

Abstract
Tauopathies are neurodegenerative diseases characterized by intraneuronal inclusions of hyperphosphorylated tau protein and abnormal expression of brain-derived neurotrophic factor (BDNF), a key modulator of neuronal survival and function. The severity of both these pathological hallmarks correlate with the degree of cognitive impairment in patients. However, how tau pathology specifically modifies BDNF signaling and affects neuronal function during early prodromal stages of tauopathy remains unclear. Here, we report that the mild tauopathy developing in retinal ganglion cells (RGCs) of the P301S tau transgenic (P301S) mouse induces functional retinal changes by disrupting BDNF signaling via the TrkB receptor. In adult P301S mice, the physiological visual response of RGCs to pattern light stimuli and retinal acuity decline significantly. As a consequence, the activity-dependent secretion of BDNF in the vitreous is impaired in P301S mice. Further, in P301S retinas, TrkB receptors are selectively upregulated, but uncoupled from downstream extracellular signal-regulated kinase (ERK) 1/2 signaling. We also show that the impairment of TrkB signaling is triggered by tau pathology and mediates the tau-induced dysfunction of visual response. Overall our results identify a neurotrophin-mediated mechanism by which tau induces neuronal dysfunction during prodromal stages of tauopathy and define tau-driven pathophysiological changes of potential value to support early diagnosis and informed therapeutic decisions.
SIGNIFICANCE STATEMENT This work highlights the potential molecular mechanisms by which initial tauopathy induces neuronal dysfunction. Combining clinically used electrophysiological techniques (i.e., electroretinography) and molecular analyses, this work shows that in a relevant model of early tauopathy, the retina of the P301S mutant human tau transgenic mouse, mild tau pathology results in functional changes of neuronal activity, likely due to selective impairment of brain-derived neurotrophic factor signaling via its receptor, TrkB. These findings may have important translational implications for early diagnosis in a subset of Alzheimer's disease patients with early visual symptoms and emphasize the need to clarify the pathophysiological changes associated with distinct tauopathy stages to support informed therapeutic decisions and guide drug discovery.
Keywords: BDNF, electroretinography, neuronal dysfunction, retinal ganglion cells, tau protein, TrkB receptor
Introduction
Intraneuronal inclusions made of filaments of hyperphosphorylated microtubule-associated tau protein are neuropathological hallmarks of a subset of neurodegenerative diseases termed tauopathies, which include Alzheimer's disease (AD), corticobasal degeneration, progressive parasupranuclear palsy, and frontotemporal dementia. In AD, levels of tau species, including total tau and phospho-tau, are altered in the CSF of subjects at risk for AD several years before putative onset of clinical symptoms (Bateman et al., 2012) and the development of tauopathy strongly correlates with the severity of cognitive impairment in patients (Arriagada et al., 1992; Gómez-Isla et al., 1997). However, the mechanisms by which tau affects neuronal physiology during prodromal stages of tauopathy remain unclear.
The P301S mutant human tau transgenic (P301S) mouse (Allen et al., 2002), a model of tauopathy, develops tau inclusions throughout the CNS, leading to neuronal loss in the spinal cord (Allen et al., 2002) and cortex (Hampton et al., 2010) and to behavioral deficits (Scattoni et al., 2010). The inner retina of P301S mice also develops mild tau pathology (Gasparini et al., 2011), resembling initial pathological stages. In P301S retinas, transgenic mutant tau is expressed only in retinal ganglion cells (RGCs); its hyperphosphorylated form accumulates in the nerve-fiber and RGC layers of the inner retina and, in a few RGCs, forms insoluble tau filament aggregates (Gasparini et al., 2011). Although none of these changes alters the overall anatomy of the retina—both inner and outer—or causes loss of neurons, P301S retinal explants in vitro display growing abnormalities. In particular, in retinal explants cultured from adult P301S mice, RGC axonal outgrowth does not respond to neurotrophic stimuli, suggesting that tau pathology alters neurotrophin signaling (Gasparini et al., 2011).
Among the neurotrophins, brain-derived neurotrophic factor (BDNF) plays a key role in promoting neuronal survival, synaptic plasticity, and memory consolidation (Lu, 2003; Binder and Scharfman, 2004; Gezen-Ak et al., 2013). Dysregulation of its levels and signaling has been linked to neurodegeneration. BDNF mRNA and protein are decreased in the blood (Pláteník et al., 2014) and brain of patients with AD (Phillips et al., 1991; Connor et al., 1997; Ferrer et al., 1999; Hock et al., 2000; Holsinger et al., 2000; Peng et al., 2005) and the extent of BDNF reduction correlates with the degree of cognitive impairment (Peng et al., 2005). In transgenic mouse AD models overexpressing mutant human β-amyloid precursor protein, administering BDNF rescues synaptic loss and cognitive dysfunction, implying that the loss of BDNF contributes to β-amyloid-induced pathological changes (Arancibia et al., 2008; Blurton-Jones et al., 2009; Nagahara et al., 2009). Decreased BDNF levels have also been found in the parietal cortex of subjects with primary tauopathies, such as corticobasal degeneration and Pick's disease (Belrose et al., 2014). While a number of studies have investigated the link between BDNF impairment and β-amyloid-induced pathological changes (Song et al., 2015), how tau pathology specifically modifies BDNF signaling and affects neuronal function during early prodromal stages of tauopathy remains unclear.
