both S. pastorianus groups indicates that at least one hybridization event involving ancestral S. cerevisiae and S. eubayanus is shared by both S. pastorianus groups.
3.5. SNV analysis against reference genomes and their chromosomal distribution
The SNVs were analysed using the S. cerevisiae S288C complete and S. eubayanus CBS12357 draft genomes (Sc and Se types, respectively) as references and classified as homo- and hetero-SNVs (Table 3). The number of homo-SNVs was similar among the strains, and the homo-SNVs were present at a density of 1/200 bp in both the Sc and Se types, indicating that the reference genomes of both species were sufficiently similar to be used for mapping-based analysis. For the Sc-type, SNVs were less prevalent in Group 1 than in Group 2; however, this did not reflect a lower SNV density because many Group 1 Sc chromosomes were missing. In contrast, the number of hetero-Sc-type SNVs in Group 2 was markedly increased compared with Se-type SNVs, along with a 10-fold higher density. In Group 1, fewer hetero-SNVs were present because most Sc-type chromosomes were haploid or missing in this group.
Table 3.
Number of SNVs in 10 S. pastorianus strains
| Strains | Sc type |
Se type |
||
|---|---|---|---|---|
| Homo-SNV | Hetero-SNV | Homo-SNV | Hetero-SNV | |
| Group 1 | ||||
| CBS1503 | 29,698 | 465 | 62,382 | 663 |
| CBS1513 | 36,371 | 258 | 62,520 | 1,636 |
| CBS1538 | 15,233 | 283 | 62,521 | 2,340 |
| CBS1174 | 15,671 | 264 | 62,044 | 2,539 |
| CBS2440 | 19,283 | 325 | 61,720 | 2,180 |
| Group 2 | ||||
| W34/70 | 48,892 | 10,900 | 59,226 | 979 |
| CBS1483 | 49,352 | 6,459 | 58,932 | 683 |
| CBS1484 | 47,551 | 7,269 | 59,479 | 887 |
| CBS2156 | 50,819 | 5,871 | 58,920 | 507 |
| CBS5832 | 49,849 | 8,022 | 60,466 | 780 |
The higher proportion of hetero-SNVs in Sc-type genomes in Group 2 compared with Se-type genomes suggests that the high heterozygosity in the former originated from an ancestral ale beer yeast.2,3 Indeed, ale beer strains—for instance, FostersB and FostersO—reportedly have high numbers of hetero-SNVs (up to 37,784 and 32,600, respectively). In comparison, lager beer yeast Sc-type genomes exhibit low heterozygosity but an uneven distribution of hetero-SNVs along Sc-type chromosomes, with some regions exhibiting many and others few or no hetero-SNVs (Fig. 4 and Supplementary Fig. S4). An even distribution of hetero-SNVs has been reported for the S. cerevisiae sake yeast strain Kyokai no. 72,4 and the industrial fuel-ethanol fermentative strain CAT-1,2,5 presumably as the result of frequent LOH.
Figure 4.

Distribution of hetero-SNVs in Sc-type chromosomes IV and VIII of Group 2 strains. The distribution of heterozygosity along Sc-type chromosomes IV and VIII in five Group 2 strains (CBS1483, CBS1484, CBS2156, CBS5832, and W34/70 from top to bottom for each chromosome) was determined by calculating a moving average of SNV density (window size = 10 kb and step size = 1 kb). Hetero-SNVs were unevenly distributed, possibly due to LOH. In many regions, low/no heterozygosity was observed in the five strains, whereas in other regions, the distribution pattern of low/no heterozygosity regions differed between strains. LOH was therefore considered a relatively frequent event, with shared low/no heterozygosity regions likely shaped in the common ancestor or by overlapping LOH events. This figure is available in black and white in print and in colour at DNA Research online.
Based on these observations and previous reports, the uneven distribution of heterozygosity may be due to the loss of variation between intrahomologous chromosomes. After a hybridization event between S. cerevisiae and S. eubayanus, variations between intrahomologous chromosomes in Sc types are thought to have gradually disappeared via LOH. Regions of high hetero-SNV density likely originated from the ancestral ale beer yeast, whereas low-density regions or those with no hetero-SNVs are likely the imprint of LOH. Low/no heterozygosity was observed in various regions in all five Group 2 strains, albeit with different distribution patterns. Therefore, LOH is presumed to have been a relatively frequent event, with low/no heterozygosity regions arising in the common ancestor or as a result of overlapping LOH events.
