

Abstract
Purpose
The presence of an ALK aberration correlates with inferior survival for patients with high-risk neuroblastoma. The emergence of ALK inhibitors such as crizotinib has provided novel treatment opportunities. However, certain ALK mutations result in de novo crizotinib resistance, and a phase I trial of crizotinib showed a lack of response in patients harboring those ALK mutations. Thus, understanding mechanisms of resistance and defining circumvention strategies for the clinic is critical.
Experimental Design
The sensitivity of human neuroblastoma-derived cell lines, cell line-derived and patient-derived xenograft (PDX) models with varying ALK statuses to crizotinib combined with topotecan and cyclophosphamide (topo/cyclo) was examined. Cultured cells and xenografts were evaluated for effects of these drugs on proliferation, signaling, and cell death, and assessment of synergy.
Results
In neuroblastoma murine xenografts harboring the most common ALK mutations, including those mutations associated with resistance to crizotinib (but not in those with wild-type ALK), crizotinib combined with topo/cyclo enhanced tumor responses and mouse event-free-survival. Crizotinib + topo/cyclo showed synergistic cytotoxicity and higher caspase-dependent apoptosis than crizotinib or topo/cyclo alone in neuroblastoma cell lines with ALK aberrations (mutation or amplification).
Conclusions
Combining crizotinib with chemotherapeutic agents commonly used in treating newly diagnosed patients with high-risk neuroblastoma restores sensitivity in preclinical models harboring both sensitive ALK aberrations and de novo resistant ALK mutations. These data support clinical testing of crizotinib and conventional chemotherapy with the goal of integrating ALK inhibition into multi-agent therapy for ALK-aberrant neuroblastoma patients.
INTRODUCTION
Neuroblastomas are embryonal tumors that arise from the malignant transformation of neural crest-derived cells and typically present in early childhood (1). Despite major enhancements in treatment approaches over the past several decades, the cure rate for patients with high-risk neuroblastoma lags significantly behind that of other common childhood cancers, and this disease contributes substantial morbidity and mortality in patients (2). In this era of more rational therapies, new treatment strategies targeting key oncogenic drivers are urgently needed. The complexities of signaling networks that modify therapeutic vulnerability in cancer cells mandate the study of drug combinations to prevent or reverse tumor drug resistance. The discovery that somatically acquired activating mutations and amplifications of the Anaplastic Lymphoma Kinase (ALK) oncogene often drive the malignant process in a subset of neuroblastomas positions ALK inhibition strategies as a promising therapeutic approach (3–6).
ALK is an orphan receptor tyrosine kinase (RTK) normally expressed only in the developing and neonatal brain with a postulated role in the regulation of neuronal differentiation (7, 8). Dysregulation of ALK signaling has been associated with the development of various cancers (9), most notably anaplastic large-cell lymphoma (ALCL) in a translocated form fused to the N-terminal of nucleophosmin (NPM) (10), and fused to the echinoderm microtubule-associated protein-like 4 (EML4) in 3–5% non-small cell lung cancers (NSCLC) (11). ALK’s oncogenic targets exert their growth advantage and anti-apoptotic effects through activation of numerous downstream pathways such as PI3K/Akt, MAPK, and STAT3 (12). Pharmacologic ALK inhibition in ALCL models is associated with decreased levels of phosphorylated ALK and its downstream effectors, and ultimately culminates with cell cycle arrest and apoptosis (13). Early-phase clinical studies of crizotinib, a dual ALK/MET small molecule tyrosine kinase inhibitor, in pretreated patients with advanced relapsed/refractory NSCLC harboring ALK rearrangements yielded dramatic response rates (14, 15). This validated ALK as a therapeutic target and led to expedited FDA approval of crizotinib in patients with ALK-translocated lung cancer.
ALK mutations are observed in 8% of neuroblastoma tumors and span the entire spectrum of patients, ranging from congenital cases to adolescents and young adults. Within the high-risk subset of patients, the frequency of ALK aberrations is 14% (10% point mutation and 4% amplification) and is independently predictive of inferior outcome, supporting the usefulness of defining ALK status at diagnosis for prognostic and therapeutic stratification of these high-risk patients (16). Somatic ALK mutations at three sites (R1275, F1174 and F1245) occur most frequently in neuroblastoma, located in key regulatory regions of the ALK kinase domain. While cell lines harboring the R1275Q mutation are initially sensitive to direct ALK kinase inhibition, cells harboring the F1174- and F1245- residue mutation are relatively resistant (16). Biochemical studies have shown that the reduced sensitivity of the F1174L mutation to crizotinib, and other ATP competitive ALK inhibitors, is due in part to an increased ATP-binding affinity that must be compensated for by higher doses of ALK inhibitors or alternative therapeutic modalities (16).
A now completed pediatric phase 1 trial and ongoing phase 2 trial of crizotinib (17), the most studied ALK inhibitor in neuroblastoma, has shown activity in a subset of neuroblastoma patients, but the frequency and duration of responses are marginal. Combining crizotinib with conventional genotoxic agents may provide superior anti-tumor activity than either approach used alone. The TP53 tumor suppressor gene is a key regulator of apoptosis, senescence, cell cycle arrest and DNA repair and, unlike in many adult cancers, retains wild-type activity in neuroblastoma (18, 19). Neuroblastomas can acquire a sustained high-level drug resistance during chemotherapy, which can be attributed to acquired p53 mutations and/or loss of p53 function (20, 21). Harnessing this central pathway and developing strategies to induce p53 functional activation may provide an opportunity to enhance therapeutic efficacy. In the present study, we sought to provide the preclinical data that support integration of crizotinib into multi-agent chemotherapy for patients with high-risk neuroblastoma.
METHODS
Mouse xenograft studies
Female CB17 SCID mice were subcutaneously implanted with human neuroblastoma NB-1643 (ALK-R1275Q mutation, TP53 WT), SH-SY5Y (ALK-F1174L mutation, TP53 WT), NB-EBc1 (ALK-WT, expresses robust phosphorylated ALK, TP53 WT), SK-N-AS (ALK-WT, TP53-H168R mutation), NB-SD (ALK-WT, TP53-C176F mutation) and NB-1691 (ALK-WT, TP53 WT, MDM2 amplified) xenografts and the Felix-PDX (ALK-F1245C, TP53 WT) patient-derived xenograft (PDX) established post-mortem from the blood of a patient who died of a progressive MYCN-non-amplified high-risk neuroblastoma. Mice with engrafted tumors that reached 200 mm3 in size were randomized into groups of 10 per condition. Tumors were measured using a digital caliper at the initiation of the study and one to two times per week during the treatment period. For all experiments mice were weighed twice weekly. The studies consisted of the following groups: 1) Vehicle control; 2) crizotinib; 3) topotecan plus cyclophosphamide (topo/cyclo); and 4) crizotinib + topo/cyclo. Mice continuously received either vehicle (solvent medium of 0.5% methyl-cellulose, 0.5% Tween-80 in water, QD, PO), crizotinib (100 mg/kg QD, PO), topo/cyclo (0.05 mg/kg and 20 mg/kg, respectively, IP), or crizotinib plus topo/cyclo treatment. The treatment schedule for the chemotherapeutic drugs was as follows: i.p. 5 days on treatment, 16 days off treatment, cycle resumed on day 21. The event resulting in mouse euthanasia was disease progression, defined as the tumor volume either reaching 4 times the initial volume or 3 cm3. In the biology studies with SH-SY5Y and Felix-PDX tumors, mice were enrolled at 300 mm3, and randomized into the same four treatment groups as described above. Mice were treated for 3 days, euthanized, and their tumors were collected 4 hours post last treatment. All animal work was conducted according to relevant national and international guidelines and steps were taken to minimize tumor burden and drug-related side effects. All animal studies were approved by The Children’s Hospital of Philadelphia IACUC (protocol # 2012-5-643).
