

Abstract
Purpose
To compare pulmonary and systemic inflammatory mediator release, pre- and poststimulation, ex vivo, in cells from Japanese patients with chronic obstructive pulmonary disease (COPD), non-COPD smoking controls, and non-COPD nonsmoking controls (NSC).
Patients and methods
This was a nontreatment study with ten subjects per group. Inflammatory biomarker release, including interleukin (IL)-6 and -8, matrix metalloproteinase-9, and tumor necrosis factor (TNF)-α, was measured in peripheral blood mononuclear cells (PBMC) and sputum cells with and without lipopolysaccharide or TNF-α stimulation.
Results
In PBMC, basal TNF-α release (mean ± standard deviation) was significantly different between COPD (81.6±111.4 pg/mL) and nonsmoking controls (9.5±5.2 pg/mL) (P<0.05). No other significant differences were observed. Poststimulation biomarker release tended to increase, with the greatest changes in the COPD group. The greatest mean increases were seen in the lipopolysaccharide-induced release of matrix metalloproteinase-9, TNF-α, and IL-6 from PBMC. Pre- and poststimulation data from sputum samples were more variable and less conclusive than from PBMC. In the COPD group, induced sputum neutrophil levels were higher and macrophage levels were lower than in either control group. Significant correlations were seen between the number of sputum cells (macrophages and neutrophils) and biomarker levels (IL-8, IL-6, and TNF-α).
Conclusion
This was the first study to compare cellular inflammatory mediator release before and after stimulation among Japanese COPD, smoking controls, and nonsmoking controls populations. Poststimulation levels tended to be higher in patients with COPD. The results suggest that PBMC are already preactivated in the circulation in COPD patients. This provides further evidence that COPD is a multicomponent disease, involving both airway and systemic inflammation.
Keywords: chronic obstructive pulmonary disease, lipopolysaccharide, peripheral blood mononuclear cells, sputum, tumor necrosis factor-α
Introduction
Japanese and overseas guidelines define chronic obstructive pulmonary disease (COPD) as an inflammatory disease of the lung.1,2 Increasing evidence suggests that COPD is a multicomponent disease involving both systemic and pulmonary inflammation.3,4 Cigarette smoking is a major risk factor for COPD and long-term smoking can cause airway inflammation characterized by neutrophil, macrophage, and activated T lymphocyte infiltration as well as increased cytokine concentrations, including tumor necrosis factor (TNF)-α and interleukin (IL)-8.5 A previous in vitro study using human monocyte-derived macrophages showed that cigarette smoke enhanced IL-8 release in a time- and concentration-dependent manner.6
Inflammatory biomarkers are becoming a useful means of characterizing the extent of both airway and systemic inflammation associated with COPD.7–9 However, serum and sputum inflammatory biomarkers have not been studied extensively in Japanese patients with COPD. Ishikawa et al were the first to conduct a study in which inflammatory markers were measured in Japanese patients with COPD, and compared against levels in smokers and nonsmoking controls (NSC).7 Trends were seen with increased levels of inflammatory cells and biomarkers in sputum and blood in COPD.7 These findings were consistent with previous reports in Western studies10,11 in which sputum neutrophils, IL-8, surfactant protein-D and Krebs von den Lungen-6, serum clara cell 16, high sensitivity C-reactive protein, and plasma fibrinogen were higher in COPD patients compared to NSC.
Chronic bronchitis in cigarette smokers shares many clinical and histological features with environmental lung diseases attributed to the bacterial endotoxin, lipopolysac-charide (LPS). LPS is a biologically active component of tobacco smoke and is one of the most potent proinflammatory stimuli known.12 Bacterial infections are one of the dominant causes of acute exacerbations in COPD with LPS being the most abundant component within the cell wall of Gram-negative bacteria.13 LPS can stimulate the release of proinflammatory cytokines, including TNF-α, leading to an acute inflammatory response toward pathogens.13,14 LPS has been extensively used in models of inflammation because it mimics inflammatory effects of cytokines.13
TNF-α is secreted by LPS-stimulated macrophages and is also produced by many different cell types including monocytes, fibroblasts, and endothelial cells.15 TNF-α is elevated in smokers5 and COPD patients.16,17 COPD and tobacco smoke may prime inflammatory cells thus increasing the inflammatory response in COPD patients and smokers compared to nonsmokers. It is possible to investigate this “priming” with the use of LPS and TNF-α stimulation since LPS induction is known to produce neutrophilic systemic and pulmonary inflammatory responses18,19 while experimental animal models show that TNF-α overexpression induces pathological changes similar to emphysema and pulmonary fibrosis.15
In this study, we compared the release of pulmonary and systemic inflammatory mediators with and without stimulation by LPS or TNF-α to investigate if inflammatory cells in COPD show an increased responsiveness compared with non-COPD smokers and non-COPD nonsmokers. This is the first study of its type in Japanese subjects. It helps to further characterize COPD-related inflammation and support treatment of the disease in Japan.