3.6. Phylogenetic analysis based on chromosomal SNVs against reference genomes
Phylogenetic trees based on SNVs in chromosomes queried against reference genomes were constructed separately for the Sc and Se types (Fig. 5). In the Sc-type tree, the strains were segregated into two clades, Groups 1 and 2, in agreement with previous studies11,12 based on partial PCR-based sequence data. In contrast, the Se-type phylogenetic tree revealed that S. pastorianus strains were mixed in nearly a single clade, with no significant phylogenetic difference between Groups 1 and 2. Indeed, the Se-type variant rate among the 10 S. pastorianus strains was only 0.063%, which was much lower than that of the Sc-type (0.348%). For the Sc-type, the tree suggests that the two groups—which are separated by geographical location—have independent origins. However, the Se-type phylogenetic tree suggests that the different aspect from that of Sc-type. Se-type genomes of the two groups look like originated from a common ancestor. Thus, the conflicting characteristics of the phylogenetic trees may be attributable to differences in genetic features between the Sc and Se type strains, namely ploidy and hetero-SNV density.
Figure 5.

Phylogenetic trees based on SNVs in chromosomes. (A and B) Phylogenetic tree of the Sc type in 10 S. pastorianus strains with S. cerevisiae S288C as a reference genome. (B) Enlarged view. (C and D) Phylogenetic tree of the Se type in S. pastorianus strains with S. eubayanus CBS12357 as a reference genome. (D) Enlarged view. In the Sc type, S. pastorianus strains are divided into two clades (Groups 1 and 2); however, in the Se type, the strains form a single mixed clade. Bootstrap values of 1,000 trials are indicated as blue numbers. This figure is available in black and white in print and in colour at DNA Research online.
3.7. Assignment of hetero-SNVs and haplotype phasing
We further validated these SNVs to address the conflicting phylogenic analysis by focusing on the hetero-SNVs in Group 2. This assessment revealed interesting results for the SNV numbers for Group 1 and Group 2 strains (Table 4). Specifically, nearly all the homo–hetero-SNVs observed between Group 1 and Group 2 are consistent relationships (i.e. Group 1's homo allele is equal to either allele of Group 2's hetero-SNV at the corresponding locus), and there are few conflicting SNVs. With few exceptions, either allele of the Group 2 hetero-type SNVs consists of the same sequence as the corresponding positions' haploid Group 1 allele. To confirm that this relationship was not coincidental, we conducted an additional analysis to solve the linkage relationships of W34/70 adjacent hetero-SNVs (‘phasing’) based on sequence data to construct two separate intrahomologous haploid chromosomes (Supplementary Fig. S5A and B). We obtained 304 blocks containing five or more consecutive hetero-SNVs each. The total size of the blocks was 1,876,249 bp (15.6% of the whole Sc type genome), and the blocks consisted of 9,765 hetero-SNVs (89.6% of all hetero-SNVs in W34/70). To maximize the length of sequences comparable to phased haplotypes of W34/70, a consensus sequence was built for the Group 1 strains. As shown in Fig. 5A, the differences between Group 1 strains are quite few and seemingly negligible for this analysis. Finally, we constructed a total of 274 blocks (1,705,313 bp including 8,731 hetero-SNVs) that could be compared with the Group 1 consensus sequence, covering 80.1% of all hetero-SNVs in W34/70.
Table 4.