Cell lines and reagents
The neuroblastoma cell lines NB-1643 (ALK-R1275Q, TP53 WT), SH-SY5Y (ALK-F1174L, TP53 WT), NB-EBc1 (ALK WT, expresses phosphorylated ALK, TP53 WT), LAN-5 (ALK-R1275Q, TP53 WT), LAN-6 (ALK-D1091N, TP53 WT), NB-1 (ALK amplified, TP53 WT), NB-SD (ALK-F1174L, TP53-C176F), KELLY (ALK-F1174L, TP53-P177T), NB-1691 (ALK WT, TP53 WT, MDM2 amplified), SK-N-AS (ALK WT, TP53-H168R), SK-N-BE(2)C (ALK WT, TP53-C135F), and NBL-S (ALK WT, TP53 WT), were obtained from the Children’s Hospital of Philadelphia cell bank and were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, 1% penicillin/streptomycin, and 0.2% gentamycin. The cell lines COG-N-295 (ALK-F1174L, TP53 WT), COG-N-415 (ALK-F1174L TP53 WT), COG-N-247 (ALK WT, TP53 WT), CHLA-122 (ALK WT, TP53 WT), and COG-N-297 (ALK WT, TP53 Mutated) were established in the laboratory of Dr. C. Patrick Reynolds and obtained through the Children’s Oncology Group Cell Culture and Xenograft Repository (www.cogcell.org). COG designated cell lines were cultured in Iscoves Modified Dulbecos Medium supplemented with 20% FBS, 1% L-glutamine, and 1% ITS (Sigma Aldrich). All cells were cultured in an atmosphere of 37°C and 5% CO2. Annual genotyping (AmpFLSTR Identifiler Kit) of these lines and a single nucleotide polymorphism array analysis (Illumina H550) were performed to ensure maintenance of cell identity using methods as previously described (22). Reagents for the in vitro studies included 4-hydroperoxycyclophosphamide (4-HC; Niomech, Bielefeld, Germany), topotecan (Sigma-Aldrich, St. Louis, MO, USA), and crizotinib (Pfizer, Inc). Identification of cell lines and xenografts were confirmed at time of experimentation by short tandem repeat (STR) analysis validated against the Children’s Hospital of Philadelphia database.
In Vitro Cytotoxicity Assays
For Cell Titer Glo® (CTG, Promega) drug combination experiments, in vitro cell viability was assessed by measuring levels of ATP using the luminescent detection assay measured on the GloMax®-Multi Microplate Multimode Reader (Promega). Cells were plated at their pre-determined seeding densities on day 1, treated 24 hours later on day 2, and then read 72 hours later on day 5. Cells were first tested with single agents and Dm (absolute IC50) values were established for each drug in each cell line. The cells were then plated and treated with single agent and the combination, and the results were analyzed.
Drug combination analysis was also conducted using the DIMSCAN assay, a semiautomatic fluorescence-based digital image microscopy system that quantifies viable cells in tissue culture multiwell plates on the basis of their selective accumulation and cleavage of fluorescein diacetate to fluorescein (23). DIMSCAN is capable of measuring cytotoxicity over a 4-log dynamic range by quantifying total fluorescence per well, which is proportional to the number of viable, clonogenic cells after eliminating background fluorescence with digital thresholding and eosin Y quenching. Briefly, cell lines were seeded into 96-well plates in 150 μL of complete medium (3000–14000 cells per well) and incubated overnight. Crizotinib, topotecan, and 4-HC (the active metabolite of cyclophosphamide) in 50 μL of complete medium were added to the wells (12 replicate wells per each concentration of drugs). Cell lines were incubated in the presence of these single agents or in combination for 96 hours, after which fluorescein diacetate in 50 μL of 0.5% eosin Y (final concentration of fluorescein diacetate 60 μg/mL) was added to each well and the cells were incubated for an additional 25 minutes at 37 °C. Total fluorescence was then measured with DIMSCAN and the results were expressed as surviving fractions of treated cells compared with control cells that were exposed to vehicle.
Determination of Synergy
Results were analyzed for synergistic, additive, or antagonistic effects using the combination index (CI) method developed by Chou and Talalay (24). The quantified CI values obtained by this method are defined as synergistic if CI<1, additive if CI=1, or antagonistic if CI>1. A confidence interval of <0.1 is represented as +++++ and indicates strong synergism. The isobologram method evaluates the interactions at a chosen effect level and is therefore useful in inspecting the drug interactions at the corresponding concentrations, often the median effect concentrations. Levels of cell viability and CI were obtained by software analysis and subsequently graphed in CI vs. Fraction affected (Fa) plots (Figure 2A–E). Using both CalcuSyn v.2.0 (Biosoft Inc., UK), and CompuSyn v.1.0 (Combo Syn, Inc., US), we determined the concentrations of crizotinib and topotecan required to produce a defined single-agent effect (e.g., IC50), and placed the values on the x and y axes in a two-coordinate plot. Second, the concentrations of the two drugs used in combination to provide the same effect were placed in the same plot. Synergy, additivity, or antagonism are indicated when the concentrations of crizotinib and topotecan are located below, on, or above the line, respectively.
Figure 2.