Here, to investigate this central question, we used the P301S retina as a model of early-stage tauopathy. We find that in the retina of P301S mice, mild tauopathy causes functional changes of visual response by disrupting the BDNF signaling via the TrkB receptor. In 5-month-old P301S mice, the activity of RGCs and retinal acuity are significantly reduced. Moreover, in the retina of these mice, BDNF signaling through TrkB receptors is significantly impaired both at the level of the ligand and receptor. Despite normal retinal expression and synthesis, the amount of BDNF in the vitreous is significantly reduced, possibly due to impaired activity-dependent secretion. Further, in P301S retinas, although TrkB receptors are selectively upregulated, they are uncoupled from the downstream extracellular signal-regulated kinase (ERK) 1/2 signaling pathway. We also show that both retinal dysfunction and TrkB upregulation are triggered by tau pathology and that tau-induced BDNF signaling impairment contributes to the reduced visual response.
Materials and Methods
Transgenic mice.
Homozygous P301S transgenic mice (Allen et al., 2002) and age-matched C57BL/6 wild-type (WT) mice of either sex were used. Animal health and comfort were veterinary controlled. Mice were housed in filtered cages in a temperature-controlled room with a 12:12 h dark/light cycle with ad libitum access to water and food. All animal experiments were performed in full compliance with the European Community Council directive 86/609/EEC and the revised directive 2010/63/EU, and were approved by the Italian Ministry of Health and by the Istituto Italiano di Tecnologia Animal Facility Committee.
Antibodies and reagents.
The following primary antibodies were used: monoclonal antibodies against BDNF (clone 35928.11; Sigma-Aldrich), βI-tubulin, actin (Sigma-Aldrich), p44/42 MAPK (3A7), p44/42 MAPK (Thr202/Tyr204; Cell Signaling Technology), tau (Tau5; Calbiochem), human tau (HT7; ThermoFisher Scientific), phospho-tau (AT8; Autogen Bioclear), phospho-Y18 tau (9G3; MédiMabs), PHF-1 and MC1 phospho-tau (kind gift from Dr. P. Davies, Albert Einstein College of Medicine, New York, NY); Trk (clone B-3), which selectively recognizes TrkA (Matrone et al., 2011; our unpublished data); fyn (Santa Cruz Biotechnology); polyclonal antisera against TrkB (clone 07-225, Millipore Biotechnology), which selectively recognizes TrkB (Cazorla et al., 2011a; our unpublished data); p75NTR (Millipore); phospho-TrkA (Tyr490)/TrkB (Tyr516); phospho-TrkA (Tyr785)/TrkB (Tyr816); phospho-p44/42 MAPK (Thr202/Tyr204); phospho(Tyr783)-PLCγ1, PLCγ1 (Cell Signaling Technology), BDNF (Santa Cruz Biotechnology). For TrkA and TrkB phosphorylation, the PathScan Phospho-TrkA (Tyr490) Sandwich ELISA and PathScan Phospho-TrkB (Tyr516) Sandwich ELISA kits (Cell Signaling Technology) were also used. HRP-conjugated secondary antibodies were from Bio-Rad Laboratories. For immunoprecipitation experiments, Western blots were developed using HRP-conjugated anti-mouse and anti-rabbit light-chain antibodies (Millipore). All reagents were from Sigma-Aldrich unless otherwise specified. Stock solution of tetrodotoxin (TTX; Tocris Bioscience) was prepared in saline at 1 mm and diluted to 5 μm. ANA12 (Sigma-Aldrich) was dissolved at 20 mg/ml in DMSO and resuspended in saline by sonication with a final DMSO concentration of 0.5%. The stock solution of the ERK inhibitor PD0325901 (Sigma-Aldrich) was 5 μm in DMSO.
Pattern electroretinogram.
Mice were anesthetized with intraperitoneal injections of 2 g/kg urethane, gently restrained using a mouth bite bar and nose holder that allowed unobstructed vision, and kept at a constant body temperature of 37°C using a feedback-controlled heating pad. Under these conditions, the eyes of mice were naturally wide open and in a stable position, with pupils pointing laterally and upward. The silver recording electrode was gently placed on the corneal surface encircling the undilated pupil without interfering with vision. The ground electrode was a small stainless steel needle inserted in the skin of the back of the head. Visual stimuli were displayed on a 17 inch monitor (Biomedica Mangoni) whose center was aligned with the projection of the pupil and presented at a distance of 20 cm from the mouse's eye. Pattern stimuli consisted of horizontal, square grating bars of constant luminance, contrast, and spatial and temporal frequencies. The standard grating contrast was 90%, with a 1 Hz reversal rate, and spatial frequency of 0.05 c/deg, which maximize pattern electroretinogram (ERG) amplitude in the mouse (Porciatti, 2007; Porciatti et al., 2007).
In selected experiments, to determine spatial visual acuity, mice were exposed to pattern stimuli with varying spatial frequencies (0.05–0.7 c/deg) at constant contrast, luminance, and temporal frequency. Spatial acuity was calculated as the spatial frequency of the intercept between the linear regression among data points in a graph, where spatial frequencies were represented on a logarithmic scale, and the noise level measured upon presentation of a blank screen.
Flash ERG.
Mice were dark adapted for ≥4 h before being anesthetized under dim red light by intraperitoneal injections of urethane (2 g/kg), and gently restrained as described above. The pupils were dilated with the mydriatic agent atropin at 1% in physiological solution. The gold recording electrode (0.25 mm diameter) was gently placed on the corneal surface encircling the dilated pupil without interfering with vision. The ground electrode was inserted in the skin of the back of the head. Visual stimuli consisted of uniform flickers of light of different luminance (0.001–30 cd s/m2) presented in scotopic conditions through a Ganzfeld dome (Biomedica Mangoni).