Materials and methods
Subjects
Japanese men and women, aged 40 years or older, were enrolled into one of three groups: 1) Patients with COPD (COPD group): subjects with a diagnosis of COPD2 who were current or ex-smokers with a ≥10 pack-year history of smoking, forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) <0.7 at 15–60 minutes following use of a salbutamol inhaler and had cough or sputum symptoms during 2 weeks before collection of the study specimens; 2) non-COPD smokers (smoking controls [SC] group): current or ex-smokers with a ≥10 pack-year history of smoking and FEV1/FVC ≥0.7 at 15–60 minutes following use of a salbutamol inhaler; and 3) non-COPD, nonsmokers (NSC group): subjects who had not smoked within 6 months before the study visit, had a ≤1 pack-year history of smoking, and a FEV1/FVC ≥0.7 at 15–60 minutes following use of a salbutamol inhaler.
Subjects were excluded if they had a diagnosis of bronchial asthma or any respiratory disorder other than COPD, had undergone lung volume reduction or lung transplant, had a chest X-ray (or computed tomography scan) indicating a major disease other than COPD which may interfere with study assessments, had a respiratory infection within 4 weeks before screening, used inhaled corticosteroids and systemic corticosteroids or low-dose xanthines (use at a regular dose was allowable) within 4 weeks before screening, had a bacterial infection, viral infection (including viral hepatitis) or systemic inflammation at enrolment, or had a known α1-antitrypsin deficiency. Undesirable medical events, including serious adverse events, were monitored during the visits.
Study design
This was a nontreatment, ex vivo study conducted between March 21, 2013 (first subject’s first visit) and December 11, 2013 (last subject’s last visit) at the Division of Respirology, Kurume University School of Medicine, Fukuoka Prefecture, Japan, in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Kurume University School of Medicine. Eligible subjects attended the hospital to provide written informed consent following the provision of study details and prior to any assessments. At this visit, cough and sputum symptoms were assessed, FEV1 and FVC were measured,20 blood and sputum samples were collected, and a chest X-ray was done if subjects had not had one within 6 months of the visit.
Sputum induction
Sputum was induced by the method described by the European Task Force21 used in other studies.7,22 Briefly, after inhaling 200 μg of salbutamol (GlaxoSmithKline, Japan), subjects inhaled an aerosol of 3%, 4%, and 5% hypertonic saline for 7 minutes each from an ultrasonic nebulizer (NE-U07; Omron, Kyoto, Japan).
Cell preparation
Induced sputum was processed as described by Pizzichini et al.23 Briefly, four-parts 0.1% dithiothreitol (Merck Millipore, Billerica, MA, USA) was added to one part sputum and incubated for 15 minutes, before four-parts phosphate-buffered saline was added.
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood using a density gradient (Accu-Prep; Bioneer Corporation, Daejeon, Korea). Total cell counts of PBMC and sputum were performed using a hemocytometer and expressed as the number of cells per milliliter.
Sputum cells were pelleted by centrifugation and the supernatant was removed. PBMC and sputum cell pellets were then resuspended in Roswell Park Memorial Institute (RPMI) 1640 (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) and 100× antibiotic-antimycotic solution (Thermo Fisher Scientific) at 0.75×106/mL (live cells were identified using trypan-blue staining). However, sputum cells after cytospin were stained using May–Giemsa method, and a 1,000-differential cell count was performed on each slide by a single observer blind to the clinical data; the mean count from two slides per subject was used for analysis.
Cell culture
PBMC and sputum cells (0.75×106 cells/mL) were incubated in RPMI 1640 with 10% FBS medium (5% carbon dioxide, 37°C) with or without LPS (Sigma-Aldrich Co., St Louis, MO, USA) or human recombinant TNF-α (R&D Systems Inc., Minneapolis, MN, USA). A concentration–response curve for LPS or TNF-α stimulation of PBMC and sputum cell IL-8 release was carried out (data on file) and LPS 100 ng/mL was selected, in each case, as the optimal concentration for cytokine release. This agrees with previous reports.24 Similarly, a time course for LPS stimulation of PBMC IL-8 was carried out (data on file) and 18 hours was selected as the optimal time for incubation. This also agrees with previous reports.25 The resulting supernatants were isolated and stored at −80°C for the enzyme-linked immunosorbent assay.
Measurement of biomarkers
Biomarkers were selected based on a previous study7 and have been used in Western population studies.26
Human IL-6, IL-8, TNF-α, and matrix metalloproteinase (MMP)-9 levels were measured by commercial enzyme-linked immunosorbent assay kits. The minimum detection limits of human IL-6 (R&D Systems), IL-8 (R&D Systems), TNF-α (R&D Systems), and MMP-9 (GE Healthcare, Amersham, Buckinghamshire, UK) were 2.0, 31.3, 15.6, and 1,000 pg/mL, respectively. A half value limit of detection for each biomarker was used for statistical analysis when levels were not detected.
Statistical methods
Thirty subjects were enrolled, as planned, with ten subjects per group. This sample size allowed for possible incompleteness of data in some subjects since obtaining sputum from NSC can be difficult; it was based on a similar study previously conducted in the UK which compared LPS-induced IL-8 expression in PBMC between COPD and SC with eight subjects per group (GlaxoSmithKline data on file).
Our primary objective was to measure the inflammatory biomarker release pre- and poststimulation by LPS or TNF-α in PBMC and sputum cells. Differences were calculated between basal and stimulated release of each biomarker to overcome variability between samples. Release of inflammatory biomarkers was analyzed as follows: Dunnett’s multicomparison, nonparametric, test to compare the COPD group versus the SC and NSC groups. The level of significance was 5%. Stratified analyses were conducted according to subject baseline factors. The per protocol set was the primary data set for analysis. Correlation between variables was assessed using Spearman’s rank correlation coefficient.
