

Abstract
While the development of second- and third-generation drug-eluting stents (DES) have significantly improved patient outcomes by reducing smooth muscle cell (SMC) proliferation, DES have also been associated with an increased risk of late-stent thrombosis due to delayed re-endothelialization and hypersensitivity reactions from the drug-polymer coating. Furthermore, DES anti-proliferative agents do not counteract the upstream oxidative stress that triggers the SMC proliferation cascade. In this study, we investigate biocompatible amphiphilic macromolecules (AMs) that address high oxidative lipoprotein microenvironments by competitively binding oxidized lipid receptors and suppressing SMC proliferation with minimal cytotoxicity. To determine the influence of nanoscale assembly on proliferation, micelles and nanoparticles were fabricated from AM unimers containing a phosphonate or carboxylate end-group, sugar-based hydrophobic domain, and a hydrophilic poly(ethylene glycol) domain. The results indicate that when SMCs are exposed to high levels of oxidized lipid stimuli, nanotherapeutics inhibit lipid uptake, downregulate scavenger receptor expression, and attenuate scavenger receptor gene transcription in SMCs, and thus significantly suppress proliferation. Although both functional end-groups were similarly efficacious, nanoparticles suppressed oxidized lipid uptake and scavenger receptor expression more effectively compared to micelles, indicating the relative importance of formulation characteristics (e.g., higher localized AM concentrations and nanotherapeutic stability) in scavenger receptor binding as compared to AM end-group functionality. Furthermore, AM coatings significantly prevented platelet adhesion to metal, demonstrating its potential as an anti-platelet therapy to treat thrombosis. Thus, AM micelles and NPs can effectively repress early stage SMC proliferation and thrombosis through non-cytotoxic mechanisms, highlighting the promise of nanomedicine for next-generation cardiovascular therapeutics.
Keywords: amphiphilic macromolecules, nanoassemblies, micelles, nanoparticles, smooth muscle cell proliferation, restenosis
INTRODUCTION
Cardiovascular disease is the primary cause of mortality in the United States, with over 600,000 deaths annually accounting for a quarter of all deaths [1]. The most prevalent type of cardiovascular disease is coronary heart disease (CHD), caused by atherosclerosis, where oxidized low-density lipoprotein (oxLDL) accumulates in macrophages, forming a lipid-laden plaque within the arterial walls. This accumulation causes the vessel to narrow and harden, thereby preventing oxygenated blood from reaching the heart [2]. For patients with severe CHD, blocked arteries can be dilated through balloon angioplasty accompanied by stent implantation. Stents are among the most common treatments for advanced stages of CHD, with over 500,000 patients annually and $15 billion in costs in the US alone [1].
Despite recent advances in stent technologies, several issues remain [3]. While bare metal stents (BMS) provide mechanical support to regain adequate circulatory flow, they have been associated with long-term restenosis, where excessive smooth muscle cell (SMC) proliferation leads to vessel re-occlusion. To address this issue, drug-eluting stents (DES) that release anti-proliferative drugs were developed and have been very effective in preventing SMC proliferation [4]. However, the indiscriminate cytotoxic and/or immunosuppressive mechanisms of these drugs can significantly delay the arterial healing process and are associated with an increased risk of thrombosis, or blood clot formation, that can re-occlude blood flow [5].
Subsequent generations of DES incorporate improved anti-restenotic agents, such as everolimus and zotarolimus, and refined polymer coatings to circumvent thrombosis; both improvements have lowered early thrombosis rates, but DES still suffer from higher rates of late-stent thrombosis relative to BMS [3, 6–8]. Late-stage stent thrombosis can occur between months to years after implantation and is caused by endothelial dysfunction and local hypersensitivity from foreign body reactions to the stent materials and localized oxidative stress [9–11]. As these issues increase the likelihood of restricted blood flow, secondary surgeries, angina, myocardial infarction, and death, recent retrospective studies have demonstrated that mortality and myocardial infarction rates are not statistically different in DES compared to BMS [3, 8]. Consequently, a significant need exists to develop improved prophylactic therapeutics that utilize a non-cytotoxic approach to minimize the risk of both restenosis and thrombosis [12].
While many treatments focus on downstream cell proliferation, an alternative strategy to reduce the risk of restenosis and thrombosis is to address upstream events, such as lowering the high oxidative stress that triggers the SMC hyperproliferation and platelet adhesion [11, 13, 14]. Elevated oxLDL levels after surgery are correlated with SMC proliferation and lead to a higher incidence of localized lipid-rich lesions, as well as restenosis six months post-stent implantation [15, 16]. Increased cholesterol loading can also cause SMCs to change to a macrophage-like state, which puts plaques at further risk of growth and rupture [17]. Additionally, a significant increase in SMC proliferation and platelet activation occurs when cells are treated with oxLDL [11, 18–21]. Given that increased levels of oxLDL often cause platelet activation and excessive SMC proliferation, targeting oxLDL receptors to inhibit oxLDL uptake is one effective strategy to lower neointimal hyperplasia and oxidative stress [22–24]. This pathway may modulate the local remodeling following stenotic injury, thereby reducing the risk of restenosis.
Recently, sugar-based amphiphilic macromolecules (AMs), consisting of a hydrophilic poly(ethylene glycol) (PEG) tail and a branched hydrophobic head, were developed as atherosclerosis therapeutics to competitively block macrophage scavenger receptor interactions with oxLDL [25–32]. AM chemical structures can be tuned to mimic the characteristics of key biomolecules with desired charge, hydrophobic domain, three-dimensional conformation, and flexibility to enable efficient targeting of scavenger receptors and inhibition of oxLDL uptake [25–28, 30, 32]. Additionally, AMs thermodynamically self-assemble into micelles or can be kinetically formulated into nanoparticles (NPs), providing two different formulations to satisfy a wide range of therapeutic requirements [33, 34]. Recent work has demonstrated that AM micelles and NPs are biocompatible, and that NPs fabricated from a carboxylate-terminated AM were able to localize to plaques in vivo after systemic administration, where they reduced cholesterol accumulation and limited inflammation [34–36].
Despite these advances, clear indications of the AMs’ ability to impact critical stages of the restenosis and thrombosis cascades have yet to be demonstrated. In this paper, AM micellar and NP formulations were developed using bioactive, biocompatible AM compounds as candidates for pro-healing restenosis and thrombosis therapeutics [37]. As AM-based nanotherapeutics are hypothesized to minimize oxLDL uptake in SMCs by blocking and downregulating the scavenger receptors macrophage scavenger receptor 1 (MSR1) and cluster of differentiation 36 (CD36), the AM net charge and formulation of synthetic nanotherapeutics was systematically varied to determine how physicochemical properties (i.e., size, charge localization, and stability) affect SMC proliferation and platelet adhesion. Unimers, micelles, and NPs were fabricated from a phosphonate-terminated AM, 1pM, and the more established carboxylate-terminated AM, 1cM (Figure 1A). Compared to carboxylates, phosphonates have an increased negative charge which may be favorable for binding to cationic scavenger receptor residues. Furthermore, a phosphonate-terminated AM previously demonstrated sustained suppression of SMC proliferation by controllably releasing from metal oxide substrates over 21 days [38]. The insights from this study provide a fundamental understanding of desirable nanotherapeutic characteristics for scavenger receptor targeting as part of a prophylactic, cytocompatible approach to target the cause of restenosis and thrombosis.
Figure 1.