Western blots.
Retinas were lysed by sonication in 150 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EGTA, 0.5% Triton X-100, and protease and phosphatase inhibitors and incubated on ice for 30 min. Samples were spun at 20,800 × g for 20 min. The supernatants were collected and protein concentration was determined using a BCA kit (Pierce). Equal amounts of protein were separated by electrophoresis on 10 or 7.5% SDS polyacrylamide gels (SDS-PAGE) as previously described (Gasparini et al., 2011). Proteins were transferred overnight at 4°C onto Protran 0.2 NC nitrocellulose membranes (GE Healthcare). Membranes were blocked for 1 h at room temperature with 5% milk in phosphate buffer (PBS), pH 7.4, containing 0.05% Tween 20, and incubated overnight at 4°C with the primary antibody in blocking buffer. Membranes were then rinsed in Tris-buffered saline, pH 7.4, with 0.05% Tween 20, incubated for 1 h at room temperature in horseradish peroxidase (HRP)-conjugated secondary antibodies (immunopure peroxidase conjugated; ThermoFisher Scientific), and visualized using SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). Chemiluminescent signals were acquired using films (Hyperfilm ECL, GE Healthcare) or an Image Quant LAS 4000 mini (GE Healthcare). Densitometric analysis was performed using the National Institutes of Health ImageJ program (Schneider et al., 2012). Equal loading was verified using MemCode (ThermoFisher Scientific) and levels of βI-tubulin or actin. Protein levels were expressed as a ratio with βI-tubulin or actin. Phosphorylated proteins were normalized to the total amount of the respective protein.
RNA isolation and quantitative real-time PCR.
Retinas were removed and snap frozen in dry ice. RNA was isolated using the RNeasy mini kit (Qiagen) and cDNA was synthesized from 1 μg of total RNA using the First-Strand cDNA Synthesis Kit (Qiagen). Quantitative real-time PCR was performed using the QuantiFast SYBR Green PCR Kit. (Qiagen) as previously described (Ferrera et al., 2014). Gene expression data were normalized to actin and GAPDH using the multiple internal control gene method with the GeNorm algorithm available in the qBasePlus software (Biogazelle). The following primers were used: TrkB, forward 5′-CCACGGCATGAGCGAGAC-3′, reverse 5′-CCTAAGCACAACGGACTCCC-3′; BDNF, forward 5′-AAGGTCTAGGATGGAGGTGG-3′, reverse 5′-CTAAGCAACTTGACACATCATTCC-3′; actin, forward 5′-AAGTGGTTACAGGAAGTCC-3′, reverse 5′-ATAATTTACACAGAAGCAATGC-3′; GAPDH, forward 5′-GAACATCATCCCTGCATCCA-3′, reverse 5′-CCAGTGAGCTTCCCGTTCA-3′.
Pharmacological treatments.
Mice were anesthetized by intraperitoneal injection of urethane (2 g/kg). Intravitreal injections were performed under a dissecting microscope with a 30 ga needle attached to a 10 μl glass syringe (Hamilton). The needle was positioned posterior to the ora serrata and 0.5 μl of the solution was slowly (3–5 s) injected into the vitreous chamber of the eye. A 10 s interval was held before removing the needle. Mice were killed at different times after injection and retinas were dissected and snap frozen in dry ice. In selected experiments, the following substances were dissolved in 0.9% NaCl physiological salt solution and injected intravitreously (0.5 μl): 2 μg of BDNF, 2 μg of nerve growth factor (NGF), TTX (500 nm final concentration), PD0325901 (20 nm final concentration), or vehicle (0.4% DMSO in the contralateral eye). In BDNF stimulation experiments, we used 2 μg of BDNF, a dosage based on the following considerations. Our initial goal was to test the potential therapeutic use of BDNF for tauopathy. To this purpose, it was important to first investigate the function of TrkB signaling. In rodents, the neuroprotective effects of BDNF on retinal ganglion neurons have been investigated by several groups using doses ranging from 0.1 to 5 μg/eye (for review, see Khalin et al., 2015), with most studies using ≥1 μg/eye both in rat and mouse. Specifically, it has been reported that a dose of 2 μg/eye of BDNF is neuroprotective and restores retinal function [assessed by pattern ERG (pERG)] in a mouse model of glaucoma (Domenici et al., 2014). We thus postulated that 2 μg of BDNF (final vitreal concentration of ≈0.2 μg/μl in the mouse) would possibly achieve a response robust enough to investigate the function of TrkB signaling and uncover even mild deficits that could affect the efficacy of a potential treatment. To control for nonspecific effects induced by the injection of proteins or vehicle, bovine serum albumin (BSA) was injected in the contralateral eye as a control. Initial experiments in WT mice indicated that intravitreal injection of BSA did not affect the levels of p[Y516]TrkB (p[Y516]TrkB/TrkB ratio, 1.0 ± 0.1), phospho-ERK1 (pERK1/ERK1 ratio, 1.6 ± 0.6), or phospho-ERK2 (pERK2/ERK2 ratio, 0.7 ± 0.1), which remained similar to those of untreated retinas (p[Y516]TrkB/TrkB ratio, 1.0 ± 0.1; pERK1/ERK1, 2.0 ± 1.1; pERK2/ERK2, 0.8 ± 0.3). In selected experiments, TrkB was inhibited by systemic administration of the TrkB inhibitor ANA12 (Cazorla et al., 2011b) by intraperitoneal injection at a 1 mg/kg dose. Vehicle (0.5% DMSO in saline) was injected in control mice.