SNV numbers between Group 1 and 2 strains
| Group 2 |
|||||
|---|---|---|---|---|---|
| W34/70 | CBS1483 | CBS1484 | CBS2156 | CBS5832 | |
| Group 1 | |||||
| CBS1503 | |||||
| SNV site # *1 | 18,783 | 17,769 | 17,323 | 17,414 | 17,673 |
| Homo/hetero site # *2 | 6,444 | 5,053 | 4,006 | 3,723 | 4,709 |
| Consistent site # *3 | 6,438 | 5,049 | 4,005 | 3,720 | 4,705 |
| Conflict site # *4 | 6 | 4 | 1 | 3 | 4 |
| CBS1513 | |||||
| SNV site # *1 | 23,791 | 21,765 | 22,912 | 21,669 | 22,308 |
| Homo/hetero site # *2 | 9,323 | 5,739 | 6,632 | 5,503 | 7,289 |
| Consistent site # *3 | 9,318 | 5,738 | 6,631 | 5,500 | 7,286 |
| Conflict site # *4 | 5 | 1 | 1 | 3 | 3 |
| CBS1538 | |||||
| SNV site # *1 | 9,117 | 8,402 | 8,638 | 8,437 | 9,225 |
| Homo/hetero site # *2 | 2,883 | 1,664 | 2,300 | 1,786 | 3,058 |
| Consistent site # *3 | 2,881 | 1,664 | 2,300 | 1,785 | 3,056 |
| Conflict site # *4 | 2 | 0 | 0 | 1 | 2 |
| CBS1174 | |||||
| SNV site # *1 | 9,550 | 8,833 | 9,111 | 8,901 | 9,641 |
| Homo/hetero site # *2 | 2,865 | 1,651 | 2,298 | 1,785 | 3,037 |
| Consistent site # *3 | 2,862 | 1,651 | 2,297 | 1,783 | 3,034 |
| Conflict site # *4 | 3 | 0 | 1 | 2 | 3 |
| CBS2440 | |||||
| SNV site # *1 | 12,223 | 11,176 | 10,287 | 11,084 | 12,242 |
| Homo/hetero site # *2 | 4,120 | 2,302 | 2,272 | 1,886 | 4,071 |
| Consistent site # *3 | 4,118 | 2,302 | 2,272 | 1,885 | 4,069 |
| Conflict site # *4 | 2 | 0 | 0 | 1 | 2 |
*1, Number of SNV sites between Group 1 and 2 strains.
*2, Number of SNV sites between Group 1 homo vs. Group 2 hetero-types.
*3, Number of consistent SNV sites, i.e. a Group 1 homo allele is included with either allele of Group 2 hetero-SNVs.
*4, Number of conflicting SNV sites.
3.8. Phylogenetic analysis based on haplotype-phased chromosomal SNVs
As the result of the analysis described above, we obtained 274 blocks consisting of three Sc-type chromosome sequences each (i.e. one Group 1 consensus chromosome and two W34/70 haplotype-phased intrahomologous chromosomes). We divided the phased chromosomes into two groups, W34/70_a and W34/70_b, based on sequence identity with Group 1 (Supplementary Fig. S5C and Fig. 6A). The same analysis results for the randomly shuffled hetero alleles of W34/70 dropping off the linkage information (Supplementary Fig. S5D and E) are presented in Fig. 6B. As shown in Fig. 6A, the Group 1 and W34/70_a identity distribution was significantly higher than that between Group 1 and W34/70_b, but no significant difference in the two distributions was observed based on randomly divided allele information (Fig. 6B). In addition, a phylogenetic tree of the consensus sequence of the Group 1 strains, the two haplotypes of W34/70 (W34/70_a and W34/70_b), and S288c is presented in Fig. 6C. The tree was described based on 14,322 sites containing 8,731 hetero-SNVs within 274 blocks. As shown in Fig. 6C, Group 1 and W34/70_a reside in the same clade. Similarly, the phylogenetic tree of the other four Group 2 strains revealed that all Group 2 ‘type a’ chromosomes reside in the same clade as Group 1 (Supplementary Fig. S6). These results support the non-coincidental nature of the observation that either of the Group 2 hetero-type SNV alleles has the same sequence with the corresponding positions' haploid Group 1 allele, thus strongly suggesting that one of Group 2's chromosomes is shared with Group 1's chromosomes.
Figure 6.

Phylogenetic relationship between haplotype-phased W34/70 sequences and Group 1 consensus sequences. (A) Identity distribution between haplotype-phased W34/70 chromosome sequences (W34/70_a, close with Group 1 sequences; W34/70_b, another chromosome sequence) and Group 1 sequences. (B) Identity distribution between haplotype-phased W34/70 chromosome sequences (hetero alleles are randomly shuffled dropping off the linkage information, average of 100 trials). (C) Phylogenetic tree based on haplotype-phased W34/70 sequences and Group 1 sequences. This figure is available in black and white in print and in colour at DNA Research online.