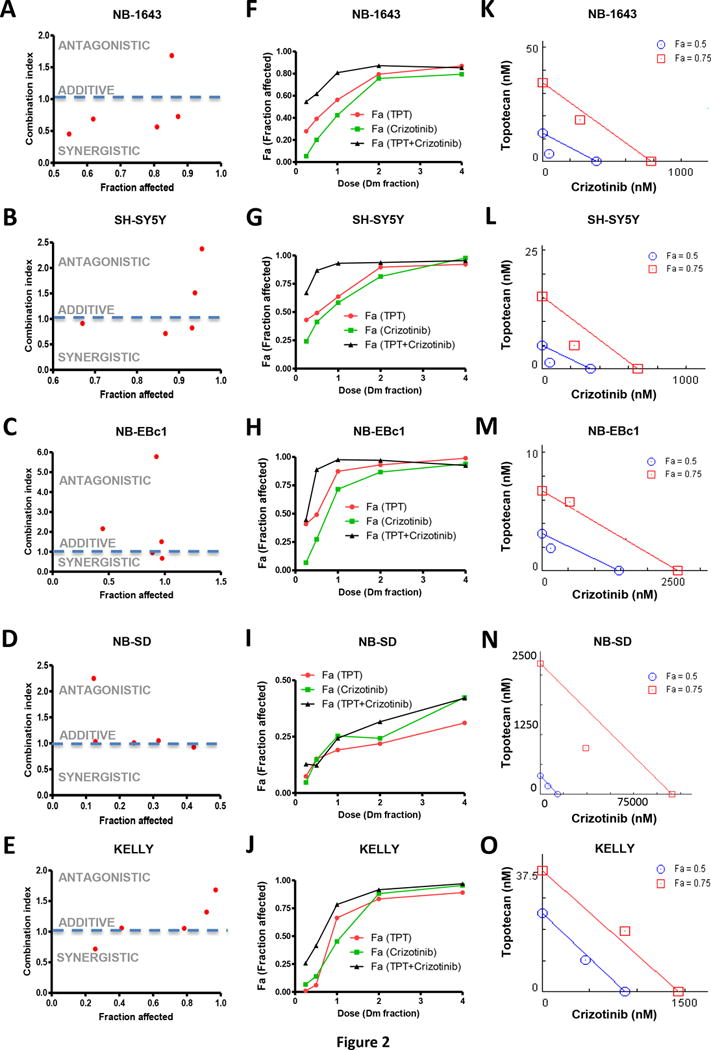
In vitro synergy analysis of crizotinib and topotecan (TPT) in neuroblastoma cells. Combination index (CI) is presented as a function of fraction affected (Fa) in NB cells: (A) NB-1643, (B) SH-SY5Y, (C) NB-EBc1, (D) NB-SD and (E) KELLY. Dose response curves are shown where the dose of drug, as a fraction of the respective Dm values from the cell lines (F) NB-1643, (G) SH-SY5Y, (H) NB-EBc1, (I) NB-SD and (J) KELLY, is plotted against the fraction affected (Fa) values. In the isobologram analysis of (K) NB-1643, (L) SH-SY5Y and (M) NB-EBc1, (N) NB-SD and (O) KELLY, the diagonal colored line indicates additivity, whereas the red and blue symbols indicate dose requirements to achieve 50% and 75% of growth inhibition with single agents and combination treatment, respectively. Data points below the line of additivity indicate synergy whereas data points above it signify antagonism.
Synergy by DIMSCAN was determined using CalcuSyn software (24).
To further evaluate the dose-dependent drug interaction of crizotinib and topotecan, isobolograms at effect levels of 50% and 75% cell growth inhibition were created. Since the single agents alone or in combination usually reach 50% inhibition, the isobologram at 50% provides an actual comparison for single drug versus combination. The 75% isobologram illustrates the utility of combining these drugs at higher effect levels, with practical implications in the clinic. Data points above or below the line of additivity indicate antagonism or synergy, respectively.
Western Blot Analysis
All cell lines were plated at their pre-determined seeding density and treated 24 hours later. Cells were harvested 8 hours post-treatment and lysed according to a previously described protocol (25). Protein concentration was calculated using the BCA method and 30–50ug of protein were separated on 4%–12% Bis-Tris gradient gels, transferred to PVDF membranes (Millipore) overnight at 4°C, and immunoblotted against pALKY1604, total ALK, Cleaved Caspase-3, Cleaved PARP, Phospho-p53S15, p21, PUMA, GAPDH, and β-Actin (Cell Signaling Technology) and MDM2 (Abcam) and total p53 (DO-1, Santa Cruz), overnight at 4°C. All primary antibodies were diluted 1:1000, except GAPDH and β-Actin, which were diluted 1:5000. Membranes were washed and probed with secondary antibodies at a dilution of 1:10,000 (Santa Cruz). When probed for multiple proteins, membranes were stripped using 0.1 M glycine, 0.5% Tween 20 (at pH 2.5) twice for 30 minutes.
Statistical Analysis
For in vivo statistical analysis, mixed-effects models were used to assess tumor volume over time between treated samples and vehicle controls. Event-free survival (EFS) was defined as the percentage of mice that survived while on therapy, where survival was defined as the lack of an “event.” An event was defined as a tumor volume reaching above 3 cm3 or 4x the initial tumor volume, at which point the mice were sacrificed and taken off study. EFS percentages were estimated using the Kaplan-Meier method and survival curves were compared by using the Log-Rank test. Statistical analyses were performed using the ‘nlme’, ‘survival’, and ’multcomp’ packages in the R statistical programming language (26–28). Statistical significance was defined as p values < 0.05.
For in vitro densitometric analysis, Image J software (1.47v; NIH) was used to quantify intensity of the bands. Results were considered statistically significant when p values were <0.05. Protein levels were estimated by quantification of the immunoblot bands. The in vitro results were analyzed by performing a linear regression on the data, with the vehicle set as the Y intercept. The values from each treatment group were averaged and then compared between groups. For the in vivo western blot quantification, since each lane represents one mouse, and there were several mice ran per group, the groups were analyzed with each mouse serving as a replicate. An ANOVA was performed on these samples, and a Tukey’s significant difference (HSD) post-hoc test was done to test the significance between the groups. Significance of results were designated as follows: *p<0.05, **p<0.01, ***p<0.001. The error reported is the standard error of the mean (SEM).
shRNA p53 Knockdown and functionality testing
NB-1643 and SH-SY5Y cells were stably transfected with retrovirus targeting p53 (Origene TL320558A and TL3200558C) or copGFP control (Origene TR30021). Infected cells were selected for and kept in a medium containing 0.5 μg/ml puromycin. Knockdown efficiency was measured by qRT-PCR, with TP53 (probe from Applied Biosystems, Hs00153349_m1) expression levels normalized to the geometric mean of two housekeeping genes, IPO8 (Applied Biosystems, Hs00183533_m1) and UBC (Applied Biosystems, HS00824723_m1). All qRT-PCR reactions were performed in triplicate. The ΔΔCT method was used to analyze fold change, and percentage knockdown was calculated compared to GFP control.
P53 functionality was tested by treating cells with 25 uM melphalan (L-PAM) for 16 hours and assaying induction of total p53, MDM2, and p21. Densitometric analysis was conducted on the p21 protein bands that were normalized to the loading control. Functional p53 was defined as having a greater than 2 fold increase of p21 upon L-PAM treatment compared to a non-treated control.
RESULTS
Crizotinib combined with standard cytotoxic agents sensitized ALK-mutant neuroblastoma xenografts with wild-type TP53
We first tested the combination of crizotinib plus topo/cyclo in the NB-1643 xenografts, which harbor the crizotinib-sensitive and most commonly occurring ALK mutation, R1275Q. Although initially responsive, the crizotinib and chemotherapy arms regrew on therapy at 5 and 8 weeks, respectively. However, crizotinib plus topo/cyclo induced complete tumor remission in the NB-1643 model for the 14 weeks of treatment (Figure 1A top), with tumors re-growing on average, 6.6 weeks after therapy had ended. Survival in the combination group was superior to survival in all the other conditions (Figure 1A bottom, Table S1). We next evaluated this combination therapy in SH-SY5Y xenografts, which harbor the second most frequent ALK mutation (F1174L), previously shown to be resistant to crizotinib (29, 30). In agreement with previous data, crizotinib alone lacked anti-tumor activity in this model, with treated tumors growing as fast as the untreated cohort. Although initially effective, topo/cyclo did not demonstrate a sustained response, with tumors re-growing on therapy at 8 weeks (Figure 1B top, Table S1). However, combining crizotinib with topo/cyclo achieved rapid and sustained complete tumor regressions for the duration of treatment. Mice treated with topo/cyclo + crizotinib showed significantly improved event-free survival (EFS) and decreased tumor growth rates compared to all groups, and maintained complete responses for 24 weeks after cessation of treatment (Figure 1B bottom, Table S1).