Intracerebral injections of brain extracts.
For intracerebral injection, brain extracts from P301S (P301SBE) and WT (WTBE) mice were prepared as described previously (Clavaguera et al., 2013). Briefly, brainstems from 5-month-old mice were dissected, homogenized in sterile PBS, and centrifuged at 3000 × g at 4°C for 5 min. The supernatant was aliquoted, snap frozen, and stored at −80°C until use. Levels of tau, transgenic human tau, and phospho-tau in P301SBE and WTBE were analyzed by Western blot (see Fig. 6A). P301S transgenic mice 1.5 months old were anesthetized with 2% isoflurane [2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane] and placed in a stereotaxic device. The skull was exposed and kept dry and clean. The bregma was identified and marked. The designated point of injection was at a depth of 1 mm from the brain surface, 2.92 mm behind the bregma on the anteroposterior axis, and 0.5 mm lateral to the midline (see Fig. 6B). A window was drilled in the scalp above the designated coordinates in the right and left hemispheres. The lysates (2 μl) were then injected using a Hamilton syringe (Hamilton) with a microinjector (injection rate, 0.5 μl/min). After injection, the needle was kept in place for an additional 1 min before gentle withdrawal. The surgical area was cleaned with sterile saline and the incision sutured. Mice were monitored until recovery from anesthesia and checked weekly after surgery. For sham-injected mice, sterile PBS was injected following the same procedure. At 3 months of age, after recording retinal activity by pERG, mice were killed and brain tissues were collected for biochemical and immunohistochemical analyses.
Figure 6.
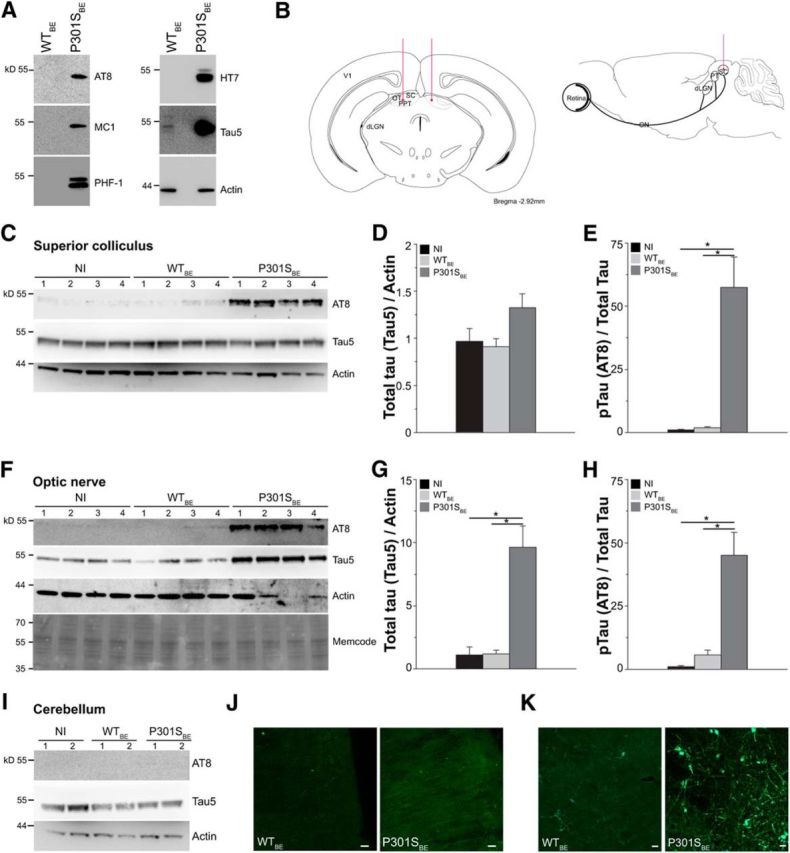
Intracollicular injection of P301S brain extract increases tau pathology in the SC and ON of 3-month-old P301S mice. WTBE or P301SBE was injected in the SC (2.92 mm behind bregma) of P301S mice at 1.5 months of age and levels of tau and phospho-tau in the SC, ON, cerebellum, and visual cortex were analyzed 6 weeks later. Red lines indicate injection sites. A, Western blot analysis of tau and phospho-tau protein in brain extracts from 5-month-old WT (WTBE) and P301S (P301SBE) mice. Phospho-tau was analyzed using the AT8, MC1, and PHF-1 monoclonal antibodies. Human tau and total tau were detected using the HT7 and Tau-5 monoclonal antibodies, respectively. Actin was analyzed as loading control. B, Schematic representation of the injection sites. WTBE or P301SBE were injected in the SC (2.92 mm behind bregma) of 1.5-month-old P301S mice. dLGN, Dorsolateral geniculate nucleus; PT, pretectum; PPT, posterior pretectal nucleus; V1, primary visual cortex. C–I, Tau levels in the SC (C, D, and E), ON (F, G, and H), and cerebellum (I) of 3-month-old P301S mice injected 6 weeks earlier with WTBE, with P301SBE, or not injected (NI). C, F, I, Western blot analysis of total tau (Tau-5) and phospho-tau (AT8) in the SC (C), ON (F), and cerebellum (I). In F, MemCode staining shows equal loading. D, E, Quantitative analysis of total tau (D) and phospho-tau (E) in the SC. *p < 0.05, Kruskal–Wallis one-way ANOVA on ranks followed by pairwise multiple-comparison procedure according to Dunn's method. G, H, Quantitative analyses of total tau (G) and phospho-tau (H) in the ON. *p < 0.05, Kruskal–Wallis one-way ANOVA on ranks followed by pairwise multiple-comparison procedure according to Dunn's method. J, K, Representative fluorescence images of AT8 immunoreactivity in the V1 primary visual cortex (J) and SC (K) of 3-month-old P301S mice injected intra-SC with WTBE or P301SBE. The images represent phospho-tau immunoreactivity in coronal sections at 3.60 mm behind bregma. Scale bars, 25 μm. In all graphs, WTBE, n = 10; P301SBE, n = 16; NI, n = 12.