3.9. Traces of frequent LOH events observed in haplotype-phased Sc-type chromosomes
As mentioned above, the uneven distribution of hetero-SNVs might be evidence of frequent LOH events (Fig. 4). In addition, we identified two examples of evidence of LOH in haplotype-phased intrahomologous Sc-type chromosomes. We also observed consecutive regions with hetero-SNVs and both types of homo alleles constituting corresponding hetero nucleotides in the Group 2 strains (Fig. 7A). All SNVs of Group 2 in these regions—which span 16.0 kb—exhibited this pattern, and there were no exclusive SNVs. Moreover, the Group 1 strains exhibited either type of homo allele. Genomes with hetero-SNVs may be more ancestral type, with LOH events likely occurring independently in each strain, thus yielding two types of homo alleles at those loci. Another example is presented in Fig. 7B; blocks spanning ∼17 kb indicate that the Group 1 sequences are nearly identical to either sequence in the hetero Group 2 strains (W34/70, CBS1483, and CBS1484). In addition, CBS2156 and CBS5832 lost heterozygosity and have the same sequences as Group 1 strains, with the exception of two continuous regions of homo-SNVs between Group 1 strains and Group 2 strains (∼3 and ∼5 kb regions shown in green boxes). The full elucidation of the evolutionary history of this 17-kb block is nearly impossible, but these regions suggest the existence of other LOH events for common Group 2 ancestor strains. These homo-SNV ‘island regions’ in the solved linkage blocks are the difference between the Group 1 and W34/70_a-type chromosome sequences. Therefore, Group 1–W34/70_a has different sequences and branch lengths remaining in the phylogenetic tree (Fig. 6A). Here, we set a threshold length and divided into separate blocks if the distance between adjacent SNV loci exceeded the threshold. The phylogenetic trees resulting from threshold lengths of 1000 and 300 bp are presented in Supplementary Fig. S7. The threshold length correlates with branch length in Group 1–W34/70_a. This phenomenon is further evidence of frequent LOH. If an LOH event occurred for a W34/70 chromosome, a homo-SNV region was established. If the opposite allele from Group 1 remained, a homo-SNV is observed in this region and caused branch length in the phylogenetic tree. If the same allele as Group 1 remained, no SNVs were detected in this continuous region. For example, in Fig. 7B, no SNV regions are observed immediately right of the left region from 396,036 to 397,000 bp, which may be the result of an LOH with the corresponding remaining alleles in Group 1. When the threshold was set shorter than this silent SNV region, the blocks were divided. Thus, shorter thresholds facilitate the detection of shorter LOH regions, resulting in shorter branch lengths.
Figure 7.

Evidence of LOH observed in haplotype-phased chromosomes. Two examples of regions in which LOH is expected to have occurred in some strains. (A) The common ancestor of the strains is considered to have had hetero-SNVs in these regions, and some strains are suspected to have undergone LOH, thus switching SNVs from the hetero to the homo type. (B) In this block, nested LOH events likely occurred. This figure is available in black and white in print and in colour at DNA Research online.
3.10. The origin of S. pastorianus mtDNA
To determine the origin of S. pastorianus mtDNA, the mtDNA of eight strains from three species was assembled into a circular scaffold. The mtDNA sizes and GC contents are presented in Table 5. In S. pastorianus strains CBS1503, CBS1513, CBS1538, and W34/70, the mtDNA was ∼69 kb, smaller than that of S. cerevisiae S288C (85,779 bp) but slightly larger than those of S. eubayanus CBS12357 (64,290 bp) and BaiFY1 (67,205 bp). The S. pastorianus strain mtDNA exhibited conserved synteny with the mtDNA of S. eubayanus and S. bayanus.
Table 5.