Figure 1.
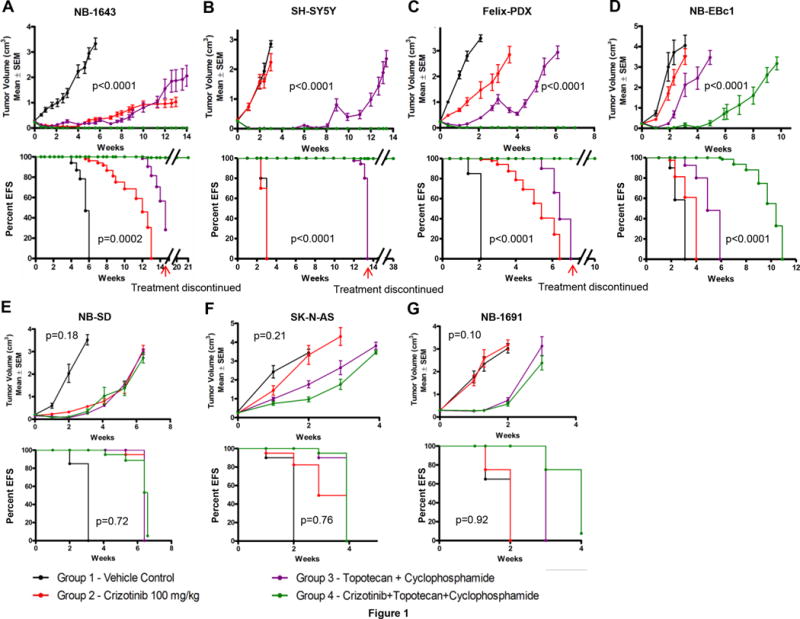
In vivo effects of crizotinib and topo/cyclo on tumor growth and event-free survival in neuroblastoma PDX and xenograft models. Female CB17 SCID mice bearing (A) NB-1643 [ALK-R1275Q, TP53-WT], (B) SH-SY5Y [ALK-F1174L, TP53-WT], (C) Felix-PDX [ALK-F1245C, TP53-WT] Patient Derived Xenograft (PDX) (D) NB-EBc1 [ALK-WT, expresses phosphorylated ALK, TP53-WT], (E) NB-SD [ALK-F1174L, TP53 mutated], (F) SK-N-AS [ALK-WT, TP53 mutated], and (G) NB-1691 [ALK-WT, TP32-WT, MDM2 amplified] human neuroblastoma xenografts were treated with vehicle, crizotinib alone (oral, 100mg/kg), topotecan (topo, 0.05 mg/kg, I.P.) plus cyclophosphamide (cyclo, 20 mg/kg, I.P.) alone, or crizotinib plus topo/cyclo. Administration of topo/cyclo was once daily for 5 days every 21 days whereas crizotinib was continuously administered on a daily basis. (Median ± S.E.M., n=10 for each data point, a mixed-effects model was used for statistical significance analysis of tumor growth delay, EFS K-M curves were compared by using a log-rank test, where p<0.05 was considered significant. The p-values reported refer to the combination treatment compared to topo/cyclo alone.)
We next assessed the combination in Felix-PDX patient-derived xenografts (PDXs), which harbor the third most common ALK mutation (R1245C) in neuroblastoma. Similarly to SH-SY5Y, these xenografts have been shown to display de novo resistance to crizotinib(21). Whereas treatment with crizotinib or the topo/cyclo alone displayed a mild tumor growth delay, Felix-PDX treated with the topo/cyclo + crizotinib achieved and maintained complete responses (Figure 1C top, Table S1), and a significant increase in EFS (Figure 1C bottom, Table S1). Combination therapy led to complete regression for the duration of treatment in Felix-PDX mice, with regrowth of tumors occurring 3 weeks after cessation of therapy.
The combination was then tested in NB-EBc1, an ALK WT xenograft that displays robust constitutive ALK activation and has been previously shown to be dependent on ALK signaling for growth (6). The combination therapy in this model was significantly more effective than vehicle, crizotinib alone, and chemotherapy alone, delaying tumor growth for 4 weeks before on-treatment progression (Figure 1D top, Table S1). While crizotinib and topo/cyclo alone resulted in significantly decreased tumor volumes compared to vehicle, neither therapy increased survival rates over control, in contrast to the prolonged survival seen with topo/cyclo + crizotinib (Figure 1D bottom, Table S1). All treatments were well tolerated by the mice, with no signs of systemic toxicity or weight loss (Figure S1).
Crizotinib + topo/cyclo was not efficacious in xenografts harboring TP53 mutations
We next investigated the activity of crizotinib in combination with cytotoxic agents against crizotinib-resistant xenograft models harboring various ALK and TP53 mutations. While the growth of NB-SD xenografts harboring ALK F1174L and TP53 mutations was significantly delayed by all treatment conditions compared to vehicle, and while the combination treated arm showed evidence of pALK abrogation (Supplementary Figure 4), all xenografts experienced rapid tumor progression while on therapy (Figure 1E, Table S1). Importantly, combining crizotinib with topo/cyclo did not increase anti-tumor activity compared to single agent crizotinib or topo/cyclo. Lastly, we sought to test the efficacy of the combination in models with wild type ALK and TP53 mutations using the xenografts SK-N-AS and NB-1691. As expected, crizotinib alone did not affect tumor growth in either xenograft model (Figure 1F top and 1G top, Table S1). Both the cytotoxic agents and the combination treatments resulted in a mild tumor growth delay, with tumors rapidly progressing on therapy. The combination showed no greater efficacy in these xenografts than therapy with topo/cyclo alone. In agreement with the tumor growth rates, EFS analysis demonstrated a similar pattern in both xenograft models (Figure 1 E–G, bottom).
The combination of crizotinib and chemotherapy was active against patient-derived ALK-mutant neuroblastoma cell lines with functional p53
To assay for synergy between crizotinib and cytotoxic agents in vitro, we used two methods of analysis: the median-effect combination index (CI) and the isobologram. We first tested crizotinib and topotecan as single agents and determined the median-effect dose (Dm) values (equivalent to the absolute IC50), in a panel of five neuroblastoma cell lines differing in their genotypic profiles (Table S3, Figure 2). Crizotinib and topotecan were the least effective as single agents and in combination in NB-SD (Figure 2D) and KELLY (Figure 2E), both cell lines that harbor a TP53 mutation. In the p53 functional cell lines, the combination therapy resulted in a greater inhibition of cell viability compared to single agent treatments, and demonstrated a high level of inhibition (high Fa). The CI analysis for NB-1643 (Figure 2A) and SH-SY5Y (Figure 2B), both ALK-mutated cell lines, revealed several values less than 1.0, indicating a synergy that was stronger at lower-dose combinations. The data was also presented in dose response curves (Figure 2F–J), where the dose of drug, as a fraction of the cell lines’ respective Dm values, was plotted against the fraction of cells affected by treatment (Fa). In all cell lines except for NB-SD, the combination treatment resulted in a greater Fa at lower doses compared to single agent crizotinib or topotecan. NB-EBc1 showed mild synergy at two doses (Figure 3C), with the higher doses reflecting antagonism. The dose-reduction indices (DRI) at Dm ranged from 2–7 for crizotinib and from 2–4 for topotecan in cell lines exhibiting synergy (Table S3), suggesting that such synergistic interactions between crizotinib and topotecan provide the opportunity to reduce the concentrations of individual drugs and thereby potentially reduce their associated toxicities. Indeed, the maximal activities of the single agents could be extended to over 90% cell growth inhibition when applied in combination.