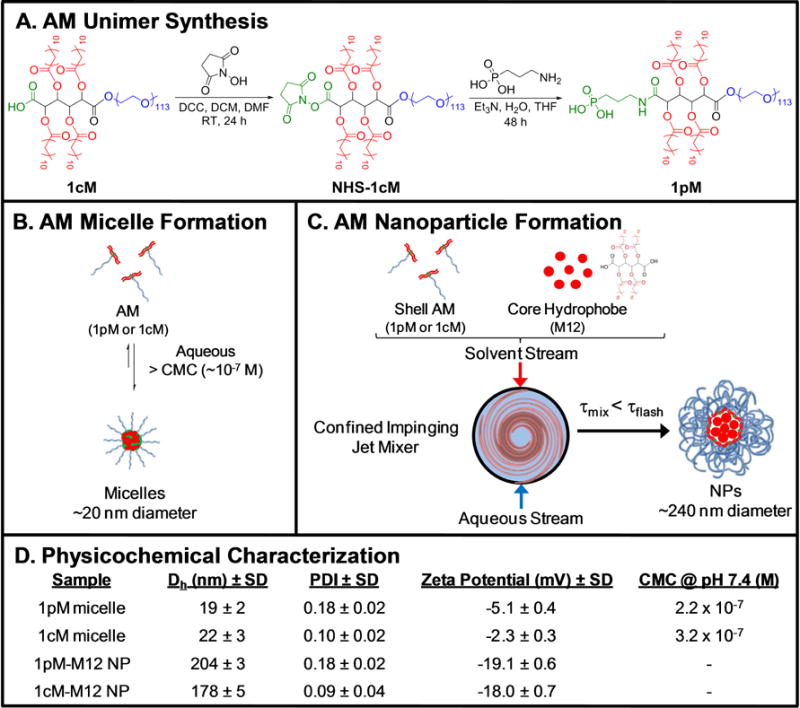
The chemical synthesis and fabrication processes used to produce AM nanotherapeutics. (A) Structure and synthesis of 1pM AM, synthesized from 1cM as previously described [40]. (B) Thermodynamic formation of 1pM and 1cM micelles in aqueous solution, when AMs are present at concentrations above their CMC (10−7 M). (C) Kinetic formation of 1pM and 1cM NPs using flash nanoprecipitation. (D) Physicochemical properties of 1pM and 1cM micelles and NPs, including hydrodynamic diameter (Dh), PDI, ζ-potential, and AM CMC values at pH 7.4 (n=3).
EXPERIMENTAL SECTION
Materials
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless otherwise noted. Hydrochloric acid (HCl, 1 N) and polytetrafluoroethylene (PTFE) syringe filters were purchased from Fisher Scientific (Fair Lawn, NJ). 4-(Dimethylamino)pyridinium-p-toluene-sulfonate (DPTS) was prepared as described by Moore and Stupp [39]. Monomethoxy-poly(ethylene glycol) (mPEG, Mn = 5000 Da) was dehydrated by azeotropic distillation with toluene prior to use. Poly(styrene15-block-ethylene glycol114) (poly(styrene-PEG)) copolymer (Mn=6600 Da, PDI=1.1) was purchased from Polymer Source Inc. and polystyrene homopolymer (Mn=1500 Da) was prepared as described previously [36]. Pluronic block copolymer P85 was provided as a gift by BASF Corporation (Parsippany, NJ). Reagents for cell culture, cytotoxicity studies, proliferation, oxLDL uptake, scavenger receptor expression, and gene regulation studies include human primary coronary artery SMCs, Smooth Muscle Basal Media, and Smooth Muscle Growth Media purchased from Lonza (Walkersville, MD), alamarBlue® assay and Hoechst 33342 purchased from Life Technologies (Carlsbad, CA), unlabeled oxLDL purchased from Biomedical Technologies Inc. (Ward Hill, MA), and 3,3′-dioctadecyloxacarbocyanine (DiO) labeled oxLDL purchased from Kalen Biomedical (Montgomery Village, MD).
AM Synthesis and Nanotherapeutic Characterization
1cM and 1pM AM Synthesis and Characterization
Synthesis and characterization of 1cM and its precursor (M12) were prepared as described previously [33]. 1cM was the starting material for the synthesis of 1pM [40]. In brief, N,N’-dicyclohexylcarbodiimide (DCC, 0.175 mL, 0.181 mmol) was added dropwise to a solution of 1cM (0.700 g, 0.118 mmol) and N-hydroxysuccinimide (NHS, 0.0868 g, 0.757 mmol) in dichloromethane (DCM, 10 mL) and dimethylformamide (DMF, 2 mL) under argon, then stirred for 24 h. The reaction mixture was filtered to remove the DCC urea byproduct, and the filtrate was washed with 0.1 N hydrochloric acid (1 × 60 mL) and 50:50 brine/H2O (2 × 60 mL), concentrated, and precipitated with diethyl ether to isolate NHS-1cM [40]. To then synthesize 1pM, 3-aminopropyl phosphonic acid (110 mg, 0.790 mmol) was dissolved in HPLC-grade H2O (6 mL), HPLC-grade tetrahydrofuran (THF, 12 mL), and triethylamine (0.380 mL, 2.70 mmol) and the solution stirred at room temperature. In a separate flask, NHS-1cM (1.10 g, 0.183 mmol) was dissolved in HPLC-grade THF (25 mL) and this solution added to the reaction flask. The solution was stirred for 20 hrs at room temperature and the THF removed in vacuo. The resulting yellow oil was dissolved in DCM and 0.1 N HCl and stirred for 20–30 min. The organic portion was washed with 0.1 N HCl (1 × 30 mL) and 50:50 brine/H2O (2 × 60 mL), dried over magnesium sulfate and concentrated to a yellow oil. White product was isolated by centrifugation with diethyl ether (5 × 40 mL). Yield: 0.896 g, 80.0%. 1H-NMR (CDCl3): δ 5.70 (m, 2H, CH), 5.00 (m, 2H, CH), 4.20 (m, 4H, CH2), 3.60 (m, ~0.45 kH, CH2O), 3.38 (s, 3H, CH3), 2.43 (m, 8H, CH2), 1.61 (m, 10H, CH2), 1.22 (m, 64H, CH2), 0.87 (t, 12H, CH3). Gel permeation chromatography: Mw: 7.0 kDa; polydispersity index (PDI): 1.1.
The critical micelle concentration (CMC) of 1pM and 1cM were determined as previously described [27]. In brief, AMs were dissolved in HPLC grade H2O and diluted to a series of concentrations ranging from 1 × 10−3 to 1 × 10−10 M. Separately, a stock solution of pyrene was prepared in HPLC grade acetone (5 × 10−6 M) and 0.5 mL of this solution was added to a series of vials. Acetone was removed in vacuo and AM solutions (5 mL) were added. AM-pyrene solutions were incubated for 48 h at 37 °C with gentle agitation (60 rpm) to allow pyrene to partition into the AM micelles. Fluorescence studies were then conducted on a RF-5301PC spectrofluorometer (Shimadzu Scientific Instruments, Columbia, MD), using pyrene as the fluorescent probe. Emission was measured from 300–360 nm, with a 390 nm excitation wavelength. Upon micelle formation, pyrene partitions into the micelle hydrophobic core and the maximum wavelength emission shifts from 332 to 334.5 nm. The ratio of absorption of pyrene in micelles (334.5 nm) to pyrene alone (332 nm) was plotted against the logarithm of AM concentration, and the inflection point of this curve was taken as the CMC.
NP Fabrication and Characterization
NPs were fabricated via a flash nanoprecipitation process (Figure 1C) described previously [34]. All equipment was sterilized in an autoclave, with 0.5 M NaOH + 1 M NaCl solution or 70% ethanol, for 30 minutes and then rinsed with sterile DI H2O. Briefly, the shell material, either 1pM, 1cM, or poly(styrene-PEG), was dissolved in THF at 40 mg/mL and the core material, either M12 or polystyrene, was dissolved in the same solution at 20 mg/mL and sterilized with a 0.22 μm nylon filter, resulting in a shell to core weight ratio of 2:1. Five hundred microliters of the THF stream was mixed with 700 μL of the aqueous stream via a confined impinging jet mixer, after which the stream was immediately dispersed into 3.8 mL of PBS in sterile cell culture hoods. The NPs were dialyzed twice against sterile PBS to remove THF and sterilized with a 0.22 μm PVDF filter. Following dialysis, three independent batches of NPs were characterized by dynamic light scattering (DLS) using a Malvern-Zetasizer Nano Series DLS detector with a 22 mW He–Ne laser operating at λ = 632.8 nm using general purpose resolution mode. Hydrodynamic diameters (peak size from intensity distribution), PDI, and ζ-potentials were determined after dialyzing the NPs against picopure water, then diluting the NPs 10-fold in picopure water (Figure 1D).