ELISA.
The BDNF levels of the vitreous were assayed using the BDNF Emax ImmunoAssay System (Promega) according to the manufacturer's instructions. Briefly, standard 96-well flat-bottom NUNC-Immuno maxisorp ELISA plates (Costar) were coated overnight at 4°C with the polyclonal anti-BDNF capturing antibody and blocked with blocking and sample buffers. Serial dilutions of known amounts of BDNF ranging from 0 to 500 pg were used for the standard curve. Fifty micrograms of retinal lysate or 1.5 μl of vitreous humor were applied in each well and incubated 2 h at room temperature with the detecting monoclonal antibody against BDNF, followed by an HRP-conjugated anti-human IgG secondary antibody for 2.5 h at room temperature and colorimetric reaction with 3,3′,5,5′-tetramethylbenzidine. After 10 min, the reaction was terminated by 1 m phosphoric acid and the absorbance at 450 nm was measured using the Victor3 V multilabel plate reader (PerkinElmer).
In selected experiments, TrkA and TrkB phosphorylation in the retina was analyzed using the PathScan Phospho-TrkA (Tyr490) and PathScan Phospho-TrkB (Tyr516) Sandwich ELISA kits (Cell Signaling Technology) according to manufacturer's instruction. The PathScan TrkA and PathScan TrkB ELISA kits (Cell Signaling Technology) were also used for total receptor levels.
Immunohistochemistry and confocal microscopy.
Retinas from homozygous P301S tau transgenic and C57BL/6 WT mice were analyzed histologically. Briefly, the mice were anesthetized with CO2 and killed by cervical dislocation. The eyecups were removed and fixed overnight in 4% PFA. Immunohistochemical detection of TrkB and phosphorylated tau was performed as previously described (Gasparini et al., 2011). Briefly, 10 μm retinal cryostat sections were blocked in 5% normal goat serum, 1% BSA, and 0.2% Triton PBS for 1 h at room temperature and incubated overnight at 4°C with primary antibodies. Immunoreactivity was visualized using secondary antibodies conjugated with Alexa fluorochromes (Invitrogen) for confocal analysis. Nuclei were counterstained with Hoechst-33342. Confocal microscopy was performed with a Leica TCS SP5 (Leica Microsystems) equipped with 63× HCX PL APO 1.40 oil and 40× HCX PL APO 1.25 oil objectives. To quantify the number of photoreceptor (PR) nuclei in the outer nuclear layer, ≥3 sections/slide adjacent to the optic nerve (ON) head were separately counted and averaged as previously described (Rex et al., 2004).
Immunohistochemical analysis of brains from mice injected with brain extracts or PBS was performed on 30 μm coronal sections collected 2.80–4.60 mm behind bregma. Sections were immunolabeled with AT8 antibody and counterstained with the Congo red analog (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB; Sato et al., 2004) as previously described (Gasparini et al., 2011). Images were collected using an automated Olympus BX51 microscope, equipped with a MBF Optonic CX9000 camera and 10× UPLFLN semi-apo fluorite 10× numerical aperture 0.3 objective and Neurolucida V11 software (MBF Bioscience) or by confocal microscopy, as described above.
Sarkosyl extraction.
Sarkosyl-insoluble tau was extracted from 200 mg of brain tissue of transgenic and WT age-matched mice as described previously (Gasparini et al., 2011). Briefly, tissues were homogenized in 10 volumes of cold extraction buffer (10 mm Tris-HCl, pH 7.4, 0.8 m NaCl, 1 mm EGTA, 10% sucrose) and the homogenates were spun for 30 min at 20,000 × g. After adding 1% sarkosyl, the samples were shaken for 1 h at room temperature and spun at 100,000 × g for 1 h at 4°C. The pellets containing insoluble tau were resuspended in 50 mm Tris-HCl, pH 7.4, and stored at 4°C until analysis by Western blot.
Coimmunoprecipitation.
Retinas were lysed by mechanical disruption on ice in 1% NP-40, 0.5 m NaCl, 10 mm Na3PO4, 2 mm EDTA, and protease and phosphatase inhibitors. The lysates were spun at 500 × g for 5 min at 4°C to remove unbroken tissue. Equal amount of proteins (250 μg) of the supernatants were immunoprecipitated by adding 2.5 μg of the antibody and protein G agarose (Pierce). Protein G agarose beads alone were used as control for immunoprecipitation. Samples were incubated overnight at 4°C while being constantly shaken. After extensive washing, the precipitates were eluted from the beads by boiling in sample buffer for 5 min. Precipitated proteins were separated by 7.5% SDS-PAGE and transferred to nitrocellulose membranes for Western blot analysis.
Statistical analysis.