Results
Subject population
All enrolled subjects complied with the protocol and were included in the analyses as the per protocol set. Table 1 summarizes the demographics and baseline characteristics. The mean FEV1/FVC (%) ± standard deviation was 39.6±17.9 in COPD, 77.0±5.6 in SC, and 79.5±4.2 in NSC, confirming that patients with COPD had been recruited. No adverse events were reported throughout the study period.
Table 1.
Summary of subject demographics and baseline characteristics
| Demographics and measurements | Non-COPD NSC group (N=10) |
Non-COPD SC group (N=10) |
COPD group (N=10) |
|---|---|---|---|
| Demographics | |||
| Age, years | 69.4±5.2 | 64.8±7.8 | 62.2±6.6 |
| Sex, males/females | 5/5 | 9/1 | 7/3 |
| Height, cm | 158.3±4.3 | 165.7±7.0 | 159.9±6.8 |
| Weight, kg | 59.3±11.2 | 63.0±10.5 | 48.9±7.3 |
| Smoking history | |||
| Current/former/never | 0/0/10 | 8/2/0 | 3/7/0 |
| Number of pack-years | 0 | 44.3±35.0 | 65.4±30.2 |
| Spirometry | |||
| FVC, L | 3.1±0.9a | 3.8±0.9b | 2.8±0.8 |
| FEV1, L | 2.4±0.7c | 2.9±0.7d | 1.2±0.9 |
| FEV1, %predicted | 79.5±4.2d | 76.6±5.8c | 40.4±17.6 |
| FEV1/FVC, % | 79.5±4.2d | 77.0±5.6c | 39.6±17.9 |
Notes:
P<0.05,
P<0.01,
P<0.005, and
P<0.0001 (difference between COPD group and non-COPD NSC group/non-COPD SC group, by Dunnett’s test).
Abbreviations: COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; SC, smoking controls; NSC, nonsmoking controls.
Stimulation of cellular release in PBMC and sputum
There was an increase in basal TNF-α release in both the COPD and SC groups (COPD: 81.6±111.4; SC: 80.3±95.7; and NSC: 9.5±5.2 pg/mL) and this was statistically significant (P<0.05) between COPD and NSC (Table S1; Figure 1). In PBMC, there was a tendency in all three subject groups for increased biomarker release following TNF-α or LPS stimulation (Table 2). The greatest changes were generally seen in COPD, with the cellular release of IL-8 and MMP-9 being higher after TNF-α stimulation and MMP-9 and TNF-α higher after LPS stimulation (Figure 2A; Table 2). No significant differences were seen in other biomarkers before or after stimulation.
Figure 1.
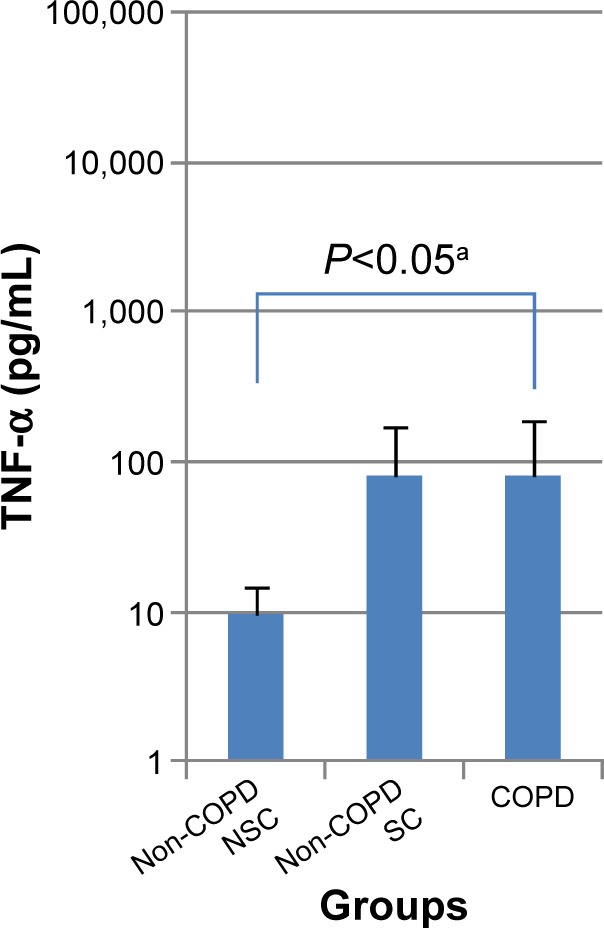
Basal release of TNF- α (pg/mL) from PBMCs in patients with COPD, non-COPD SC, and non-COPD NSC.
Notes: Data are presented as mean ± standard deviation; n=10 in each group. aDifference between COPD and non-COPD NSC groups by Dunnett’s test.
Abbreviations: TNF-α, tumor necrosis factor-alpha; LPS, lipopolysaccharide; COPD, chronic obstructive pulmonary disease; SC, smoking controls; NSC, nonsmoking controls; PBMCs, peripheral blood mononuclear cells.
Table 2.