Smooth Muscle Cell Culture
Human primary coronary artery SMCs (Lonza) between passages 3–6 were cultured in cultured in Smooth Muscle Basal Media (SmBM, Lonza) supplemented with 5% FBS and Smooth Muscle Growth Media-2 SingleQuots (Lonza) and maintained in T75 flasks (BD Falcon) at 37 °C in a humidified atmosphere containing 5% CO2. Media was changed every 2 days until cells were passaged (~80% confluency) and plated at a seeding density of 5,000 cells/cm2 in either a 12-well plate (BD Falcon) for flow cytometry or an 8-well Nunc Labtek chamber slide system (Sigma) treated with poly-D-lysine for microscope imaging.
oxLDL-induced SMC Proliferation
Cells were serum-starved for 24 h to synchronize mitosis and arrest cell proliferation before the media was aspirated and replaced with co-treatments consisting of 10 μg/mL of oxLDL to induce proliferation as specified below and either SmBM alone for the basal condition or nanotherapeutic treatments diluted in basal media to the appropriate concentration. After 24 or 48 h, cell proliferation was assessed using a CyQUANT NF Cell Proliferation Assay Kit (Life Technologies). A design of experiments with varying seeding density and oxLDL concentration was used to determine optimal conditions for inducing SMC proliferation (Supporting Information, Figure S5).
OxLDL Uptake
To evaluate the influence of AM nanotherapeutics on oxLDL uptake, SMCs were incubated in base media for 24 or 48 h with fluorescent DiO oxLDL (5 μg/mL, Kalen Biomedical) and unlabeled oxLDL (5 μg/mL, Biomedical Technologies), with and without unimers, micelles, or NPs (10−5, 10−6, and 10−7 M) of each end group chemistry. Studies were conducted with an oxLDL possessing a high relative electrophoretic mobility (1.8–2.1 cm2), as it is representative of the highly oxidative states of LDL encountered by SMCs in restenosis [41]. Cells were prepared for flow cytometry analysis by incubating on ice for 15 min, detached by vigorous pipetting, transferred to 5 mL tubes, centrifuged at 1000 rpm for 10 min, and then fixed with 1% paraformaldehyde (PFA). Fluorescence was quantified using a FACSCalibur flow cytometer (BD Biosciences) by collecting 8,000 events per sample and analyzed with FlowJo software (Treestar).
Scavenger Receptor Surface Expression
To evaluate the influence of NPs on cell surface scavenger receptor expression, SMCs were incubated with unlabeled oxLDL (10 μg/mL) with or without micelles or NPs of each end group chemistry for 24 h. Following the incubation, treatments were removed, and cells were prepared for evaluation via flow cytometry or microscopy. For flow cytometry preparation, the cells were washed in blocking buffer (0.5% bovine serum albumin (BSA), 0.1% sodium azide, and 1% normal goat serum in PBS), and placed on ice for 15 min. Cells were detached by vigorous pipetting, transferred to 5 mL tubes and centrifuged at 1000 rpm for 10 min. Supernatants were then decanted, and the cells incubated for 1 h at 4 °C with monoclonal mouse anti-human MSR1-phycoerythrin (PE) antibody (clone 351615, R&D Systems) and monoclonal mouse anti-human CD36-allophycocyanin (APC) antibody (clone: 5–271, Biolegend) or their corresponding isotype control mouse IgG2β PE (clone 133303, R&D systems) and mouse IgG2α, κ APC (clone: MOPC-173, Biolegend). Following antibody incubation, the cells were washed twice and then fixed with 1% PFA. Flow cytometry analysis was conducted similarly to oxLDL uptake quantification.
For microscopy, treated cells were washed with PBS, fixed with 4% PFA and blocked with 0.5% BSA, 0.1% sodium azide, 10% normal goat serum, and 0.1% Tween-20 in PBS for 15 min at 25 °C. Next, cells were incubated for 24 h at 4 °C with monoclonal mouse anti-human MSR1-PE antibody (clone 351615, R&D Systems) and monoclonal mouse anti-human CD36-APC antibody (clone: 5–271, Biolegend) or their corresponding isotype control mouse IgG2β PE (clone 133303, R&D systems) and mouse IgG2α, κ APC (clone: MOPC-173, Biolegend). Following incubation, samples were washed twice and incubated with the secondary antibody, goat anti-mouse IgG2α Alexa Fluor 488 (CD36 conditions) or goat anti-mouse IgG2β alexa fluor 647 (MSR1 conditions) for 1 h at 25 °C. They were then washed, fixed in 4% PFA, and counterstained with nuclear Hoechst 33342 before imaging on a Leica TCS SP2 confocal microscope. Representative images of receptor expression were obtained and processed with ImageJ software (NIH).
Gene Transcription Studies using Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
To evaluate the influence of nanotherapeutics on gene regulation, SMCs were incubated with unlabeled oxLDL (10 μg/mL) with or without AM treatments in SmBM for 24 h. Following the incubation, RNA was extracted with the RNeasy Plus Mini Kit following the manufacturer’s protocol. RNA was reverse transcribed to cDNA with High Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s protocol using a BioRad thermal cycler. qRT-PCR was performed on a Lightcycler 480 (Roche Life Science) with Fast SYBR Green Master Mix (Life Technologies) for 40 cycles. Fold change was calculated using ∆∆Ct with β-actin and GAPDH as endogenous control genes. Primer sequences are listed in Supporting Information.
AM Coating Fabrication
Coated coupons were fabricated following a method described previously [38]. Briefly, polished stainless steel 316L coupons were coated using tethering by aggregation and growth, where coupons were suspended in a 1 mM 1pM or 1cM solution in THF and the solvent was allowed to fully evaporate. Following coating, coupons were annealed in a vacuum oven (140 C, 2 torr, 3 h) and rinsed twice with THF to remove untethered molecules. The coupons were dried in a stream of nitrogen, and successful coating was confirmed using X-ray photoelectron spectroscopy, as previously described [38].
Platelet Isolation
Whole human blood (NY Blood Center) was separated by centrifugation (150 × g for 15 min, room temperature) in a tube containing 1/10 volume of ACD anticoagulant buffer (39 mM citric acid, 75 mM sodium citrate, and 135 mM dextrose in nanopure water, pH 7.4). The upper phase of platelet-rich plasma was transferred to a new tube and centrifuged with 1/10 ACD anticoagulant buffer at 900 × g for 5 min at room temperature to pellet platelets. The platelets were resuspended in 1 mL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-Tyrode buffer (134 mM sodium chloride, 12 mM sodium bicarbonate, 2.9 mM potassium chloride, 0.34 mM sodium phosphate monobasic, 5 mM HEPES, 5 mM glucose, 1% bovine serum albumin, pH 7.4) and cell count adjusted with HEPES-Tyrode buffer to a suspension of 108 platelets/mL.
Platelet Adhesion Assay
To evaluate the influence of AM nanoassemblies on platelet adhesion, 1pM and 1cM coated and uncoated stainless steel 316L coupons were incubated with platelets suspended in buffer for 90 min (37 C, 5% CO2) with gentle shaking. Following the incubation, the coupons were rinsed three times with HEPES, and adherent platelets on the coupon fixed using a 2.5% glutaraldehyde in HEPES solution. Samples were dried under high vacuum, sputter coated with gold, and imaged using field emission scanning electron microscopy (Zeiss Sigma Field Emission SEM with Oxford EDS, Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Images were recorded with 5.0 kV accelerating voltage from three independent sets of samples.
Statistical Analysis
Unless otherwise noted, all results represent the average of three independent experiments with two technical replicates per experiment. Statistical analyses were performed using JMP by SAS. Statistical significance (p ≤ 0.05) was determined using a one-way ANOVA with Tukey’s posthoc test for comparisons between multiple groups. Statistical analysis was conducted over the full set of data to make comparisons between all treatment groups. In the figures, treatments with the same letter are not statistically significantly different from one another.
RESULTS
AM Synthesis and Characterization of Nanotherapeutics
Carboxylate- and phosphonate-AM unimers were synthesized following a previously published procedure (Figure 1A) [40]. Micelles and NPs comprised of AMs were prepared as previously described via thermodynamic self-assembly and kinetic flash nanoprecipitation processes, respectively (Figure 1B and 1C) [33, 34, 40]. The hydrodynamic diameters of 1pM and 1cM nanotherapeutics revealed monodisperse size populations (Figure 1D and Supporting Information, Figure S1). The sizes of 1pM and 1cM micelles were similar (19–22 nm), whereas the size of 1pM and 1cM NPs were about 10-fold larger and ranged between 178–204 nm.
The NP ζ-potential values were generally more negative than their micelle counterparts. NPs have a higher negative surface charge density, indicating a higher localized charge and greater stability (Figure 1D) [34]. The NPs also have less variability in ζ-potential due to their more stable form, whereas the variability of the micelles was higher due to the dynamic equilibrium between the micellar and unimer forms.