Statistical analysis of groups with normal distributions was performed using a Student's t test for two groups or ANOVA in case of multiple comparisons. When the normality test failed, the analysis was performed using nonparametric tests, such as the Mann–Whitney rank-sum test or the Kruskal–Wallis one-way ANOVA on ranks followed by a post hoc test according to Dunn's or Student–Newman–Keuls methods. A two-way ANOVA followed by multiple comparisons with the Holm–Sidak method was performed in some instances. Differences among groups were considered statistically significant when p < 0.05. Data throughout the text are reported as average values ± SEM, except when otherwise specified.
Results
RGC activity is reduced in P301S mice
To investigate how a mild degree of tauopathy affects retinal physiology in P301S mice, we used ERG to measure the response of the retina to patterned light stimuli that specifically elicit RGC activity (pERG; Porciatti, 2007) or to flickered uniform light [flash ERG (fERG)] that elicits responses from PRs and the outer retina (mainly bipolar cells; Porciatti, 2007). For pERG, anesthetized mice were shown squared pattern stimuli alternating in counter phase at 1 Hz, at saturating contrast and spatial frequency. In WT mice, the typical waveform of pERG consists of a positive P1 wave (amplitude ± SD, 5.9 ± 3.1 μV; latency ± SD, 74.9 ± 16.4 ms) and a negative N2 component (amplitude ± SD, 2.7 ± 1.5 μV; latency ± SD, 209.2 ± 35.2 ms; Fig. 1A–C). In transgenic P301S mice, the peak amplitudes of both the P1 and N2 components were significantly reduced by 39 and 48%, respectively (Fig. 1B), and their peak latency was increased (Fig. 1C). Consistent with a delayed response and increased rise time, the amplitude of the P1 wave detected at the median latency of WT (i.e., 72.5 ms) was reduced by 51% in P301S mice (average ± SD: WT, 4.3 ± 3.2 μV; P301S, 2.1 ± 1.3 μV; p < 0.01 vs WT, Mann–Whitney rank-sum test). To determine the impact on visual acuity (Fig. 1D), WT and P301S mice were exposed to pattern stimuli with spatial frequencies ranging from 0.05 to 0.7 c/deg. In agreement with steady-state pERG results, retinal acuity was significantly lower in P301S mice than in WT mice (Fig. 1E). These findings indicate that despite anatomical preservation of RGC number (Gasparini et al., 2011), mild levels of tau pathology are sufficient to trigger significant RGC dysfunction.
Figure 1.

RGC activity is reduced in P301S mice. A, Representation of pERG assessment in the mouse. Anesthetized mice were presented a square pattern of black and white stripes reversing at a rate of 1 Hz, with constant luminance, maximal contrast, and spatial frequency (inset). Retinal activity was measured through a corneal electrode. Average pERG waveform recorded in 5-month-old P301S mice and age-matched WT mice are shown. B, C, Box plots of P1 and N2 peak amplitude (B) and latency (C) values in P301S (n = 15) and WT (n = 16) mice. Average values of individual mice are overlaid on each plot (gray circles). In each box, median (white line) and mean (dotted gray line) values are indicated. *p < 0.05, **p < 0.01, Mann–Whitney rank-sum test. D, Representative visual acuity determination in one WT mouse. The dotted line represents the noise level. The shaded area represents the SD of noise measurements. E, Visual acuity of P301S and WT mice. Bars represent mean ± SEM. **p < 0.01, Student's t test. WT, n = 5; P301S, n = 4. F, Representative mouse fERG response upon exposure to light flicker of maximal luminance 30 cd s/m2. The negative response (a wave) and positive (b wave) waves represent the response of PRs and bipolar cells, respectively. G, Peak amplitude of a and b waves in P301S and WT mice. Bars represent the average amplitude ± SEM. n = 12 per group.
To investigate whether tau pathology in RGCs was associated with general retinal impairment, we evaluated the anatomical and functional integrity of the outer retina and bipolar cells. Using sections from the central retina, we examined the integrity of PRs by counting the number of nuclei in the outer plexiform layer of WT and P301S retinas. The number of rows of PR nuclei was similar in WT (9.8 ± 0.8) and P301S (9.3 ± 0.5; p = 0.61 vs WT, Student's t test) retinas. To examine the functional status of the outer retina, WT and P301S mice were exposed to uniform flickers of light of increasing luminance generated by a Ganzfeld dome and the retinal response was recorded through fERG. A typical fERG waveform includes a and b waves, which mainly represent the activity of PRs and bipolar cells, respectively (Fig. 1F). In WT mice, the PR response (i.e., a wave) is either undetectable or very low at luminance ranging from 0.001 to 0.03 cd s/m2 and increases at luminance >0.03 cd s/m2, reaching amplitudes comparable to those of the b wave at the maximal luminance, i.e., 30 cd s/m2 (Fig. 1F). To compare fERG responses of WT and P301S retinas, mice were exposed to maximal stimuli (luminance 30 cd s/m2), representing the most challenging setting, to highlight any subtle differences between genotypes. In these conditions, the amplitudes of both a and b waves were similar in WT and P301S mice (Fig. 1G). Likewise, the ratio between a and b waves was not statistically different in WT (1.1 ± 0.2, n = 12) and P301S mice (1.1 ± 0.1, n = 12; p = 0.9 vs WT, Mann–Whitney rank-sum test), indicating that the overall activity of the outer retina is preserved in transgenic mice.
Overall, these findings indicate that in P301S transgenic mice, tau pathology in RGCs is associated with a specific impairment of their activity and reduced retinal acuity, with preserved function of other retinal neurons, which are devoid of tau pathology.