Figure 3.
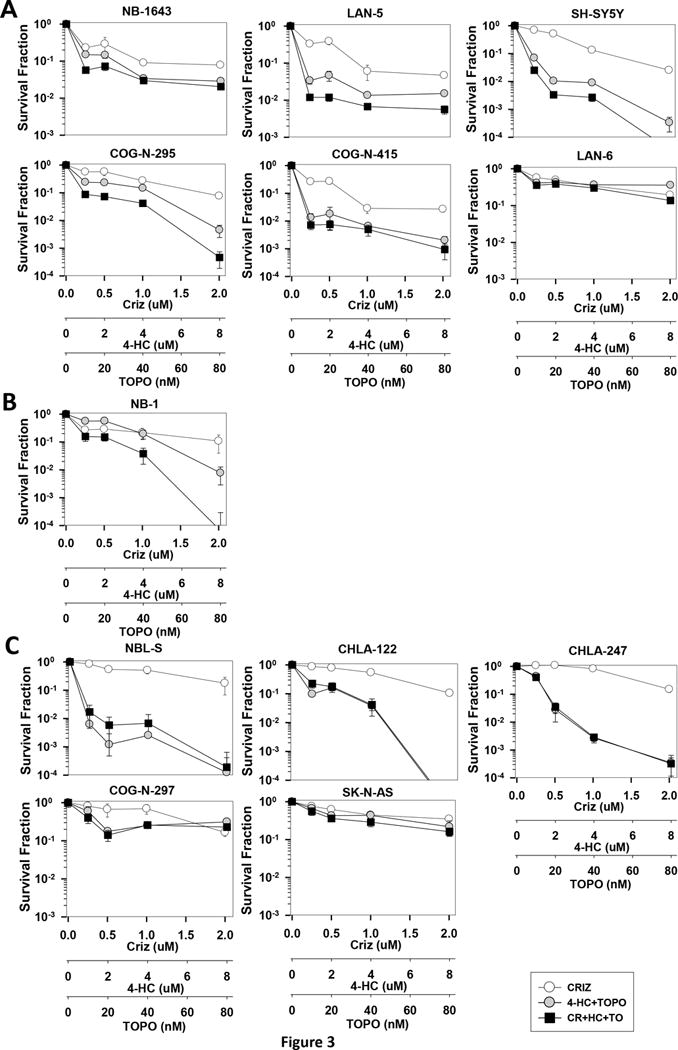
In vitro DIMSCAN cytotoxicity analysis of crizotinib, 4-HC (an active metabolite of cyclophosphamide), and topotecan in neuroblastoma cells. Dose-response curves of cells treated with crizotinib (0–2μM) in combination with 4-HC (0–8μM) and topotecan (0–80nM). (A) Synergy was observed in ALK mutated, TP53 WT lines and in an (B) ALK amplified, TP53 WT line. (B) No synergy was observed in the following lines with WT ALK, mutant TP53, or both: NBL-S [ALK-F1174L, TP53 non-functional], CHLA-122 [ALK–WT, TP53-WT], CHLA-247 [ALK–WT, TP53-WT], COG-N-297 [ALK–WT, TP53-mutated], and SK-N-AS [ALK–WT, TP53-mutated].
To further evaluate the dose-dependent drug interaction of crizotinib and topotecan, isobolograms at effect levels of 50% and 75% cell growth inhibition were created and analyzed (Figure 2K–O). As shown in Figure 2, the isobole of the combination was below the line of additivity at 50% inhibition in cell lines with functional p53 (Figure 2K, L, M). Importantly, in NB-SD and KELLY, TP53 mutant cell lines, additivity and antagonism was observed at most concentrations tested, in agreement with the CI plots (Figure 2 I–J). In line with our in vivo data, these results further suggest that the synergy of this combination relies, at least in part, on functional p53.
We employed the DIMSCAN assay as a second method to assess cell viability and synergy and included 4-HC, the active metabolite of cyclophosphamide, in this assay. In accordance with the CTG assay results, the DIMSCAN assay showed synergy at clinically achievable doses in a broader panel of neuroblastoma cell lines with ALK mutations and functional p53: NB-1643, LAN-5, SH-SY5Y, COG-N-295, COG-N-415, and LAN-6 (Figure 3A, Table S3). The combination was also synergistic in NB-1, an ALK amplified line with functional p53 (Figure 3B, Table S3, Figure S2B). In several cell lines, the combination of crizotinib with topotecan+4-HC resulted in a 3-log (99.9%) cell kill (Figure 3A). Notably, this combination displays CI values that range from synergistic to additive in all seven ALK aberrant, p53 functional cell lines tested (Figure S2A–B) and potently inhibits a number of cell lines with ALK-F1174L mutations, which confer preferential ATP binding affinity and innate resistance to single agent crizotinib. Thus, these data support our in vivo findings and predict greater efficacy when combining crizotinib with cytotoxic agents in p53 functional models where crizotinib monotherapy is insufficient. Further, we also tested the combination therapy by DIMSCAN in the ALK WT and TP53 WT lines, NBL-S, CHLA-122, and CHLA-247 and the ALK WT, TP53 mutated lines, COG-N-297 and SK-N-AS (Figure 3C and S2C). Synergy between crizotinib and topo/4-HC was not observed in any of these cell lines, highlighting the requirement of aberrant or activated ALK for the efficacy of this combination therapy.
Crizotinib potentiated downstream signals of p53 activation in topotecan-treated neuroblastoma cells in vitro
To next evaluate the potential role of crizotinib in combination with chemotherapy on the p53 pathway, we treated NB-1643 (Figure 4A), SH-SY5Y (Figure 4C), and NB-EBc1 (Figure 4E) cells with vehicle, crizotinib, topotecan, or crizotinib plus topotecan at fractional doses of each cell lines’ respective IC50s. To assess the impact of the single agents and combination therapy on ALK signaling, cells were treated for 8 hours prior to collection for immunoblot analysis. Crizotinib individually or in combination with topotecan resulted in a dose-dependent abrogation of phosphorylated ALKY1604 in all three cell lines, as well as upregulation of cleaved caspase-3 at one- and two-times the IC50 doses of single agent and combination therapy (Figure 4A, 4C, and 4E). We also evaluated levels of phosphorylated p53s15, and total p53, as well as the downstream markers of p53 activation, p21 and PUMA, in the treated cells. Expectedly, as a DNA damaging agent, topotecan induced phosphorylated and total p53 upregulation across all cell lines (Figure 4A, 4C, and 4E). Whereas crizotinib increased levels of phosphorylated p53s15 in the crizotinib-sensitive line, NB-1643 (Figure 4A and 4B), it did not in the crizotinib insensitive SH-SY5Y (Figure 4C and 4D) and NB-EBc1 (Figure 4E and 4F) cells, irrespective of caspase-3 activation. Nonetheless, in combination treated cells, both forms of p53, as well as p21 and PUMA, were found to be highly upregulated, to levels suggestive of greater than additivity. In fact, densitometric analysis of the bands revealed a significantly higher increase in phospho-p53S15 (Figure 4B, 4D, and 4F, top), p21 (Figure 4B, 4D, and 4F, bottom) and cleaved caspase-3 (data not shown) in the combination treated cells, suggesting that these agents cooperate with one another to induce greater apoptosis.