As previous studies have established that negatively charged functional groups provide enhanced binding to oxLDL receptors on macrophages, it was important to compare these end-groups to determine the effects of the singly charged carboxylic acid relative to the doubly charged phosphonic acid [42]. In comparing the end-group chemistries, the 1pM micelles had an increased ζ-potential due to the higher net negative charge (−2 net charge for 1pM compared to −1 net charge for 1cM) on the anionic end group (Figure 1D), yet the ζ-potential of phosphonate-and carboxylate-terminated AM micelles and NPs were indistinguishable, likely due to the negative AM termini being shielded by the PEG corona to varying extents.
Nanotherapeutics Attenuate SMC Proliferation
As nanoscale presentation is a critical determinant of atheroprotective efficacy, AM unimers, micelles, and NPs were evaluated to determine formulation effects on reducing SMC proliferation induced by oxLDL. In these studies, AM treatments were co-incubated with oxLDL for either a 24- or 48-h treatment period before proliferation was quantified using a fluorescence-based assay to determine the cell count.
Both AM micelles and NPs demonstrate a significant reduction in oxLDL-induced SMC proliferation. Above the CMC, at 10−5 M and 10−6 M, 1pM and 1cM micelles reduced SMC proliferation by 40–60% compared to an oxLDL-only control (not shown, normalized to 0% reduction in SMC proliferation), while 1pM and 1cM NPs across all concentrations demonstrated a 55–60% reduction in SMC proliferation (Figure 2). By evaluating concentrations above (10−5 M and 10−6 M) and below (10−7 M) the AM CMC values (~2.5 × 10−7 M), the structure-activity relationships between unimers and AM micelles were elucidated. Micelles exhibited a concentration-dependent effect; levels above the CMC were highly effective at reducing SMC proliferation while levels below the CMC (i.e., AM unimers) exhibited slight bioactivity, suggesting that the micellar form is critical for bioactivity (Figure 2A). Additionally, at the highest concentration (10−5 M) and 48 h treatment duration, 1pM micelles reduced proliferation to a greater extent compared to 1cM micelles, but 1pM and 1cM NPs exhibited similar results across the other concentrations and treatment periods (Figure 2), suggesting that increased localized density of charge and/or hydrophobicity (relative to unimers) is crucial to minimizing proliferation. The 48 h treatments generally reduced proliferation between 3–15% more than 24 h treatments.
Figure 2.
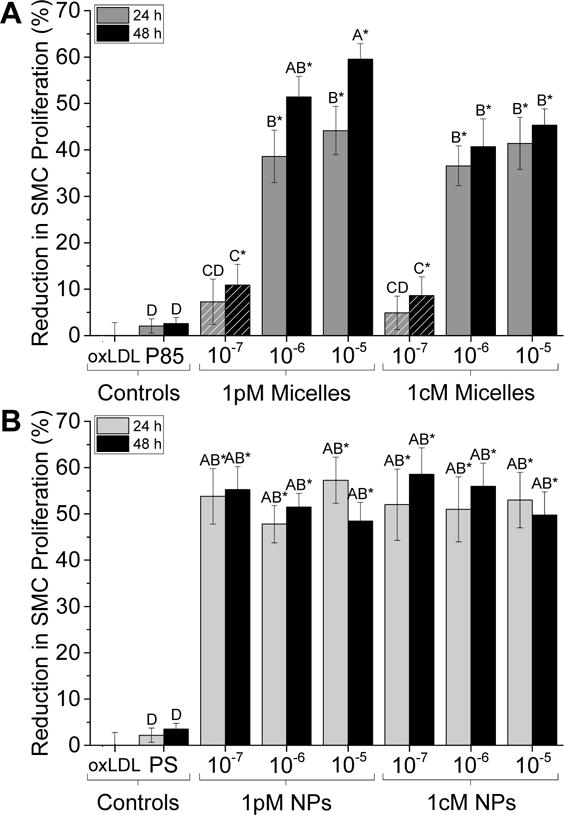
AM nanotherapeutics significantly reduce levels of SMC proliferation after 24- and 48-h treatments relative to Pluronic P85 control. (A) 1pM and 1cM micelle treatments at concentrations above the CMC (solid color) reduce SMC proliferation by up to 60%. Below the CMC value (10−7 M, hatched pattern), SMC proliferation is significantly reduced by ~10% compared to the oxLDL-treated control. (B) 1pM and 1cM NP treatments across all concentrations reduce SMC proliferation by ~50–60%. The asterisk (*) indicates statistical significance from the control (Mean ± SD; n = 3; p ≤ 0.05).
The control micelles and NPs did not appreciably decrease the degree of SMC proliferation after 24 or 48 h compared to the oxLDL only control. The control amphiphile used to form micelles, Pluronic P85, is a triblock PEG-PPG-PEG copolymer that has a comparable molecular weight and is structurally similar in the hydrophilic region to AMs. The control polystyrene-based (PS) NPs were formed using the same flash nanoprecipitation process as the experimental NPs and previously shown to be non-bioactive [36]. The NP shell contains poly(styrene-PEG) copolymer and the core consists of polystyrene homopolymer, both of which have a similar molecular weight to that of the experimental NP shells and cores, respectively.
Nanotherapeutics Suppress Oxidized Low-Density Lipoprotein Internalization
To elucidate the mechanism by which nanotherapeutics inhibit SMC proliferation, oxLDL uptake was examined. OxLDL has been shown to induce SMC proliferation and restenosis, which may be due to the higher local oxidative stress causing healthy SMCs to develop a proliferative phenotype [16, 21, 43]. To this end, SMCs were co-incubated with fluorescently-labeled oxLDL in the presence of either micelles or NPs for 24 h, after which oxLDL uptake was quantified via flow cytometry or visualized via confocal microscopy. SMCs co-incubated with oxLDL and AM micelles (Figure 3A) or NPs (Figure 3B) demonstrated significant reduction in oxLDL uptake compared to the oxLDL control. 1pM and 1cM micelles at concentrations above the CMC and NPs across all concentrations reduced oxLDL internalization by up to 90% compared to the positive oxLDL control. Microscopy of SMC oxLDL uptake confirmed low levels of internalized oxLDL (Figure 3C and Supporting Information, Figure S2).
Figure 3.
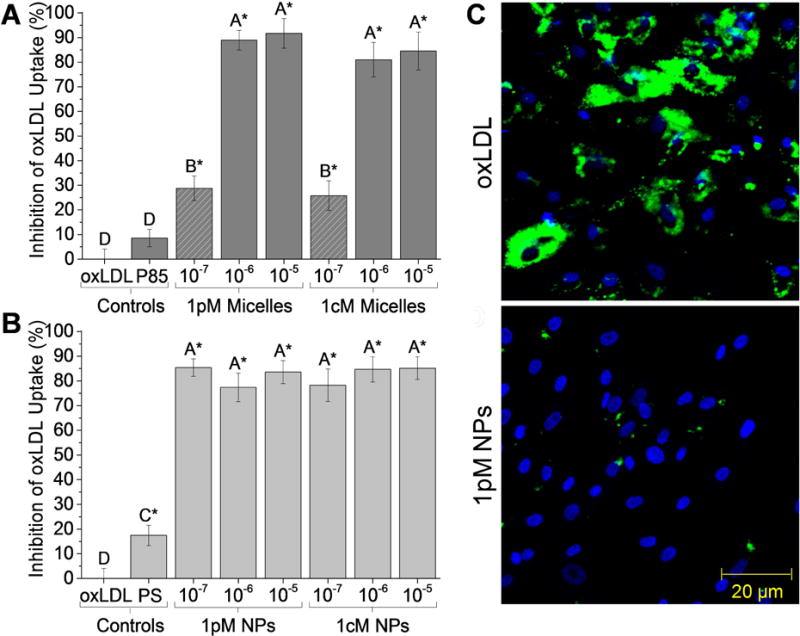
AM nanotherapeutics significantly reduce the accumulation of oxLDL in SMCs. Flow cytometry data demonstrates significant reduction in oxLDL internalization after incubation with AM concentrations above the CMC (10−5 M and 10−6 M, solid dark gray) (A) and 10−5 M to 10−7 M NP treatments (B) in serum-free media for 24 h. The asterisk (*) indicates statistical significance from the oxLDL control (Mean ± SD; n = 3; p ≤ 0.05). (C) SMCs co-treated with 10−5 M 1pM NPs and oxLDL for 24 h demonstrate significant reductions in fluorescently labeled oxLDL uptake (green) compared to the oxLDL-only control. Nuclei were counterstained with Hoechst 33342 (blue).