BDNF secretion in the vitreous is impaired in P301S mice
We have previously demonstrated that in retinal explants cultured from adult P301S transgenic mice, axonal outgrowth is impaired in response to a mixture of neurotrophic factors, including BDNF (Gasparini et al., 2011). BDNF plays a key role in retinal plasticity and visual acuity refinement during development (Landi et al., 2007, 2009; Liu et al., 2007; Schwartz et al., 2011) and, in cultured neurons, its synthesis and secretion is regulated by neuronal activity (Hardingham et al., 2002; Matsumoto et al., 2008). We therefore investigated whether in P301S retinas the reduced retinal activity was associated with altered BDNF metabolism or secretion.
BDNF forms upon cleavage of the precursor pro-BDNF in the retina (data not shown), is mainly expressed in the RGC and inner plexiform layers (data not shown; Grishanin et al., 2008), and is secreted into the vitreous. We found that mRNA levels of BDNF were unchanged in P301S retinas compared with WT (Fig. 2A). Likewise, protein levels of both pro-BDNF (Fig. 2B,C) and mature BDNF (Fig. 2D) were similar in P301S and WT retinas, indicating that BDNF synthesis is preserved in transgenic mice.
Figure 2.

BDNF levels are reduced in the P301S vitreous. A, mRNA expression of BDNF in the retina of P301S and WT mice. BDNF mRNA levels were analyzed by real-time PCR and normalized to the housekeeping genes actin and GAPDH. Bars represent the average relative expression ± SEM. WT, n = 7; P301S, n = 8. B, C, Pro-BDNF protein levels in the retinas of P301S and WT mice. B, Western blot analysis of pro-BDNF. Each lane corresponds to retinal lysate from one mouse. Tubulin was analyzed as loading control. C, Quantitative analysis of pro-BDNF protein levels. Values were normalized to tubulin levels. Bars represent the average ratio ± SEM. WT, n = 6; P301S, n = 11. D, E, BDNF levels in the retinas (D) and vitreous (E) from WT and P301S mice, determined by ELISA. The bars represent the average BDNF level ± SEM. **p < 0.01, Mann–Whitney rank-sum test. In D, WT, n = 10; P301S, n = 9. In E, n = 8 per group. F, Vitreal levels of BDNF in WT mice injected intravitreally with TTX or vehicle, determined by ELISA. Data represent vitreal concentrations (nanograms per milliliter) ± SEM, n = 12 per group. *p < 0.05, Mann–Whitney rank-sum test.
To determine whether there were changes in BDNF secretion, we measured vitreal levels of the protein. In P301S mice, the concentration of BDNF in the vitreous was reduced by 72% compared with WT (Fig. 2E), indicating a strong impairment of BDNF release. One possible explanation for the reduction in vitreal BDNF levels is impaired RGC activity. To test this hypothesis, we inhibited retinal activity in WT mice through intravitreous injection with the sodium channel blocker TTX (Miura et al., 2009) and measured BDNF in the vitreous 2.5 h later. BDNF levels were significantly reduced in TTX-injected mice (Fig. 2F), suggesting that impaired RGC activity explains the reduced vitreal levels of BDNF in P301S mice.
TrkB receptor is upregulated in RGCs of P301S transgenic mice
To examine how tauopathy affects BDNF signaling in P301S RGCs, we next analyzed the expression of the BDNF receptor TrkB, and the related neurotrophin receptors P75NTR and TrkA. In the retina, TrkB is mainly expressed in RGC and inner plexiform layers (Grishanin et al., 2008; data not shown). In P301S mice, TrkB protein levels were significantly upregulated compared with WT mice (Fig. 3A,B), while P75NTR and TrkA were unchanged (Fig. 3A,B). To determine whether TrkB upregulation was mediated through an effect on gene expression, we evaluated TrkB mRNA levels by quantitative real-time PCR. mRNA levels were similar in P301S (1.1 ± 0.1) and WT (1.0 ± 0.1; p = 0.73, Student's t test) retinas, indicating unchanged TrkB gene transcription.
Figure 3.

TrkB receptor is uncoupled from ERK signaling in the P301S retina. A, Western blot analysis of the TrkB, TrkA, and P75NTR receptors in WT and P301S retinas. Tubulin was analyzed as loading control. The asterisk indicates the band of truncated TrkB. Each lane corresponds to retinal lysate from one mouse. B, Quantitative analysis of the protein levels of TrkB, P75NTR, and TrkA receptors in the retinas of WT and P301S mice. Values were normalized to tubulin. Bars represent the average ratio ± SEM. **p < 0.01, Student's t test. For TrkB: WT, n = 12; P301S, n = 13; for p75NTR and TrkA: n = 6 per group. C, Western blot analysis of the p[Y816]TrkB, TrkB, phosphorylated PLCγ (pPLCγ), and PLCγ in WT and P301S retinas. Tubulin was analyzed as loading control. The asterisk indicates the band of truncated TrkB. Each lane corresponds to retinal lysate from one mouse. D, Phosphorylation of TrkB on Y816 in the retinas of WT and P301S mice. p[Y816]TrkB and TrkB levels were analyzed by Western blot and normalized on total receptor expression. Bars represent the average ratio ± SEM. n = 7 per group. E, F, Protein levels of total PLCγ (E) and pPLCγ (F) in WT and P301S retinas. Levels of PLCγ and pPLCγ were analyzed by Western blot. PLCγ was normalized on tubulin. pPLCγ was normalized on the expression of the unphosphorylated protein. Bars represent the average ratio ± SEM. n = 7 per group. G–J, P301S and WT mice were injected intravitreously with 2 μg of BDNF or BSA; retinas were dissected 10 min later and analyzed by Western blot as described in Materials and Methods. G, Western blot analysis of the p[Y516]TrkB, TrkB, pERK1/2, and ERK1/2 in WT and P301S retinas. Tubulin was analyzed as loading control. The asterisk indicates the band of truncated TrkB. Each lane corresponds to retinal lysate from one mouse. H, Phosphorylation of TrkB on Y516 in retinas of P301S and WT mice in basal and BDNF-stimulated conditions. Phospho-TrkB was normalized to total TrkB. Box plots of the ratio values are shown. I, J, Phosphorylation of ERK1 (I) and ERK2 (J) in retinas of P301S and WT mice in basal and BDNF-stimulated conditions. Phospho-ERK1/2 values were normalized to total ERK1/2 proteins. Box plots of the ratio values are shown. In H–J, values of individual mice are overlaid on each plot (gray circles). In each box, median (white line) and mean (dotted gray line) values are indicated. In H–J: *p < 0.05, **p < 0.01 two-way ANOVA followed by multiple comparison with the Holm–Sidak method. In H: P301S, n = 10; WT, n = 14. In I and J: n = 10 per group.