Summary of the differences in biomarker levels before and after stimulation in PMBC and induced sputum cells (all subjects)
| Inflammatory biomarkers | Non-COPD NSC group (N=10) | Non-COPD SC group (N=10) | COPD group (N=10) |
|---|---|---|---|
| PBMC: Mean ± SD difference (pre- and poststimulation with TNF-α) | |||
| IL-8, pg/mL | 3,520.3±2,979.1 | 2,630.0±1,977.2 | 4,147.0±2,974.5 |
| IL-6, pg/mL | 41.4±59.7 | −36.7±92.2 | 40.7±85.7 |
| MMP-9, pg/mL | 268.3±442.2 | 443.9±679.4 | 2,633.2±4,630.7 |
| PBMC: Mean ± SD difference (pre- and poststimulation with LPS) | |||
| IL-8, pg/mL | 46,881.7±24,615.3 | 34,948.2±9,743.3 | 34,192.7±15,365.8 |
| IL-6, pg/mL | 7,309.7±5,511.0 | 7,979.4±3,285.2 | 8,393.5±3,200.5 |
| MMP-9, pg/mL | 292.3±508.3 | 446.0±697.3 | 2,997.4±5,267.0 |
| TNF-α, pg/mL | 1,296.0±841.6 | 4,419.9±4,421.3 | 8,146.9±20,195.6 |
| Sputum: Mean ± SD difference (pre- and poststimulation with TNF-α) | |||
| IL-8, pg/mL | −9,191.8±22,715.9 | −619.1±17,886.7 | 2,714.0 ± 11,359.2 |
| IL-6, pg/mL | −446.4±1,223.5 | 556.3±2,855.3 | 62.1±716.4 |
| MMP-9, pg/mL | −801.4±2,643.3 | −726.9±1,971.3 | −311.3±974.9 |
| Sputum: Mean ± SD difference (pre- and poststimulation with LPS) | |||
| IL-8, pg/mL | 20,785.4±48,223.6 | 1,175.8±2,180.1 | 2,885.3±15,438.1 |
| IL-6, pg/mL | 1,702.2±2,549.0 | −45.3±173.5 | 1,496.9±2,615.2 |
| MMP-9, pg/mL | −1,011.5±2,926.7 | −39.0±321.4 | 132.6±2,715.0 |
| TNF-α, pg/mL | 9,773.4±26,966.8 | 2,533.8±14,710.9 | 3,589.3±8,590.8 |
Abbreviations: COPD, chronic obstructive pulmonary disease; IL-8, interleukin-8; IL-6, interleukin-6; LPS, lipopolysaccharide; MMP-9, matrix metalloproteinase-9; SC, smoking controls; NSC, nonsmoking controls; PBMC, peripheral blood mononuclear cells; SD, standard deviation; TNF-α, tumor necrosis factor-alpha.
Figure 2.
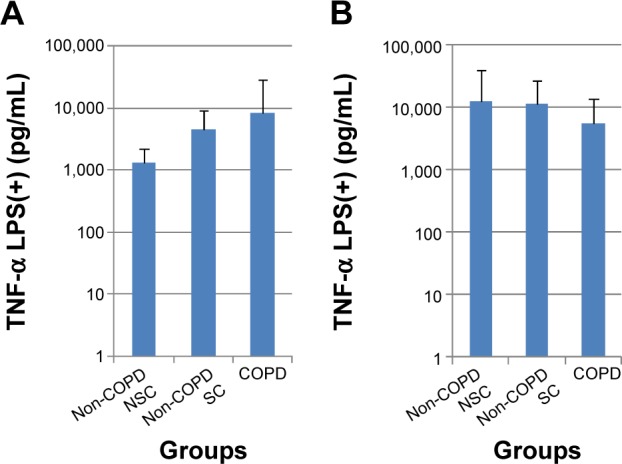
LPS-induced TNF-α release (pg/mL) from PBMC (A) and sputum cells (B) in patients with COPD, non-COPD SC, and non-COPD NSC.
Notes: Data are presented as mean ± standard deviation; N=10 in each group.
Abbreviations: TNF-α, tumor necrosis factor-alpha; LPS, lipopolysaccharide; COPD, chronic obstructive pulmonary disease; SC, smoking controls; NSC, nonsmoking controls; PBMC, peripheral blood mononuclear cells.
In sputum cells, there were inconsistent findings pre- and poststimulation with either TNF-α or LPS. Indeed, following LPS stimulation, the greatest mean release of IL-8 and TNF-α tended to be in the NSC group compared with COPD and SC groups (Figure 2B; Table 2). No significant differences were seen between subject groups across all biomarkers with or without stimulation.
Sputum inflammatory cell counts and correlations
There were no significant differences in total cell counts and the numbers and percentages of neutrophils, macrophages, lymphocytes, and eosinophils in induced sputum among the COPD, SC, and NSC groups (Table 3). However, the mean total cell count and mean number of neutrophils and eosinophils tended to be the highest in COPD followed by SC and then NSC (Table 3). In contrast, the highest number of macrophages was seen in NSC followed by SC and then COPD.
Table 3.