The oxLDL uptake results followed similar trends to the SMC proliferation results: AM micelles exhibited a concentration-dependence, whereas AM NPs were highly effective at reducing uptake across all concentrations. With respect to end-group chemistry, 1pM and 1cM micelles and NPs exhibited similar ability to inhibit oxLDL accumulation.
Nanotherapeutics Downregulate Scavenger Receptor Expression
Scavenger receptors, particularly MSR1 and CD36, have been implicated in the progression of restenosis and SMC proliferation [44–47]. To further examine the mechanism for the reduction in SMC proliferation and oxLDL uptake, the expression of two scavenger receptors integral in this pathway (MSR1 and CD36) were evaluated after 24-h incubation with oxLDL and micelles or NPs (Figure 4A–D). Nanotherapeutic treatments downregulated scavenger receptor expression of MSR1 by up to 48% and CD36 by up to 33% as compared to the positive oxLDL-only control. Specifically, 1pM and 1cM micelles at 10−5 M and 10−6 M (Figures 4A and 4B) and NPs across all concentrations (Figures 4C and 4D) significantly lowered scavenger receptor expression to basal expression levels. Similar to the SMC proliferation and oxLDL uptake results described above, AM micelles exhibited a concentration-dependence, whereas AM NPs were highly effective at reducing scavenger receptor expression across all concentrations. Visualization of scavenger receptor expression via confocal microscopy revealed SMCs co-incubated with oxLDL and AM micelles or NPs at 10−5 M exhibited MSR1 and CD36 expression similar to basal expression levels (Figure 4E and Supporting Information, Figure S3).
Figure 4.
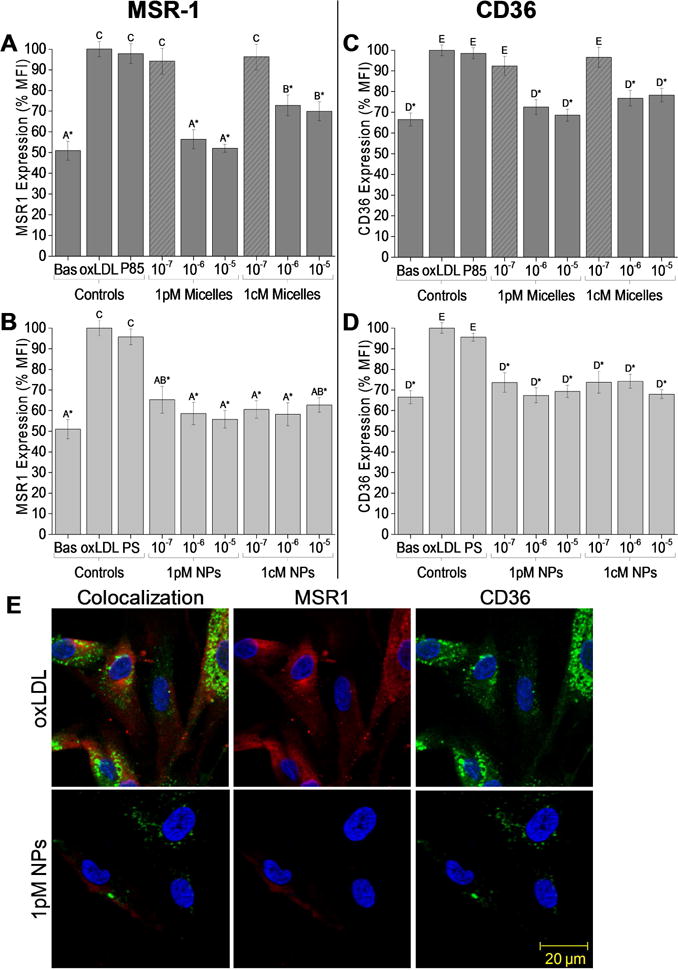
OxLDL significantly increases SMC scavenger receptor surface expression of MSR1 (A,B) and CD36 (C,D), which is attenuated by AM treatment. Specifically, 24 h co-incubation with inflammatory oxLDL and AM treatments of either 10−5 M or 10−6 M 1pM micelle concentration (A,C, solid dark gray) or at all NP concentrations tested (B,D, light gray) lowers the scavenger receptor expression to basal levels. The asterisk (*) indicates statistical significance from the oxLDL control (Mean ± SD; n = 3; p ≤ 0.05). (E) SMCs co-treated with 10−5 M 1cM NPs and oxLDL for 24 h demonstrate significant reductions in MSR1 (red) and CD36 (green) expression compared to the oxLDL-only control. Nuclei were counterstained with Hoechst 33342 (blue).
Nanotherapeutics Downregulate Gene Transcription
As restenosis is due to a cascade of events triggered by a change in SMC from a contractile phenotype to more proliferative phenotypes, the gene regulation of key pro-inflammatory markers (IL-1β, IL-6, IL-8, TNF-α, MCP-1, MMP9) and scavenger receptors (CD36, MSR1, LOX1) was examined following oxLDL exposure and AM treatment.
The fold-change in mRNA levels is shown in Figure 5. AM micelles and AM NPs reduced the oxLDL-induced increase in inflammatory and scavenger receptor transcription back to near-basal levels. Both 1pM and 1cM NPs exhibited increased downregulation of genes upregulated by oxLDL compared to their micellar AM counterparts, notably those for scavenger receptors MSR1 and CD36. Comparing end group chemistries, 1cM micelles and NPs slightly but significantly downregulated genes to a lesser extent than 1pM micelles and both NPs. At concentrations below the CMC, AM unimers exhibited gene transcription levels that are not statistically significantly different than the basal levels, which were normalized to 1.0 (data not shown).
Figure 5.

SEM images of platelet adhesion assay indicate significant platelet attachment after uncoated coupons were incubated with a platelet suspension for 90 min (A), but minimal platelet attachment in coupons coated with 1pM (B) and 1cM (C).
AM Nanoassembly Coatings Decrease Platelet Adhesion to Limit Thrombosis
The end group chemistry of the AMs studied in these experiments can be further exploited to functionalize surfaces for bioactive medical device coatings. For example, the phosphonate and carboxylate AM end groups have previously been shown to form strong coordination bonds to metal oxide substrates, yielding uniform self-assembled monolayers using a simple grafting process [38]. As DES exhibit post-implantation complications associated with thrombosis, the coatings’ ability to minimize the surface platelet adhesion was evaluated through a platelet adhesion assay. Stainless steel coupons were coated in solutions of 1pM and 1cM and immersed in a platelet suspension for 2 h before adhered platelets were fixed and imaged using SEM. While uncoated coupons exhibited significant platelet adhesion, 1pM and 1cM coated coupons displayed significantly fewer attached platelets (Figure 5).
DISCUSSION
Although vascular stents are the gold standard of interventional devices in treating severe CHD, restenosis and thrombosis remain significant challenges [2, 3, 7]. Restenotic complications result from intimal hyperplasia and platelet adhesion following localized injury, and the local sites are frequently accompanied by high levels of lipid burden. We developed a novel strategy to counteract these phenomena and promote a pro-healing, non-cytotoxic remodeling approach for the repression of SMC lipid uptake and hyperproliferation, which could be effective in preventing further oxidative stress, SMC proliferation, and platelet adhesion within stented arteries.
Previous research has shown the importance of downregulating macrophage scavenger receptors, as they exacerbate the inflammatory pathway in atherosclerosis [29]. As oxLDL uptake by scavenger receptor is also a critical step in the SMC proliferation pathway, we sought to specifically design AM systems to counteract oxidative stress leading to restenosis and thrombosis. OxLDL displays anionic characteristics in the physiological environment and binds to cationic residues of scavenger receptors. Thus, AMs with two different net charges were synthesized and evaluated as three different types of nanotherapeutic formulations (unimers, micelles, and NPs). These studies evaluated the effects of physicochemical properties of synthetic nanoassemblies (i.e., size, charge localization, and stability) on the resulting SMC proliferation, oxLDL uptake, scavenger receptor expression, gene transcription, and anti-platelet properties.