Mitochondrial genome in S. pastorianus strains and related species
| Length (bp) | GC content (%) | |
|---|---|---|
| S. pastorianus | ||
| CBS1503 | 68,790 | 18.97 |
| CBS1513 | 69,294 | 19.41 |
| CBS1538 | 68,699 | 19.24 |
| W34/70 | 68,862 | 19.04 |
| S. eubayanus | ||
| CBS12357 | 64,290 | 17.48 |
| BaiFY1 | 67,205 | 18.43 |
| S.bayanus | ||
| CBS380 | 64,736 | 16.32 |
| NBRC1948 | 65,795 | 19.51 |
| S. cerevisiae | ||
| S288c | 85,779 | 17.11 |
Phylogenetic analysis based on the mitochondrial ORFs revealed that the mtDNA of S. pastorianus strains shares a common origin between Groups 1 and 2, with BaiFY1 exhibiting greater similarity in mtDNA to S. pastorianus (Fig. 8). The mtDNA of interspecies hybrids can be inherited from either parental species. Although it was previously demonstrated that S. pastorianus mtDNA is not of the Sc type,2,6 it could not be confirmed as Se type because the mtDNA sequence of S. eubayanus had not been determined. Interestingly, BaiFY1 mtDNA (isolated in Tibet)4 differs considerably from that of CBS12357 (isolated in Patagonia),3 with a sequence identity of 90–95% between the two strains despite being pure strains of the same species. Therefore, the Se-type genome of S. pastorianus strains is thought to originate from Tibet or its surrounding regions, consistent with previous studies.4
Figure 8.

Phylogenetic tree based on mitochondrial ORFs. Shown are the phylogenetic relationships among S. pastorianus (CBS1503, CBS1513, CBS1538, and W34/70), its parental species S. cerevisiae (S288C) and S. eubayanus (CBS12357 and BaiFY1), and the related species S. bayanus (CBS380 and NBRC1948) based on mitochondrial ORFs. Four S. pastorianus strains (from Groups 1 and 2) are located in the same clade. Additionally, the mtDNA of S. eubayanus BaiFY1 is most closely related to that of S. pastorianus, suggesting that the latter originated from a single parental species (S. eubayanus). This figure is available in black and white in print and in colour at DNA Research online.
3.11. Evolutionary history of S. pastorianus
On the basis of these results, we discuss whether the S. pastorianus strains originated from independent hybridization events or a common event. Translocation analysis revealed that the Group 1 and 2 strains share at least three interchromosomal translocations at the nucleotide level, indicative of common ancestors for both the Sc- and Se-type parental strains. The phylogenetic mtDNA genome analysis provided further support for a common origin (Tibet). In contrast, ploidy and phylogenetic analyses using SNVs against reference sequences suggested that the groups have different Sc-type ancestors and therefore independent evolutionary origins. The frequency of translocations is known to be much higher than that of point mutations;2,7 therefore, SNVs provide more robust evidence. However, hetero-SNV analysis indicated that 17.0–39.2% of SNV sites between Groups 1 and 2 could be accounted for by consistent SNV relationships (Table 4). Furthermore, phylogenetic analysis using haplotype-phased sequences suggested that Group 1 and Group 2 strains may share haploid Sc-type chromosomes as well as a common ancestor.
Based on these analyses, we hypothesized that at least one hybridization event was shared between Group 1 and 2 strains. Following this hypothesis, the difference in ploidy between the two groups can be explained by the chromosomal deletions in Group 1 (Fig. 9A) or an additional hybrid Sc-type genome in Group 2 (Fig. 9B). In the former scenario, a common ancestor originating from both Sc- and Se-types had a diploid genome, with subsequent chromosomal translocations; the ancestral Group 1 strains then experienced numerous chromosomal deletions in the Sc-type genome, leaving only some haploid chromosomes in the current Group 1 strains. In the latter scenario, a common ancestor originated from the first hybrid between Sc-type haploid and Se-type diploid genomes, followed by the formation of two groups after chromosomal translocations. In the Group 1 strains, chromosomal deletions in the Sc-type genome occurred, whereas the ancestral Group 2 strain interbred with another Sc-type species to produce a second hybrid.
Figure 9.

Two hypotheses regarding S. pastorianus origins based on shared chromosomal translocations and differences in ploidy between Groups 1 and 2. (A) Hybridization between diploid Sc and Se types occurred before chromosomal translocations, whereas chromosomal deletions occurred only in ancestral Group 1 strains. (B) After hybridization between haploid Sc and diploid Se types and chromosomal translocations, ancestral Group 2 strains gained another Sc type (i.e. a second hybridization event occurred). This figure is available in black and white in print and in colour at DNA Research online.