Figure 4.
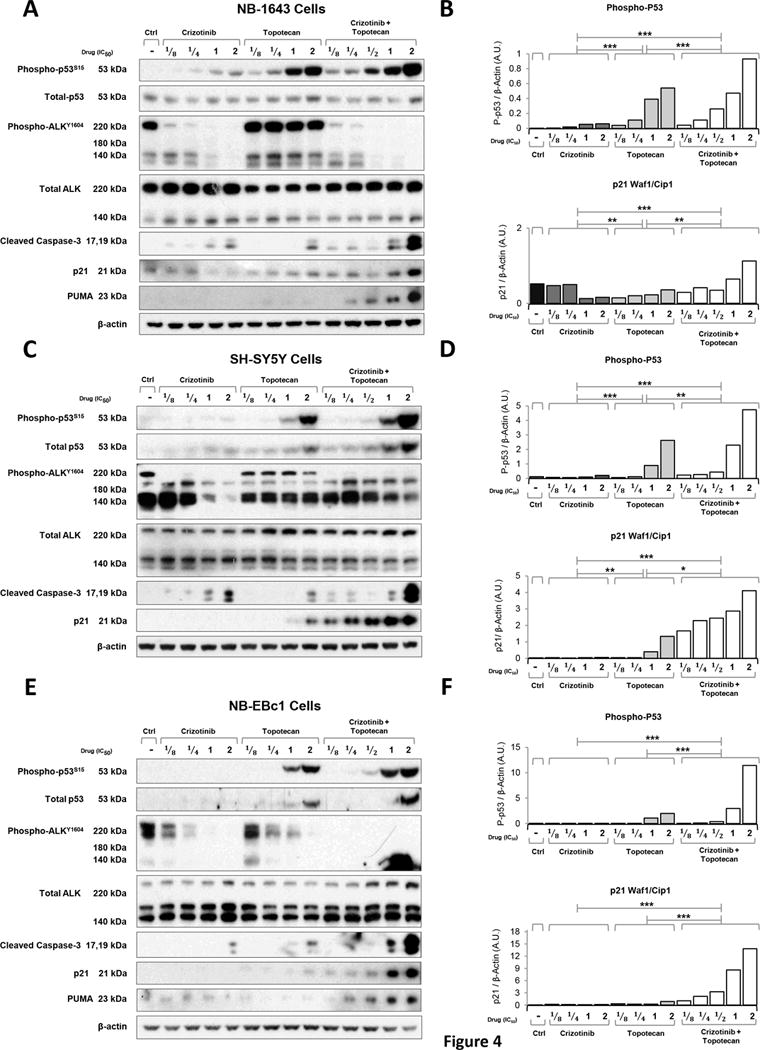
Combination of crizotinib with topotecan induces downstream markers of cell death in vitro. Protein levels were assessed by immunoblot analysis of (A) NB-1643, (C) SH-SY5Y and (E) NB-EBc1 cells treated with crizotinib, topotecan or the combination of both for 8 hours with fractional doses of predetermined IC50s. Cell lysates were normalized for total protein content and subjected to immunoblot analysis using the indicated antibodies. Bands corresponding to phosphorylated-p53-ser15 (P-p53, top) and p21 Waf1/Cip1 (p21, bottom) of NB-1643 (B), SH-SY5Y (D) and NB-EBc1 (F) were quantified by densitometric analysis using Image J. Results are expressed as Arbitrary Units (A.U.) and were normalized to the loading control β-actin. Linear regression models with the vehicle set as the y-intercept were used to compare the treatment groups. *p<0.05, **p<0.01, ***p<0.001.
Crizotinib potentiated p53 activation in chemotherapy treated neuroblastoma xenografts
To next explore the in vivo effects of chemotherapy and crizotinib on p53 activation, mice bearing SH-SY5Y and Felix-PDX tumors were treated with crizotinib, topo/cyclo and the combination for 3 days. Tumors were collected 4 hours post last treatment, lysed and analyzed by immunoblotting. In SH-SY5Y, the standard dose of crizotinib (100 mg/kg) did not alter levels of phosphorylated-ALK (Figure 5A), in agreement with previous results indicating that this dose is insufficient for abrogating ALK signaling (16). Unexpectedly, topo/cyclo alone reduced protein levels of both total and phosphorylated ALK (Figure 5A and 5B). Mirroring the effects described in vitro, levels of total, phosphorylated p53 and cleaved caspase-3 were greater in the combination therapy compared to topo/cyclo and crizotinib agent alone (Figure 5A). Quantitative analysis showed that the combination induced significantly greater levels of phospho-p53 compared to all other treatments, including topo/cyclo alone (Figure 5B top).
Figure 5.
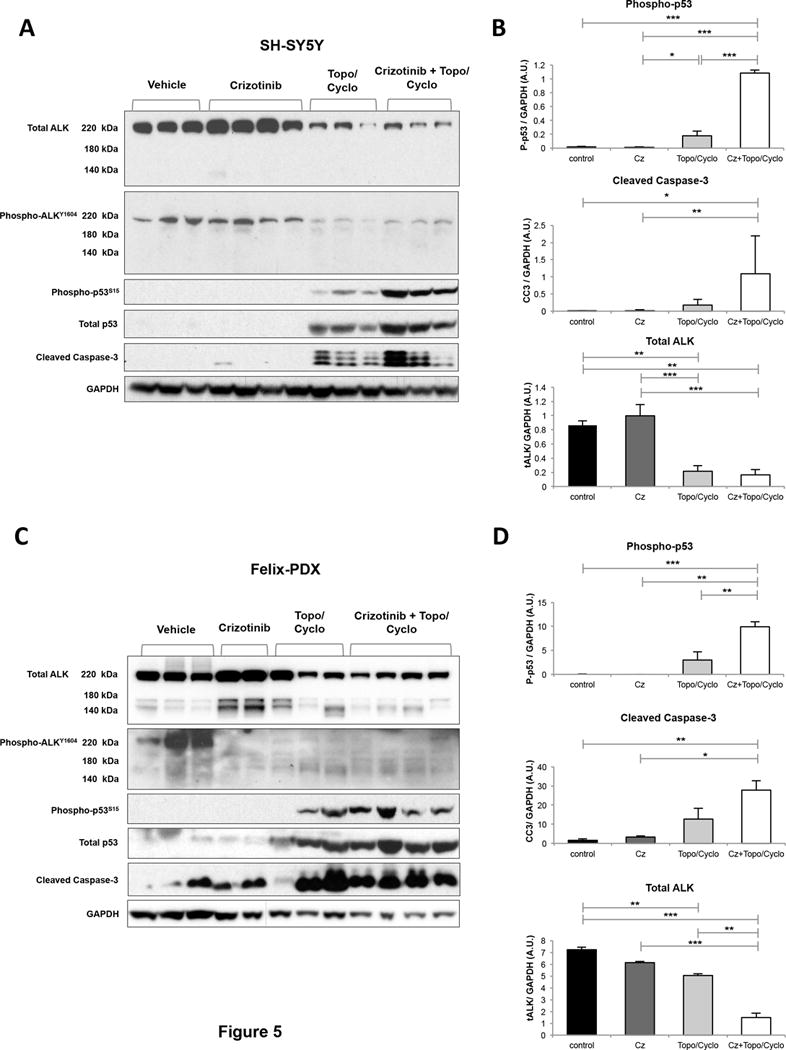
Combination of crizotinib with topo/cyclo induces downstream markers of cell death in vivo. Once tumors reached 200 mm3 mice bearing (A) SH-SY5Y and (C) Felix-PDX human neuroblastoma xenografts were treated for three consecutive days as follows: vehicle, crizotinib (Cz, 100 mg/kg, oral), topotecan (Topo, 0.05 mg/kg) and cyclophosphamide (Cyclo, 20 mg/kg) and combination of both. Tumors were harvested 4 hours post last treatment. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Bands corresponding to phosphorylated-p53-ser15 (P-p53, top), cleaved caspase-3 (cc3, middle), and total ALK (bottom) of (B) SH-SY5Y and (D) Felix-PDX were quantified by densitometric analysis using the software Image J. Results are expressed as Arbitrary Units (A.U.) and were normalized against the loading control GAPDH. The bands from each treatment group were averaged and compared using an ANOVA and a Tukey’s HSD test. *p<0.05, **p<0.01, ***p<0.001.