Inflammatory cell counts in sputum
| Inflammatory cells | Sputum cell count
|
||
|---|---|---|---|
| Non-COPD NSC group (N=10) | Non-COPD SC group (N=10) | COPD group (N=10) | |
| Total cell count, ×105 cells/mL | 8,433.0±4,822.1 | 19,027.6±20,716.7 | 26,059.1±48,773.9 |
| Neutrophils, ×105 cells/mL | 7,235.9±4,727.2 | 17,573.1±20,508.7 | 24,528.6±48,376.7 |
| Macrophages, ×105 cells/mL | 1,029.4±780.7 | 1,191.8±725.7 | 831.7±775.1 |
| Lymphocytes, ×105 cells/mL | 0.7±0.8 | 0.5±0.5 | 0.6±1.0 |
| Eosinophils, ×105 cells/mL | 0.9±1.3 | 2.2±5.2 | 6.4±12.2 |
Note: All data are expressed as mean ± standard deviation.
Abbreviations: COPD, chronic obstructive pulmonary disease; SC, smoking controls; NSC, nonsmoking controls.
Statistically significant correlations were seen between inflammatory cells and biomarkers across all three subject groups (Table 3). Significant correlations were seen between the number of macrophages and %macrophages and IL-8, IL-6, and TNF-α (Table 4). Significant correlations were also seen between %neutrophils and IL-8, IL-6, and TNF-α. No correlations were seen with MMP-9 (Table 4).
Table 4.
Correlations between sputum inflammatory cell counts and sputum biomarkers in all subjects
| Inflammatory cells | Sputum
|
|||
|---|---|---|---|---|
| IL-8 | IL-6 | MMP-9 | TNF-α | |
| Absolute number | ||||
| Total cells | −0.28 | −0.27 | 0.20 | −0.10 |
| Neutrophils | −0.34 | −0.31 | 0.22 | −0.13 |
| Macrophages | 0.56c | 0.48b | 0.19 | 0.49b |
| Cell differentiation | ||||
| Percentage of neutrophils | −0.69e | −0.63d | 0.18 | −0.41a |
| Percentage of macrophages | 0.72e | 0.64d | −0.12 | 0.49b |
Notes:
P<0.05,
P<0.01,
P<0.005,
P<0.0005, and
P<0.0001 (correlation between variables, by Spearman’s rank correlation coefficient).
Abbreviations: IL-8, interleukin-8; IL-6, interleukin-6; MMP-9, matrix metalloproteinase-9; TNF-α, tumor necrosis factor-alpha.
Discussion
The objective of our study was to compare inflammatory cell responsiveness to stimulation between Japanese patients with COPD, SC, and NSC. We measured the release of inflammatory biomarkers from PBMC and sputum cells before and after stimulation to investigate whether there were differences between these three subject groups.
Firstly, there was a significant difference in basal TNF-α release from PBMC between the COPD and NSC groups. Secondly, there was increased biomarker release following stimulation with TNF-α or LPS, with the highest increases tending to be in the COPD group: For example, after stimulation, there was a small increase in MMP-9 release in cells from the SC and NSC groups, but an approximately tenfold increase in the COPD group. This was also apparent in LPS-induced TNF-α release, for which there was a greater than sixfold increase in PBMC from the COPD group, compared with NSC (Table 2). Together, these observations suggest that PBMC may be primed in the circulation in COPD, which agrees with previous findings in Western populations27,28 and supports the concept of systemic inflammation being important in COPD.
Sputum cell data were more variable and less conclusive than those from PBMC. This may be because sputum samples are more heterogeneous and contain a variety of cells, which may have increased the variability in biomarker release. Of particular interest is that following LPS stimulation, the greatest increase in TNF-α and IL-8 release was seen in the NSC group rather than in either of the COPD and SC groups (Table 2).
The apparent contrast between the response to LPS stimulation of circulating PBMC and resident sputum cells in both the SC and COPD groups may be explained by the fact that sputum cells, unlike PBMC, may have been de-sensitized possibly by prior exposure to LPS in cigarette smoke in the airways. Alternatively, as our results show, the predominant cells in sputum, in both SC and COPD, are neutrophils, which have been reported to show only a modest response to LPS stimulation.29
Following stimulation, the release of inflammatory mediators from PBMC was increased in both SC and COPD, compared with NSC. This suggests that circulating cells may be activated by smoking, with or without COPD, and that smoking, per se, may directly or indirectly increase systemic inflammation.
COPD is associated with systemic and pulmonary inflammation,3,4 but it remains unclear how the systemic inflammation arises: Is it due to the spillover of inflammation from the lungs? Or, does systemic inflammation occur in parallel to the pulmonary inflammation and over time may even enhance it?3,16 Our results show that inflammation is present in both the blood and airways of smokers and patients with COPD, but we cannot determine the order in which systemic and pulmonary inflammatory components arise.