However, the remaining 60–80% of SNVs between these two groups are difficult to explain when only accounting for hetero–homo-SNV relationships. To address this point, we assumed that LOH events occurred with high frequency and could produce two homozygous alleles when occurring at a heterozygous location. Therefore, if Group 1 and 2 strains share common chromosomes (dark blue in Fig. 9) and LOH occurred in the diploid ancestor of Group 2 strains (early S. pastorianus in Fig. 9A or after the second hybridization strain in Fig. 9B), these two alleles may be observed randomly. If the opposite allele of Group 1 strains was retained, this site would display a homo-SNV/homo-SNV relationship in the two groups (light blue region for Group 2 in Fig. 9). However, the difference at this site would disappear if the same allele of Group 1 was retained. Consequently, homo-SNVs between the two groups were observed despite their shared common ancestral chromosomes. This speculation is supported by the distribution of hetero- and homo-SNVs between the Group 1 and 2 strains (Supplementary Fig. S8). Hetero-SNVs, homo-SNV regions, and regions with no SNVs co-segregated as continuous blocks, possibly as a result of LOH corresponding to these regions. Furthermore, when we examined individual SNV allele sequences present in the blocks (i.e. at least five continuous SNV sites) in each strain—for which at least one of the alleles of the Group 2 strain exhibited hetero-SNVs—we determined that despite 512 blocks with 11,178 sites expanding to 2.34 Mb, there were only nine conflicting sites (Supplementary Table S2). These observations strongly suggest that the common ancestor of Group 2 had highly heterozygous chromosomes, of which one pair was shared with Group 1. After repeated chromosomal deletions and LOH in continuous regions, some blocks exhibited the homo-SNV/homo-SNV relationship; other blocks displayed no SNVs, and hetero-SNV/homo-SNVs were observed in the remaining 17–39% of the genome.
4. Conclusions
Saccharomyces pastorianus is a useful species for investigating the evolutionary history of an interspecies hybrid. The genome sequences of the two parental species, S. cerevisiae and S. eubayanus, are available as reference genomes for analysis and can provide details of genome rearrangement events, including interchromosomal translocations, as well as chromosomal deletions and duplications following hybridization. We sequenced 14 strains of three species, including 10 strains of S. pastorianus, by NGS, and we compared the sequences to determine the phylogenetic relationships between S. pastorianus and its relatives. There are two conflicting hypotheses about whether the two S. pastorianus groups are derived from separate or a single hybridization event. Our analyses provide strong evidence that the haploid Sc-type chromosomes of Group 1 genomes and one of the intrahomologous diploid Sc-type chromosomes of Group 2 genomes were shared. Thus, the two S. pastorianus groups are not independent; they may have originated from the same parental species (S. cerevisiae and S. eubayanus) and share at least one hybridization event. Many of the SNVs shared by the two groups may have resulted from chromosomal deletion with LOH as an adaptive strategy following hybridization. This evolutionary scenario resolves all of the apparent contradictions, yet the genome sequence alone cannot reveal the driving force behind chromosomal deletions and LOH. Experimental approaches—such as the detection of LOH after hybridization between genomes exhibiting high vs. low heterozygosity—may provide additional insights into these processes.
5. Availability
Genome sequences were submitted to the DDBJ, EMBL, and GenBank DNA databases. The accession numbers are BBYU01000000, BBYV01000000, BBYW01000000, and BBYY01000000.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
Funding to pay the Open Access publication charges for this article was provided by the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Supplementary Material
Acknowledgements
We thank the members of the laboratories of H. Iwasaki and T. Itoh for their valuable participation in scientific discussions. The expert technical assistance of M. Ogawa is gratefully acknowledged.
References
- 1.P, L. 1909, Mikroskopische Betriebskontrolle in den Garungswerben 6th edn.