Nanotherapeutics Competitively Inhibit oxLDL to Regulate SMC Proliferation
The AM nanotherapeutics were effective at reducing SMC proliferation triggered by oxLDL. Based on oxLDL uptake, scavenger receptor expression, and gene transcription studies, this bioactivity is likely due to a competitive inhibition mechanism, where the nanoassemblies prevent oxLDL from binding to scavenger receptors and thus reduce oxLDL internalization. This hypothesis is further supported by previous modeling and experimental studies, which demonstrated that negatively charged micelles and NPs preferentially bind to scavenger receptor domains on macrophages [25, 32, 36]. These studies confirmed the mechanisms of receptor blocking and cellular internalization rather than AM complexation with oxLDL, as AMs did not bind to oxLDL [25, 29, 34, 36]. Additionally, these therapeutic benefits occur by cellular mechanisms distinct from current cytotoxic or immunosuppressive mechanisms, as the nanotherapeutics demonstrate low cytotoxicity (Supporting Information, Figure S4).
AM Formulation Influences Bioactivity
In general, AM nanotherapeutics demonstrate a therapeutically relevant effect by reducing SMC proliferation in vitro. Nanoscale AM micelles and NPs have ideal characteristics for therapeutics, as they possess favorable size and surface charge for specific receptor interactions, and may also evade metabolic deactivation and clearance by the mononuclear phagocyte system due to PEG shielding [31]. The influence of the nanotherapeutic formulation, which has implications on how the size, localized charge and/or hydrophobicity, and stability affects efficacy, was studied by using concentrations above and below the CMC values to compare unimers, micelles, and the concentration-dependence of NPs. The data demonstrate that for micelles, concentrations above the CMC (in which the assemblies are in a micellar form) yield significantly higher efficacy of reduced SMC proliferation, attenuated oxLDL uptake, and a downregulation of inflammatory markers and scavenger receptor expression. The higher efficacy of AMs in the self-assembled, micellar form as compared to unimer form may arise from the higher local density of anionic charge and hydrophobic domains in the micelles, which promote enhanced binding to the cationic, lipophilic scavenger receptors [28, 29]. Below the CMC, AMs provided smaller reductions in SMC proliferation and oxLDL uptake, which suggests that while AM unimers have some inherent efficacy, they lack sufficient local concentrations, size, and/or localized charge and hydrophobicity to bind to scavenger receptors.
The AMs formulated as NPs had higher efficacy than their analogous micellar form; this result is likely due to the increased local concentration of the negatively charged hydrophobic domain that is critical for binding to scavenger receptors [34]. The proliferation, oxLDL uptake, and scavenger receptor expression were similarly effective for all NP concentrations tested, suggesting that the scavenger receptors may be locally saturated with nanotherapeutics, even at the lowest concentration tested.
Direct comparison of varying treatments (nanotherapeutic formulation, end-group chemistry, and concentration) illustrates that concentrated charge and/or hydrophobicity in NPs has a more significant impact on bioactivity than changes in AM net charge and end-group chemistry for SMC proliferation, oxLDL uptake, scavenger receptor expression, and gene transcription. The specific binding affinity of micelles and NPs to scavenger receptors is under further investigation and will be discussed in a future publication.
While 1pM and 1cM NPs behaved similarly, 1pM in micellar form demonstrated increased reduction in scavenger receptor expression at 10−5 M compared to 1cM. At the higher concentrations, the phosphonate-AM micelles may provide enhanced binding to the cationic MSR1 and CD36 residues, thereby inhibiting oxLDL internalization and moderating scavenger receptor expression [36, 42]. In previous work, anionic AMs with a greater net negative charge were more efficacious in counteracting macrophage oxLDL uptake in silico and in vitro due to their favorable binding energies [25–27]. This study demonstrates that similar principles can be applied to repress SMC proliferation through scavenger receptor interactions.
Unique Mechanism of Action to Reduce Thrombosis in Addition to Restenosis
Prior to this work, the interactions between oxLDL and SMC gene transcription were not well established. The qRT-PCR results demonstrate that oxLDL significantly upregulated TNF-α, MCP-1, MMP9, CD36, MSR1, and OLR1, genes actively involved in the restenosis cascade. This data suggests that oxLDL contributes to the change from a contractile to a synthetic SMC phenotype, leading to excessive SMC proliferation. TNF-α, oxLDL, and other inflammatory triggers lead to elevated levels of matrix metalloproteinases (MMP-9), which is critical to the SMC migration and extracellular matrix remodeling processes [48, 49]. A previous study demonstrated that inhibition of MMPs resulted in 97% decrease in SMCs migrating into the intima within 4 days post-surgery, which suggests that the ~9.5-fold downregulation from oxLDL-induced proliferation in MMP-9 transcription by AM nanotherapeutics could be a promising strategy to counteract restenosis [50]. The proposed mechanisms by which the AMs inhibit SMC hyperproliferation, as elucidated by this study, are illustrated in Figure 6.
Figure 6.
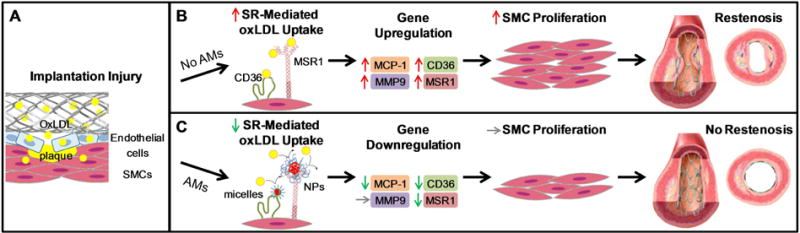
Proposed model for effect of AM nanotherapeutics on SMC proliferation dynamics and its consequent outcomes. (A) The force of stent implantation releases oxLDL accumulated in atherosclerotic plaque and elicits a foreign material response, increasing localized inflammation and provoking an immune response from the native tissue. (B) Unregulated scavenger receptor-(SR-) mediated oxLDL uptake causes MCP-1, MMP-9, CD36, and MSR1 gene upregulation, leading to excessive SMC proliferation and in-stent restenosis. (C) Optimized AM nanotherapeutic formulations and end group functionalities competitively inhibit oxLDL internalization by binding to scavenger receptors, thus downregulating MCP-1, CD36, and MSR1 and reducing SMC proliferation.
1pM and 1cM coated coupons significantly reduced the number of attached platelets (Figure 5), which suggests that coatings of AMs may minimize thrombosis events in the early restenosis cascade. The decreased platelet adhesion may be due to the molecular orientation of 1pM and 1cM on the surface, where the hydrophobic domain forms a base layer and a PEG layer is presented on the outermost surface, as proposed previously [38]. PEG has demonstrated non-fouling properties, where PEG-coated samples significantly reduce protein and cell adhesion [51]. For stents, thrombosis remains a significant issue. While PEG-coated stents have previously shown promising clinical results due to their ability to reduce thrombosis,[52] AMs are unique in that they have inherent ability to concurrently reduce SMC proliferation, thereby eliminating the need for patients to take anti-platelet therapies after stent implantation.
As the first study of its kind on the ability of AMs to reduce SMC proliferation, this effort builds further on the previous structure-activity relationships elucidated in macrophages for a broad range of AM-based nanotherapeutics [25–30, 32, 34, 36]. In those studies, both AM micelles and NPs significantly reduced inflammatory atherosclerosis both in vitro and in vivo through similar interactions with macrophage scavenger receptors [25–30, 32, 34–36]. The ability of AMs to change macrophage phenotype on a cellular level is highly relevant to this current study, as macrophages are also implicated in the restenosis cascade, where they promote inflammation that commences directly after stent implantation and continues to three or more weeks after surgery [53]. Specifically, oxidative stress causes upregulated expression of inflammatory macrophage scavenger receptors and consequently leads to rapid SMC proliferation and extracellular matrix formation [2]. As AMs preferentially bind to scavenger receptors, AM nanotherapeutics may provide a broad-based lesion-targeted treatment, thereby enabling lower doses compared to non-specific therapeutics [27, 28]. Additionally, the AMs possess the benefit of a tunable structure, in which the end group can be modified to promote self-assembly to cardiovascular stents, catheter balloons or other devices, allowing for a wide range of functional coatings. Localization to arterial plaques could also occur due to the targeting capability of AM nanoassemblies, which could be further enhanced by infusion catheter delivery [35].