The levels of sputum inflammatory total cells, neutrophils, and macrophages tended to vary across the subject groups (Table 3). Neutrophil levels were higher in the COPD group than either control group indicating that neutrophils are part of the inflammatory response in COPD. This concurs with previous data in Japanese subjects.7 In contrast, macrophage levels were lower in patients with COPD than either control group suggesting that macrophages may be consumed by the inflammatory process and this confirms previous observations.7,30,31
Significant correlations were seen among inflammatory cells, %macrophages, and %neutrophils with biomarkers IL-8, IL-6, and TNF-α across all three subject groups, which agrees with previous data in a Japanese COPD population in which significant correlations were observed between IL-8 and neutrophils and IL-8 and macrophages.7 Neutrophils are a source of IL-810,32 and our data provide further evidence that IL-8 may be secreted into sputum to act as a chemoattractant for neutrophils migrating into the airway. Furthermore, IL-8 is synthesized by various cell types including neutrophils and macrophages10 and so the correlation seen in our study is not surprising. TNF-α has a broad spectrum of inflammatory effects in COPD resulting in the activation of various cells, including neutrophils and macrophage precursors, monocytes.10 IL-6 exerts proinflammatory effects in COPD, initiating the acute phase response, while IL-6 trans-signaling controls macrophage recruitment during acute inflammation.33
Study limitations and strengths
Our study design had a number of limitations. The number of subjects studied was low and this may have prevented us from clearly defining differences between the groups. Biomarker data are known to be variable and our sample size was insufficient to overcome this variability. In addition, direct measurement of inflammatory biomarkers in peripheral blood is known to be difficult due to their short half-lives, low concentrations, and their binding to soluble receptors and the effects of renal clearance.34,35 In addition, the bio-marker assay kits were originally developed for plasma and serum. Larger sample sizes would be needed to investigate these effects further. We did not assess sputum cell (largely comprising neutrophils) viability after incubation, which is known to be affected by TNF-α and LPS and this omission may have been additionally responsible for the variability in the sputum data. Furthermore, a comparison of sputum and blood cellular data was difficult because of the differences in cell populations between the two sample types. Isolating specific inflammatory cell types from each medium would enable comparisons to be made and correlations explored. As a single-center study, consistency in the sampling and analysis methodology was a possible strength.
Conclusion
This was the first study in which inflammatory cell mediator release before and after stimulation was compared among Japanese COPD, SC, and NSC groups. Our results suggest preactivation of circulating inflammatory cells such as PBMC in patients with COPD. In addition, this finding provides further evidence that COPD is a multicomponent disease, involving both airway and systemic inflammation.
Supplementary material
Table S1.
Summary of biomarkers in peripheral blood mononuclear cells (stimulated by TNF-α or LPS)
| Inflammatory cells | Non-COPD NSC group (N=10) |
Non-COPD SC group (N=10) |
COPD group (N=10) |
|||
|---|---|---|---|---|---|---|
| (−)TNF-α | (+)TNF-α | (−)TNF-α | (+)TNF-α | (−)TNF-α | (+)TNF-α | |
| IL-8, pg/mL | 421.8±361.5 | 3,942.1±3,293.4 | 525.5±360.0 | 3,155.5±1,796.6 | 315.8±155.0 | 4,462.8±3,087.4 |
| IL-6, pg/mL | 3.0±4.7 | 44.4±63.1 | 50.6±89.2 | 13.9±12.0 | 3.6±6.6 | 44.25±91.9 |
| MMP-9, pg/mL | 500.0±0.0b | 768.3±442.2 | 500.0±0.0b | 943.9±679.4 | 500.0±0.0b | 3,133.2±4,630.7 |
| (−)LPS | (+)LPS | (−)LPS | (+)LPS | (−)LPS | (+)LPS | |
| IL-8, pg/mL | 421.8±361.5 | 47,303.5±24,858 | 525.5±360.0 | 35,473.7±9,802.1 | 315.8±155.0 | 34,508.5±15,412.7 |
| IL-6, pg/mL | 3.0±4.7 | 7,312.7±5,512.9 | 50.6±89.2 | 8,030.0±3,340.4 | 3.6±6.6 | 8,397.1±3,203.8 |
| MMP-9, pg/mL | 500.0±0.0b | 792.3±508.3 | 500.0±0.0b | 946.0±697.3 | 500.0±0.0b | 3,497.4±5,267.0 |
| TNF-α, pg/mL | 9.5±5.2a | 1,305.4±840.7 | 80.3±95.7 | 4,500.2±4,453.8 | 81.6±111.4 | 8,228.5±20,171.3 |
Notes: All data are expressed as mean ± standard deviation.
P<0.05 (difference between COPD groups and non-smokers, by the Dunnett’s test); (+) with stimulation, (−) without stimulation.
The levels of MMP-9 before stimulation with TNF-α were below the limit of detection (1,000 pg/mL).
Abbreviations: COPD, chronic obstructive pulmonary disease; IL-8, interleukin-8; IL-6, interleukin-6; LPS, lipopolysaccharide; MMP-9, matrix metalloproteinase-9; SC, smoking controls; NSC, nonsmoking controls; TNF-α, tumor necrosis factor-alpha.
Acknowledgments
Funding for this study was provided by GlaxoSmithKline. The authors would like to thank the following people for their contributions: members of Kurume University who participated in subject recruitment – Kyoko Yamaguchi and Emiko Kuma for performing the biomarker analyses. The authors would also like to thank the following: members of the GlaxoSmithKline team who developed the study protocol – Etsuko Hayashi, Hideo Kikkawa, and Daisuke Yoshimoto; and The Institute of Japanese Union of Scientists & Engineers for performing the data management and statistical analysis. Medical writing and editorial support in the form of development of draft outline, development of manuscript (all drafts), assembling tables, and collating author comments was provided by Dr Kathryn White of Cathean Ltd, and was funded by GlaxoSmithKline.