- 2.Gibson B.R., Storgårds E., Krogerus K., Vidgren V.. 2013, Comparative physiology and fermentation performance of Saaz and Frohberg lager yeast strains and the parental species Saccharomyces eubayanus, Yeast, 30, 255–66. [DOI] [PubMed] [Google Scholar]
- 3.Libkind D., Hittinger C.T., Valério E. et al. 2011, Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast, Proc. Natl Acad. Sci. USA, 108, 14539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bing J., Han P.J., Liu W.Q., Wang Q.M., Bai F.Y.. 2014, Evidence for a Far East Asian origin of lager beer yeast, Curr. Biol., 24, R380–1. [DOI] [PubMed] [Google Scholar]
- 5.Hewitt S.K., Donaldson I.J., Lovell S.C., Delneri D.. 2014, Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse, PLoS One, 9, e92203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakao Y., Kanamori T., Itoh T. et al. 2009, Genome sequence of the lager brewing yeast, an interspecies hybrid, DNA Res., 16, 115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louis V.L., Despons L., Friedrich A. et al. 2012, Pichia sorbitophila, an interspecies yeast hybrid, reveals early steps of genome resolution after polyploidization, G3 Genes, Genomes, Genet., 2, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mira N.P., Münsterkötter M., Dias-Valada F. et al. 2014, The genome sequence of the highly acetic acid-tolerant Zygosaccharomyces bailii-derived interspecies hybrid strain ISA1307, isolated from a sparkling wine plant, DNA Res., 21, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borneman A.R., Desany B.A., Riches D. et al. 2012, The genome sequence of the wine yeast VIN7 reveals an allotriploid hybrid genome with Saccharomyces cerevisiae and Saccharomyces kudriavzevii origins, FEMS Yeast Res., 12, 88–96. [DOI] [PubMed] [Google Scholar]
- 10.Tadami H., Shikata-Miyoshi M., Ogata T.. 2014, Aneuploidy, copy number variation and unique chromosomal structures in bottom-fermenting yeast revealed by array-CGH, J. Inst. Brew., 120, 27–37. [Google Scholar]
- 11.Dunn B., Sherlock G.. 2008, Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus, Genome Res., 18, 1610–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liti G., Peruffo A., James S.A., Roberts I.N., Louis E.J.. 2005, Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex, Yeast, 22, 177–92. [DOI] [PubMed] [Google Scholar]
- 13.Monerawela C., James T.C., Wolfe K.H., Bond U.. 2015, Loss of lager specific genes and subtelomeric regions define two different Saccharomyces cerevisiae lineages for Saccharomyces pastorianus Group I and II strains, FEMS Yeast Res., 15, fou008. [DOI] [PubMed] [Google Scholar]
- 14.Kajitani R., Toshimoto K., Noguchi H. et al. 2014, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads, Genome Res., 24, 1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Durbin R.. 2009, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinformatics, 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul S.F., Madden T.L., Schäffer A.A. et al. 1997, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebly M., Brickwedde A., Bolat I. et al. 2015, S. cerevisiae × S. eubayanus interspecific hybrid, the best of both worlds and beyond, FEMS Yeast Res., 15, fov005. [DOI] [PubMed] [Google Scholar]
- 18.Krzywinski M., Schein J., Birol I. et al. 2009, Circos: an information aesthetic for comparative genomics, Genome Res., 19, 1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A., Hanna M., Banks E. et al. 2010, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data, Genome Res., 20, 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Handsaker B., Wysoker A. et al. 2009, The Sequence Alignment/Map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guindon S., Lethiec F., Duroux P., Gascuel O.. 2005, PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference, Nucleic Acids Res., 33, 557–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walther A., Hesselbart A., Wendland J.. 2014, Genome sequence of Saccharomyces carlsbergensis, the world's first pure culture lager yeast, G3 Genes, Genomes, Genet., 4, 783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borneman A.R., Desany B.A., Riches D. et al. 2011, Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae, PLoS Genet., 7, e1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akao T., Yashiro I., Hosoyama A. et al. 2011, Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7, DNA Res., 18, 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babrzadeh F., Jalili R., Wang C. et al. 2012, Whole-genome sequencing of the efficient industrial fuel-ethanol fermentative Saccharomyces cerevisiae strain CAT-1, Mol. Genet. Genomics, 287, 485–94. [DOI] [PubMed] [Google Scholar]
- 26.Rainieri S., Kodama Y., Nakao Y., Pulvirenti A., Giudici P.. 2008, The inheritance of mtDNA in lager brewing strains, FEMS Yeast Res., 8, 586–96. [DOI] [PubMed] [Google Scholar]
- 27.Hiraoka M., Watanabe K., Umezu K., Maki H.. 2000, Spontaneous loss of heterozygosity in diploid Saccharomyces cerevisiae cells, Genetics, 156, 1531–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.