CONCLUSION
In this study, phosphonate- and carboxylate-terminated AM unimers, micelles, and NPs were evaluated in an oxidized lipid-induced SMC proliferation model to determine the efficacy of these nanotherapeutics as potential non-cytotoxic treatments for restenosis. Additionally, AM coatings were evaluated to determine the ability to prevent platelet attachment on a metal surface. AM micelles and NP treatments demonstrated significant reduction in SMC proliferation and subsequent mechanistic studies suggest that this effect is due to inhibition of oxLDL uptake, and concomitant scavenger receptor expression downregulation. AM formulation is a significant factor in the efficacy, where the unimeric forms of the AMs had decreased efficacy in mediating SMC proliferation compared to micelles and NPs. Additionally, the nanotherapeutics’ anionic character promotes binding to the cationic residues of MSR1 and CD36 scavenger receptors, thereby inhibiting oxLDL internalization and moderating scavenger receptor expression. Additionally, AM coatings significantly minimized platelet attachment to a metal substrate, which suggests that AM coatings limit thrombosis and highlighting that AMs could be readily delivered via stent coatings. These results indicate that AMs could serve as a new class of pro-healing therapeutics for restenosis and thrombosis, with mechanisms-of-action that can bypass the current cytotoxic or immunosuppressive approaches. Thus, this work offers a novel approach to address the critical determinants of SMC proliferation in conjunction with both localized injury and hyperlipidemia. Potential applications include integration of the AMs examined within surface modified post-intervention cardiovascular devices, such as coated stents or balloons, and intravenous injected solutions.
Supplementary Material
Table 1.
qRT-PCR Gene Transcription Levels (shown as fold-change in expression vs. basal)
| Sample | Inflammatory Markers | Scavenger Receptors | |||||||
|---|---|---|---|---|---|---|---|---|---|
| IL-1β | IL-6 | IL-8 | TNF-α | MCP-1 | MMP9 | MSR1 | CD36 | LOX1 | |
| Basal | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| oxLDL | 0.6 | 0.9 | 0.6 | 4.7 | 6.3 | 9.5 | 12.4 | 10.0 | 10.1 |
| P85 | 0.8 | 0.9 | 1.2 | 3.5 | 7.8 | 11.4 | 10.7 | 8.3 | 7.9 |
| 1pM micelles | −0.5 | 0.1 | −0.6 | −0.7 | 0.3 | 0.6 | −0.7 | −0.9 | 0.5 |
| 1cM micelles | −1.1 | −0.5 | −1.3 | −0.4 | 0.1 | 1.1 | −0.7 | −1.5 | 0.4 |
| PS | 0.3 | 1.2 | 0.8 | 4.2 | 6.1 | 10.3 | 12.8 | 9.5 | 10.2 |
| 1pM NPs | 0.0 | −0.4 | −1.4 | −0.5 | −1.4 | 0.3 | −1.8 | −1.8 | −0.5 |
| 1cM NPs | 0.4 | −0.4 | −0.5 | −1.3 | −1.4 | −0.5 | −1.9 | −1.5 | 0.4 |
Acknowledgments
The authors thank gratefully acknowledge Rebecca Chmielowski, Margot Zevon, and Daniel Martin for technical assistance. This work was supported with funding provided by the Coulter Biomedical Engineering Research Grant to PVM, National Institutes of Health (NIH R01 HL107913 to PVM and KEU) and the National Science Foundation Graduate Research Fellowship (NSF GRF, JWC).
APPENDIX A: SUPPLEMENTARY DATA
AM micelle and NP size distributions, cell viability results, proliferation assay development details, gene expression primer sequences, oxLDL uptake images, and scavenger receptor expression images can be found online.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. Heart Disease and Stroke Statistics—2009 Update A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–6. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 2.Orford JL, Selwyn AP, Ganz P, Popma JJ, Rogers C. The comparative pathobiology of atherosclerosis and restenosis. The American journal of cardiology. 2000;86:6H–11H. doi: 10.1016/s0002-9149(00)01094-8. [DOI] [PubMed] [Google Scholar]
- 3.Akin I, Schneider H, Ince H, Kische S, Rehders TC, Chatterjee T, et al. Second- and third-generation drug-eluting coronary stents: progress and safety. Herz. 2011;36:190–6. doi: 10.1007/s00059-011-3458-z. [DOI] [PubMed] [Google Scholar]
- 4.Farooq V, Gogas BD, Serruys PW. Restenosis: Delineating the Numerous Causes of Drug-Eluting Stent Restenosis. Circ Cardiovasc Interv. 2011;4:195–205. doi: 10.1161/CIRCINTERVENTIONS.110.959882. [DOI] [PubMed] [Google Scholar]
- 5.Mitra A, Agrawal D. In stent restenosis: Bane of the stent era. J Clin Pathol. 2006;59:232–9. doi: 10.1136/jcp.2005.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bangalore S, Amoroso N, Fusaro M, Kumar S, Feit F. Outcomes With Various Drug-Eluting or Bare Metal Stents in Patients With ST-Segment–Elevation Myocardial Infarction: A Mixed Treatment Comparison Analysis of Trial Level Data From 34 068 Patient-Years of Follow-up From Randomized Trials. Circ Cardiovasc Interv. 2013;6:378–90. doi: 10.1161/CIRCINTERVENTIONS.113.000415. [DOI] [PubMed] [Google Scholar]
- 7.Jukema JW, Ahmed TA, Verschuren JJ, Quax PH. Restenosis after PCI. Part 2: prevention and therapy. Nature Reviews Cardiology. 2012;9:79–90. doi: 10.1038/nrcardio.2011.148. [DOI] [PubMed] [Google Scholar]
- 8.Kazi DS, Leong TK, Chang TI, Solomon MD, Hlatky MA, Go AS. Association of Spontaneous Bleeding and Myocardial Infarction With Long-Term Mortality After Percutaneous Coronary Intervention. J Am Coll Cardiol. 2015;65:1411–20. doi: 10.1016/j.jacc.2015.01.047. [DOI] [PubMed] [Google Scholar]
- 9.Kalesan B, Pilgrim T, Heinimann K, Räber L, Stefanini GG, Valgimigli M, et al. Comparison of drug-eluting stents with bare metal stents in patients with ST-segment elevation myocardial infarction. European heart journal. 2012;33:977–87. doi: 10.1093/eurheartj/ehs036. [DOI] [PubMed] [Google Scholar]
- 10.Finn AV, Nakazawa G, Joner M, Kolodgie FD, Mont EK, Gold HK, et al. Vascular responses to drug eluting stents importance of delayed healing. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1500–10. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- 11.Jackson SP, Calkin AC. The clot thickens[mdash]oxidized lipids and thrombosis. Nat Med. 2007;13:1015–6. doi: 10.1038/nm0907-1015. [DOI] [PubMed] [Google Scholar]
- 12.Kirtane AJ, Stone GW. How to Minimize Stent Thrombosis. Circulation. 2011;124:1283–7. doi: 10.1161/CIRCULATIONAHA.110.976829. [DOI] [PubMed] [Google Scholar]
- 13.Puranik R, Bao S, Nobecourt E, Nicholls SJ, Dusting GJ, Barter PJ, et al. Low dose apolipoprotein AI rescues carotid arteries from inflammation in vivo. Atherosclerosis. 2008;196:240–7. doi: 10.1016/j.atherosclerosis.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Azevedo LC, Pedro MdA, Souza LC, de Souza HP, Janiszewski M, da Luz PL, et al. Oxidative stress as a signaling mechanism of the vascular response to injury The redox hypothesis of restenosis. Cardiovasc Res. 2000;47:436–45. doi: 10.1016/s0008-6363(00)00091-2. [DOI] [PubMed] [Google Scholar]
- 15.Yoon NS, Jeong MH, Kim YA, Lee WS, Hwang SH, Lee SR, et al. The Clinical Value of Modified Low Density Lipoprotein-Cholesterol in Patients Who Underwent Percutaneous Coronary Intervention. Korean Circulation Journal. 2008;38:475–82. [Google Scholar]
- 16.Naruko T, Ueda M, Ehara S, Itoh A, Haze K, Shirai N, et al. Persistent high levels of plasma oxidized low-density lipoprotein after acute myocardial infarction predict stent restenosis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:877–83. doi: 10.1161/01.ATV.0000209886.31510.7f. [DOI] [PubMed] [Google Scholar]
- 17.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13531–6. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S-J, Yen H-T, Chen Y-H, Ku H-H, Lin F-Y, Chen Y-L. Expression of interleukin-1β and interleukin-1 receptor antagonist in oxLDL-treated human aortic smooth muscle cells and in the neointima of cholesterol-fed endothelia-denuded rabbits. J Cell Biochem. 2003;88:836–47. doi: 10.1002/jcb.10431. [DOI] [PubMed] [Google Scholar]