Footnotes
Author contributions
All authors were involved in the acquisition and analysis of data, drafting and critical revision of the manuscript, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Akihiro Kobayashi and Tomoyuki Hayamizu were employees of GlaxoSmithKline at the time of conducting the study, and report no other relationships or activities that could appear to have influenced the submitted work. Tomoaki Hoshino discloses that his university (Kurume University) has received research grants from GlaxoSmithKline, who sponsored the study. Kazuko Matsunaga reports no conflicts of interest in this work. Takashi Kinoshita discloses his university (Kurume University) has received research grants from GlaxoSmithKline, who sponsored the study. Tomotaka Kawayama discloses having received honoraria/consulting fees from GlaxoSmithKline for participating in advisory board meetings and his university (Kurume University) has received research grants from GlaxoSmithKline, who sponsored the study. Malcolm Johnson is an independent respiratory consultant employed by GlaxoSmithKline to help in the design and conduct of the study. Malcolm Johnson is a shareholder in GlaxoSmithKline, but has no other relationships or activities that could appear to have influenced the submitted work. The authors report no other conflicts of interest in this work.
References
- 1.Japanese Respiratory Society COPD Guideline ver. 4. Tokyo Incorporated Association, JRS; 2013. [Google Scholar]
- 2.Global Strategy for Diagnosis, Management, and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2014. Available from: http://www.goldcopd.org/
- 3.Agusti A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should) Proc Am Torac Soc. 2007;4(7):522–525. doi: 10.1513/pats.200701-004FM. [DOI] [PubMed] [Google Scholar]
- 4.Agusti AGN. COPD, a multicomponent disease: implications for management. Respir Med. 2005;99(6):670–682. doi: 10.1016/j.rmed.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Tanni SE, Pelegrino NRG, Angeleli AYO, Correa C, Godoy I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. J Inflamm (Lond) 2010;7:29. doi: 10.1186/1476-9255-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarir Hi, Mortaz E, Karimi K, et al. Combination of fluticasone propionate and salmeterol potentiates the suppression of cigarette smoke-induced IL-8 production by macrophages. Eur J Pharmacol. 2007;571(1):55–61. doi: 10.1016/j.ejphar.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 7.Ishikawa N, Hattori N, Kohno N, Kobayashi A, Haymizu T, Johnson M. Airway inflammation in Japanese COPD patients compared with smoking and non-smoking controls. Int J Chron Obstruct Pulmon Dis. 2015;10(1):185–192. doi: 10.2147/COPD.S74557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes NC, Qiu YS, Pavord ID, et al. Antiinflammatory effects of salmeterol/fluticasone propionate in chronic obstructive lung disease. Am J Respir Crit Care Med. 2006;173(7):736–743. doi: 10.1164/rccm.200508-1321OC. [DOI] [PubMed] [Google Scholar]
- 9.Lomas DA, Silverman EK, Edwards LD, Miller BE, Coxson HO, Tal-Singer R. Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax. 2008;63(12):1058–1063. doi: 10.1136/thx.2008.102574. [DOI] [PubMed] [Google Scholar]
- 10.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):51–58. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- 11.Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res. 2011;12:146. doi: 10.1186/1465-9921-12-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. 1999;115:829–835. doi: 10.1378/chest.115.3.829. [DOI] [PubMed] [Google Scholar]
- 13.Ngkelo A, Meja K, Yeadon M, Adcock I, Kirkham PA. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3kinase signaling. J Inflamm (Lond) 2012;9(1):1. doi: 10.1186/1476-9255-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60(1):8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 15.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNF alpha in pulmonary pathophysiology. Respir Res. 2006;7:125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacNee W. Systemic inflammatory biomarkers and co-morbidities of chronic obstructive pulmonary disease. Ann Med. 2013;45(3):291–300. doi: 10.3109/07853890.2012.732703. [DOI] [PubMed] [Google Scholar]
- 17.Hacievliyagil SS, Mutlu LC, Temel I. Airway inflammatory markers in chronic obstructive pulmonary disease patients and healthy smokers. Niger J Clin Pract. 2013;16(1):76–81. doi: 10.4103/1119-3077.106771. [DOI] [PubMed] [Google Scholar]
- 18.Korsgren M, Linden M, Entwistle N, et al. Inhalation of LPS induces inflammatory airway responses mimicking characteristics of chronic obstructive pulmonary disease. Clin Physiol Funct Imaging. 2012;32(1):71–79. doi: 10.1111/j.1475-097X.2011.01058.x. [DOI] [PubMed] [Google Scholar]