Lastly, in line with the SH-SY5Y results, treatment with topo/cyclo +/− crizotinib showed a reduction in total ALK expression in the Felix-PDX patient derived-tumors (Figure 5C and 5D). Upregulation of both phosphorylated p53 and caspase-3 was observed in topo/cyclo and topo/cyclo + crizotinib-treated tumors but levels of these two proteins were not markedly greater in the combination therapy compared to topo/cyclo alone (Figure 5C and 5D).
The combination of crizotinib and chemotherapy enhanced cytotoxicity in neuroblastoma cell lines carrying wild-type p53
To explore the potential role of p53 in mediating the synergy between crizotinib and chemotherapy, we established p53 knockdown NB-1643 and SH-SY5Y cells using two different shRNA vectors (referred to as shRNA “A” and “B”). Both cell lines showed greater than 60% p53 knockdown compared to their respective GFP-transduced control with both vectors (Figure 6A and 6B). Testing the combination of crizotinib + topotecan/4-HC in these lines by DIMSCAN revealed synergy in the GFP-transduced control lines (Figure 6C and 6E left), consistent with the previous DIMSCAN results using the parental NB-1643 and SH-SY5Y lines (Figure 3A). Notably, synergy was abrogated in the NB-1643 or SH-SY5Y p53 knockdown lines (Figure 6C–D, and 6E–F, respectively).
Figure 6.
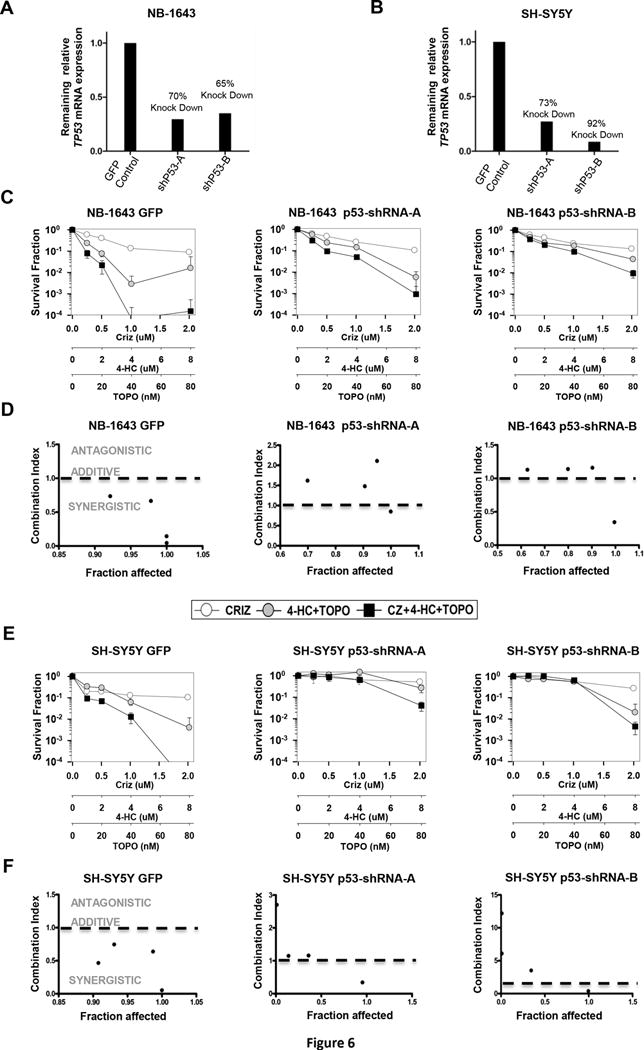
Synergy between crizotinib and topo/cyclo is abrogated in ALK mutated cells with stable knockdown of p53. Cells were treated with shRNA lentivirus against p53 or a copGFP control and levels of p53 knockdown compared to the GFP control are shown for (A) NB-1643 and (B) SH-SY5Y cells. The GFP control infected cells, and two different shRNA infected cells, were treated with vehicle, crizotinib, topotecan plus 4-HC, or crizotinib and topotecan/4-HC and synergy was analyzed by DIMSCAN. Survival curves and CI vs. Fa plots are shown for NB-1643 (C) and (D) and SH-SY5Y (E) and (F).
DISCUSSION
Neuroblastoma remains a leading cause of childhood cancer death despite progress made in the field of chemo-radiotherapy, surgery and immunotherapy. Optimizing treatment for these patients has become increasingly complex, requiring an approach that allows us to determine when it is best to incorporate targeted drugs and with what chemotherapy to combine targeted agents, and in which patients. Rapid advances in the development of targeted therapies have provided the unprecedented ability to therapeutically exploit oncogenic drivers. ALK is a tractable molecular target in neuroblastoma, with aberrations in this gene accounting for over 80% of hereditary cases, and present in 14% of high-risk cases (6, 29). We previously reported differential sensitivity of the most common ALK kinase domain mutations in neuroblastoma (29, 30), suggesting that ALK inhibitors as a single agent are likely to fail in such patients. However, there remains the potential for ALK inhibitors to enhance traditional cytotoxic agents in tumors carrying ALK mutations that confer single-agent ALK inhibitor resistance.