- 19.Segev A, Strauss BH, Witztum JL, Lau HK, Tsimikas S. Relationship of a comprehensive panel of plasma oxidized low-density lipoprotein markers to angiographic restenosis in patients undergoing percutaneous coronary intervention for stable angina. Am Heart J. 2005;150:1007–14. doi: 10.1016/j.ahj.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 20.George J, Harats D, Bakshi E, Adler Y, Levy Y, Gilburd B, et al. Anti-oxidized low density lipoprotein antibody determination as a predictor of restenosis following percutaneous transluminal coronary angioplasty. Immunol Lett. 1999;68:263–6. doi: 10.1016/s0165-2478(99)00050-4. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee S. Role of oxidized human plasma low density lipoproteins in atherosclerosis: Effects on smooth muscle cell proliferation. Mol Cell Biochem. 1992;111:143–7. doi: 10.1007/BF00229586. [DOI] [PubMed] [Google Scholar]
- 22.Ishigami M, Swertfeger DK, Granholm NA, Hui DY. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J Biol Chem. 1998;273:20156–61. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- 23.Zettler ME, Prociuk MA, Austria JA, Massaeli H, Zhong G, Pierce GN. OxLDL stimulates cell proliferation through a general induction of cell cycle proteins. 2003 doi: 10.1152/ajpheart.00494.2001. [DOI] [PubMed] [Google Scholar]
- 24.Hinagata J-i, Kakutani M, Fujii T, Naruko T, Inoue N, Fujita Y, et al. Oxidized LDL receptor LOX-1 is involved in neointimal hyperplasia after balloon arterial injury in a rat model. Cardiovascular research. 2006;69:263–71. doi: 10.1016/j.cardiores.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Chnari E, Lari HB, Tian L, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules for selective retention of low-density lipoproteins. Biomaterials. 2005;26:3749–58. doi: 10.1016/j.biomaterials.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 26.Chnari E, Nikitczuk JS, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules can inhibit cellular uptake of differentially oxidized LDL. Biomacromolecules. 2006;7:597–603. doi: 10.1021/bm0506905. [DOI] [PubMed] [Google Scholar]
- 27.Hehir S, Plourde NM, Gu L, Poree DE, Welsh WJ, Moghe PV, et al. Carbohydrate composition of amphiphilic macromolecules influences physicochemical properties and binding to atherogenic scavenger receptor A. Acta Biomaterialia. 2012;8:3956–62. doi: 10.1016/j.actbio.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis DR, Kholodovych V, Tomasini MD, Abdelhamid D, Petersen LK, Welsh WJ, et al. In silico design of anti-atherogenic biomaterials. Biomaterials. 2013;34:7950–9. doi: 10.1016/j.biomaterials.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plourde NM, Kortagere S, Welsh W, Moghe PV. Structure–Activity Relations of Nanolipoblockers with the Atherogenic Domain of Human Macrophage Scavenger Receptor A. Biomacromolecules. 2009;10:1381–91. doi: 10.1021/bm8014522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Plourde NM, Iverson N, Moghe P, Uhrich KE. Nanoscale amphiphilic macromolecules as lipoprotein inhibitors: the role of charge and architecture. Int J Nanomed. 2007;2:697–705. [PMC free article] [PubMed] [Google Scholar]
- 31.Gu L, Faig A, Abdelhamid D, Uhrich K. Sugar-based amphiphilic polymers for biomedical applications: From nanocarriers to therapeutics. Acc Chem Res. 2014;47:2867–77. doi: 10.1021/ar4003009. [DOI] [PubMed] [Google Scholar]
- 32.Iverson NM, Sparks SM, Demirdirek B, Uhrich KE, Moghe PV. Controllable inhibition of cellular uptake of oxidized low-density lipoprotein: Structure–function relationships for nanoscale amphiphilic polymers. Acta Biomater. 2010;6:3081–91. doi: 10.1016/j.actbio.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian L, Yam L, Zhou N, Tat H, Uhrich KE. Amphiphilic scorpion-like macromolecules: Design, synthesis, and characterization. Macromolecules. 2003;37:538–43. [Google Scholar]
- 34.York AW, Zablocki KR, Lewis DR, Gu L, Uhrich KE, Prud’homme RK, et al. Kinetically Assembled Nanoparticles of Bioactive Macromolecules Exhibit Enhanced Stability and Cell-Targeted Biological Efficacy. Adv Mater. 2012;24:733–9. doi: 10.1002/adma.201103348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis DR, Petersen LK, York AW, Zablocki KR, Joseph LB, Kholodovych V, et al. Sugar-based amphiphilic nanoparticles arrest atherosclerosis in vivo. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1424594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen LK, York AW, Lewis DR, Ahuja S, Uhrich KE, Prud’homme RK, et al. Amphiphilic Nanoparticles Repress Macrophage Atherogenesis: Novel Core/Shell Designs for Scavenger Receptor Targeting and Down-Regulation. Mol Pharm. 2014;11:2815–24. doi: 10.1021/mp500188g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harmon AM, Uhrich KE. In Vitro Evaluation of Amphiphilic Macromolecular Nanocarriers for Systemic Drug Delivery. J Bioact Compat Polym. 2009;24:185–97. [Google Scholar]
- 38.Chan JW, Zhang Y, Uhrich KE. Amphiphilic Macromolecule Self-Assembled Monolayers Suppress Smooth Muscle Cell Proliferation. Bioconjug Chem. 2015 doi: 10.1021/acs.bioconjchem.5b00208. [DOI] [PubMed] [Google Scholar]
- 39.Moore JS, Stupp SI. Room temperature polyesterification. Macromolecules. 1990;23:65–70. [Google Scholar]
- 40.Sparks SM. Thesis. Rutgers University. 2011. Design, synthesis, and utility of fuctionalized nanoscale amphiphilic macromolecules for biomedical applications. [Google Scholar]
- 41.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–6. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- 42.Šolínová V, Kašička V, Sázelová P, Holý A. Chiral analysis of anti-acquired immunodeficiency syndrome drug, 9-(R)-[2-(phosphonomethoxy) propyl] adenine (tenofovir), and related antiviral acyclic nucleoside phosphonates by CE using β-CD as chiral selector. Electrophoresis. 2009;30:2245–54. doi: 10.1002/elps.200800790. [DOI] [PubMed] [Google Scholar]
- 43.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–11. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricciarelli R, Zingg J-M, Azzi A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–7. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- 46.Allahverdian S, Pannu PS, Francis GA. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. 2012 doi: 10.1093/cvr/cvs094. [DOI] [PubMed] [Google Scholar]
- 47.Proudfoot D, Davies JD, Skepper JN, Weissberg PL, Shanahan CM. Acetylated Low-Density Lipoprotein Stimulates Human Vascular Smooth Muscle Cell Calcification by Promoting Osteoblastic Differentiation and Inhibiting Phagocytosis. Circulation. 2002;106:3044–50. doi: 10.1161/01.cir.0000041429.83465.41. [DOI] [PubMed] [Google Scholar]
- 48.Lee S-O, Jeong Y-J, Yu MH, Lee J-W, Hwangbo MH, Kim C-H, et al. Wogonin suppresses TNF-α-induced MMP-9 expression by blocking the NF-κB activation via MAPK signaling pathways in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2006;351:118–25. doi: 10.1016/j.bbrc.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 49.Chen Q, Jin M, Yang F, Zhu J, Xiao Q, Zhang L. Matrix Metalloproteinases: Inflammatory Regulators of Cell Behaviors in Vascular Formation and Remodeling. Mediators Inflamm. 2013;2013:14. doi: 10.1155/2013/928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–45. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 51.Gölander CG, Herron J, Lim K, Claesson P, Stenius P, Andrade JD. Properties of immobilized PEG films and the interaction with proteins. In: Harris JM, editor. Poly(ethylene glycol) Chemistry. Springer; US: 1992. pp. 221–45. [Google Scholar]
- 52.Billinger M, Buddeberg F, Hubbell JA, Elbert DL, Schaffner T, Mettler D, et al. Polymer stent coating for prevention of neointimal hyperplasia. J Invasive Cardiol. 2006;18:423–6. [PubMed] [Google Scholar]
- 53.Moreno PR, Bernardi VcH, López-Cuéllar J, Newell JB, McMellon C, Gold HK, et al. Macrophage Infiltration Predicts Restenosis After Coronary Intervention in Patients With Unstable Angina. Circulation. 1996;94:3098–102. doi: 10.1161/01.cir.94.12.3098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.