- 19.Leaker BR, Barnes PJ, O’Connor B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir Res. 2013;14:137. doi: 10.1186/1465-9921-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller MR, Hankinson J, Brusasco F, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–338. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 21.Paggiaro PL, Chanez P, Holz O, et al. Sputum induction. Eur Respir J Suppl. 2002;37:3s–8s. doi: 10.1183/09031936.02.00000302. [DOI] [PubMed] [Google Scholar]
- 22.Asai K, Kobayashi A, Makihara Y, Johnson M. Anti-inflammatory effects of salmeterol/fluticasone propionate 50/250 mcg combination therapy in Japanese patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10(1):803–811. doi: 10.2147/COPD.S79842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pizzichini E, Pizzichini MM, Efthimiadis A, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid-phase measurements. Am J Respir Crit Care Med. 1996;154(2 pt 1):308–317. doi: 10.1164/ajrccm.154.2.8756799. [DOI] [PubMed] [Google Scholar]
- 24.Ayer JG, Song C, Steinbeck K, Celermajer DS, Freedman SB. Increased tissue factor activity in monocytes from obese young adults. Clin Exp Pharmacol Physiol. 2010;37(11):1049–1054. doi: 10.1111/j.1440-1681.2010.05430.x. [DOI] [PubMed] [Google Scholar]
- 25.Jansky L, Reymanov P, Kopecky J. Dynamics of cytokine production in human peripheral blood mononuclear cells stimulated by LPS or Infected by Borrelia. Physiol Res. 2003;52:593–598. [PubMed] [Google Scholar]
- 26.Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2014;69(7):666–672. doi: 10.1136/thoraxjnl-2013-204778. [DOI] [PubMed] [Google Scholar]
- 27.Oudjik EJ, Gerritsen WBM, Nijhuis EHJ, et al. Expression of priming-associated cellular markers on neutrophils during an exacerbation of COPD. Respir Med. 2006;100(10):1791–1799. doi: 10.1016/j.rmed.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Koethe SM, Kuhnmuench JR, Becker CG. Neutrophil priming by cigarette smoke condensate and a tobacco anti-idiotypic antibody. Am J Pathol. 2000;157(5):1735–1743. doi: 10.1016/S0002-9440(10)64810-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aida Y, Pabst MJ. Neutrophil responses to lipopolysaccharide. Effect of adherence on triggering and priming of the respiratory burst. J Immunol. 1991;146(4):1271–1276. [PubMed] [Google Scholar]
- 30.Stănescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51(3):267–271. doi: 10.1136/thx.51.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malerba M, Ricciardolo F, Radaeli A, et al. Neutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjects. Thorax. 2006;61(2):129–133. doi: 10.1136/thx.2005.043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bazzoni F, Cassatella MA, Rossi F, Ceska M, Dewald B, Baggiolini M. Pagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. J Exp Med. 1991;173:771–774. doi: 10.1084/jem.173.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravi AK, Khurana S, Lemon J, et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res. 2014;15:103. doi: 10.1186/s12931-014-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takabatake N, Nakamura H, Abe S, et al. The relationship between chronic hypoxemia and activation of the tumor necrosis factor-alpha system in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1179–1184. doi: 10.1164/ajrccm.161.4.9903022. [DOI] [PubMed] [Google Scholar]
- 35.Oudijk EJ, Nijhuis EH, Zwank MD, et al. Systemic inflammation in COPD visualised by gene profiling in peripheral blood neutrophils. Thorax. 2005;60:538–544. doi: 10.1136/thx.2004.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Summary of biomarkers in peripheral blood mononuclear cells (stimulated by TNF-α or LPS)
| Inflammatory cells | Non-COPD NSC group (N=10) |
Non-COPD SC group (N=10) |
COPD group (N=10) |
|||
|---|---|---|---|---|---|---|
| (−)TNF-α | (+)TNF-α | (−)TNF-α | (+)TNF-α | (−)TNF-α | (+)TNF-α | |
| IL-8, pg/mL | 421.8±361.5 | 3,942.1±3,293.4 | 525.5±360.0 | 3,155.5±1,796.6 | 315.8±155.0 | 4,462.8±3,087.4 |
| IL-6, pg/mL | 3.0±4.7 | 44.4±63.1 | 50.6±89.2 | 13.9±12.0 | 3.6±6.6 | 44.25±91.9 |
| MMP-9, pg/mL | 500.0±0.0b | 768.3±442.2 | 500.0±0.0b | 943.9±679.4 | 500.0±0.0b | 3,133.2±4,630.7 |
| (−)LPS | (+)LPS | (−)LPS | (+)LPS | (−)LPS | (+)LPS | |
| IL-8, pg/mL | 421.8±361.5 | 47,303.5±24,858 | 525.5±360.0 | 35,473.7±9,802.1 | 315.8±155.0 | 34,508.5±15,412.7 |
| IL-6, pg/mL | 3.0±4.7 | 7,312.7±5,512.9 | 50.6±89.2 | 8,030.0±3,340.4 | 3.6±6.6 | 8,397.1±3,203.8 |
| MMP-9, pg/mL | 500.0±0.0b | 792.3±508.3 | 500.0±0.0b | 946.0±697.3 | 500.0±0.0b | 3,497.4±5,267.0 |
| TNF-α, pg/mL | 9.5±5.2a | 1,305.4±840.7 | 80.3±95.7 | 4,500.2±4,453.8 | 81.6±111.4 | 8,228.5±20,171.3 |
Notes: All data are expressed as mean ± standard deviation.
P<0.05 (difference between COPD groups and non-smokers, by the Dunnett’s test); (+) with stimulation, (−) without stimulation.
The levels of MMP-9 before stimulation with TNF-α were below the limit of detection (1,000 pg/mL).
Abbreviations: COPD, chronic obstructive pulmonary disease; IL-8, interleukin-8; IL-6, interleukin-6; LPS, lipopolysaccharide; MMP-9, matrix metalloproteinase-9; SC, smoking controls; NSC, nonsmoking controls; TNF-α, tumor necrosis factor-alpha.