Here we report that a novel therapeutic combination of the ALK inhibitor, crizotinib, with conventional chemotherapeutics (topotecan plus cyclophosphamide), has marked anti-tumor activity and substantially prolongs event-free survival in both crizotinib-sensitive and crizotinib-resistant models of ALK-mutant neuroblastoma. We also show evidence for in vitro synergy with this combination in one neuroblastoma cell line harboring high-level ALK amplification. While our data suggest that ALK-amplifed tumors will be responsive to crizotinib combined with topotecan and cyclophosphamide, additional non-clinical studies are warranted in models that represent this infrequent subset of neuroblastomas with ALK aberrations. Importantly, synergy was seen with the combination in settings where crizotinib or chemotherapy alone induced only mild or no activity. For instance, xenografts harboring the resistant F1174L mutation showed complete remission of tumors for an additional 24 weeks after discontinuation of therapy. Interestingly, this sustained antitumor activity was accompanied by striking upregulation of the tumor suppressor protein, p53, and its downstream effectors p21, PUMA, and cleaved-caspase 3 in ALK aberrant and p53 functional neuroblastoma models treated with combination therapy. This observation prompted us to investigate whether activation of the p53 pathway could be a determinant of the synergy observed when combining crizotinib and genotoxic agents. The lack of response to the combination therapy in neuroblastoma models carrying mutated TP53 provided the initial evidence that this synergy relies, at least in part, on a functional p53 pathway. Additionally, in two ALK-mutated neuroblastoma cells with genetically ablated p53, we found an abrogation of synergy upon combination treatment compared to the respective control lines. Taken together, we show here that activation of the p53 pathway plays a role in mediating the synergy observed between crizotinib and chemotherapy in neuroblastoma models with activated ALK.
Our findings are supported by recent studies suggesting a correlation between inhibition of ALK signaling and reactivation of p53-mediated cell death. Additionally, synthetic peptides corresponding to the proapoptotic domain of ALK caused p53-mediated cytotoxicity in ALCL and neuroblastoma cells (31). In the same study, ALK peptides interacted with proteins that have been previously reported to interact with the p53 gene and protein. Similar to our findings, p53 knockdown rescued both ALCL and neuroblastoma cells from ALK peptide mediated cell death. The precise mechanism of how ALK inhibition impacts the p53 pathway and promotes synergy between crizotinib and chemotherapy has not been fully elucidated. Studies are ongoing to determine whether ALK inhibitor-mediated cell death is due to a dual function of ALK in regulating not only the canonical receptor tyrosine kinase pathway but also the p53 pathway. This could inform the design of drug combinations targeting ALK and other specific p53-targeted therapies such as MDM2 inhibitors. We observed a marked reduction in total ALK levels in the chemotherapy and combination treated xenograft tumors of unclear significance, but we postulate this could be secondary to decreased transcription upon chemotherapy treatment or a potential feedback mechanism between ALK and p53 in which ALK, serving as a negative regulator of p53, is then decreased by transcriptional activity of p53.
Altogether, we demonstrate that combining crizotinib with conventional chemotherapeutic agents is effective in ALK-driven models of neuroblastoma that show differential crizotinib sensitivity, providing the rationale for the currently accruing COG phase 1 trial (NCT01606878). Within the high-risk subset of newly diagnosed neuroblastoma patients, 14% will harbor an ALK aberration and will have inferior outcome, providing a unique opportunity for clinical studies testing theintegration of crizotinib into the backbone of contemporary chemotherapy regimens for these patients. The absence of synergy in neuroblastoma models harboring wild type ALK and/or loss-of-function TP53 mutations supports a responder hypothesis that ALK status in addition to a functional p53 pathway is a determinant of response to this therapeutic strategy. Systematic investigation of crizotinib and other ALK inhibitors with DNA-damaging chemotherapy combinations is ongoing to rigorously evaluate the importance of timing and sequence of these regimens in order to best guide clinical trial design.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Despite intensive, multimodal therapies, patients with high-risk neuroblastoma still face unacceptably high mortality rates. We have previously shown that ALK activation is present in 14% of patients with high-risk disease and that the presence of an ALK aberration (point mutation or amplification) correlates with inferior survival in these patients. Importantly, the F1174L and F1245C amino acid substitutions, which comprise two of the hot spot mutations, confer intrinsic resistance to crizotinib and present a significant obstacle in the clinic. In an effort to develop novel and more effective ALK inhibition strategies, we evaluated the in vitro and in vivo response of ALK aberrant neuroblastoma models to the ALK inhibitor, crizotinib, in combination with conventional chemotherapy agents, topotecan and cyclophosphamide. We provide the preclinical rationale for clinical testing of crizotinib with conventional chemotherapy with the goal of integrating ALK inhibition into multi-agent therapy for patients with high-risk ALK aberrant neuroblastoma.
Acknowledgments
Grant Support: This study was supported by NIH Grant R01CA140198 (to Y.P.M).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM. Recent advances in neuroblastoma. The New England journal of medicine. 2010;362(23):2202–11. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 4.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455(7215):967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 6.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):439–49. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 8.Yao S, Cheng M, Zhang Q, Wasik M, Kelsh R, Winkler C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PloS one. 2013;8(5):e63757. doi: 10.1371/journal.pone.0063757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 10.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 11.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 12.Mosse YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009;15(18):5609–14. doi: 10.1158/1078-0432.CCR-08-2762. [DOI] [PubMed] [Google Scholar]
- 13.Christensen JG. Proof of principle for crizotinib in anaplastic lymphoma kinase-positive malignancies was achieved in ALK-positive nonclinical models. Molecular cancer therapeutics. 2011;10(11):2024. doi: 10.1158/1535-7163.MCT-11-0721. [DOI] [PubMed] [Google Scholar]
- 14.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. The lancet oncology. 2012;13(10):1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Science translational medicine. 2011;3(108):108ra14. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–80. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komuro H, Hayashi Y, Kawamura M, Hayashi K, Kaneko Y, Kamoshita S, et al. Mutations of the p53 gene are involved in Ewing’s sarcomas but not in neuroblastomas. Cancer Research. 1993;53(21):5284–8. [PubMed] [Google Scholar]
- 19.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nature genetics. 2013;45(3):279–84. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carr-Wilkinson J, O’Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(4):1108–18. doi: 10.1158/1078-0432.CCR-09-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61(16):6185–93. [PubMed] [Google Scholar]
- 22.Attiyeh EF, Diskin SJ, Attiyeh MA, Mosse YP, Hou C, Jackson EM, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome research. 2009;19(2):276–83. doi: 10.1101/gr.075671.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frgala T, Kalous O, Proffitt RT, Reynolds CP. A fluorescence microplate cytotoxicity assay with a 4-log dynamic range that identifies synergistic drug combinations. Mol Cancer Ther. 2007;6(3):886–97. doi: 10.1158/1535-7163.MCT-04-0331. [DOI] [PubMed] [Google Scholar]
- 24.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. The Journal of biological chemistry. 1977;252(18):6438–42. [PubMed] [Google Scholar]
- 25.Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M, et al. Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes & development. 2012;26(10):1041–54. doi: 10.1101/gad.184325.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 27.Pinheiro J, Bates D, DebRoy S, Sarkar D, Team RC. nlme: Linear and Nonlinear Mixed Effects Models. R package version R package version. 3:1–120 ed2015. [Google Scholar]
- 28.Therneau T, Grambsch P. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer; 2000. [Google Scholar]
- 29.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell. 2014;26(5):682–94. doi: 10.1016/j.ccell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3(108):108ra14. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aubry A, Galiacy S, Ceccato L, Marchand C, Tricoire C, Lopez F, et al. Peptides derived from the dependence receptor ALK are proapoptotic for ALK-positive tumors. Cell Death Dis. 2015;6:e1736. doi: 10.1038/cddis.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.