We next asked whether TrkB upregulation could be caused by impaired retinal activity. To test this hypothesis, we intravitreally injected WT mice with TTX to inhibit retinal activity and, 2.5 h later, dissected the retinas and analyzed TrkB levels by Western blot. TrkB levels were unchanged in TTX-treated retinas (TrkB/tubulin, average of saline injected retinas: saline, 0.6 ± 0.1; TTX, 0.5 ± 0.1), suggesting that impaired RGC activity per se does not acutely affect TrkB levels in RGCs.
TrkB receptor is uncoupled from downstream ERK1/2 signaling in the P301S transgenic retina
To investigate whether higher levels of TrkB functionally compensate for lower levels of BDNF ligand, or whether signaling is impaired, we examined TrkB's basal activation state and its ability to activate upon BDNF stimulation. TrkB signals through two distinct pathways. One pathway is activated by phosphorylation of TrkB on tyrosine 816 (Y816) and is mediated by phospholipase Cγ (PLCγ) and calcium elevation, while the second is activated by phosphorylation on tyrosine 516 (Y516) and leads to activation of ERK1 and ERK2 (Minichiello, 2009). To investigate the basal activity of the PLCγ-mediated pathway, we first examined the phosphorylation of TrkB on Y816 (p[Y816]TrkB) and the levels of PLCγ phosphorylation on Y783 (p[Y783]PLCγ), which is indicative of its activation (Choi et al., 2007). There was no significant difference in the basal levels of p[Y816]TrkB in the retinas of P301S and WT mice (Fig. 3C,D). Furthermore, the retinal expression of PLCγ (Fig. 3C,E) and levels of activated PLCγ (Fig. 3C,F) were similar, indicating that this pathway is not significantly altered in P301S mice.
We next investigated TrkB signaling through ERK1/2. We found that phosphorylation of Y516 on TrkB (p[Y516]TrkB) was significantly reduced in naive P301S retinas (p[Y516]TrkB/TrkB, 0.6 ± 0.1) compared with WT retinas (1.0 ± 0.01; p < 0.01, Mann–Whitney rank-sum test, n = 6 per group), while basal levels of ERK1/2 were unchanged (ERK1/tubulin: WT, 0.18 ± 0.05; P301S, 0.13 ± 0.05; p = 0.46, Mann–Whitney rank-sum test, n = 7. ERK2/tubulin: WT, 0.82 ± 0.03; P301S, 0.78 ± 0.09; p = 1.0, Mann–Whitney rank-sum test, n = 7). To determine the level to which TrkB activates ERK1/2 signaling upon ligand binding, we stimulated retinas by intravitreal injections of BDNF or an equal amount of BSA as a control. To elicit a robust response of the receptor and highlight any subtle differences between genotypes, we used 2 μg of BDNF. This dose effectively rescues the neurodegenerative phenotype and retinal dysfunction in a mouse model of glaucoma (Domenici et al., 2014) and is consistent with doses previously used in the majority of studies on neuroprotective properties of BDNF in the retina of rodents (Khalin et al., 2015). Ten minutes after intravitreous injection, mice were killed and retinas dissected for Western blot analysis of p[Y516]TrkB and ERK1/2 phosphorylation (Fig. 3G). In BSA-injected P301S retinas, p[Y516]TrkB was reduced (Fig. 3G,H), while phospho-ERKs were unchanged compared with WT (Fig. 3G,I,J). In WT mice, BDNF stimulation increased the phosphorylation of [Y516]TrkB by ∼69% (Fig. 3G), and the phosphorylation of ERK1 and ERK2 by ∼8-fold and ∼4-fold, respectively (Fig. 3G,I,J). However, in P301S mice, BDNF stimulation did not cause any significant activation of TrkB signaling or phosphorylation of ERK1/2. In fact, levels of p[Y516]TrkB and phospho-ERK1/2 were similar in P301S retinas stimulated with BDNF or BSA (Fig. 3G,H–J). Overall, these results indicate that TrkB signaling through ERK1/2 is specifically impaired in P301S retinas and that the signaling machinery cannot compensate for ligand deficiency.
Inhibition of TrkB/ERK signaling impairs RGC activity
We next investigated whether defective TrkB signaling per se reduces RGC activity. To inhibit TrkB in WT mice, we treated mice systemically with the TrkB inhibitor ANA12 (Cazorla et al., 2011b). We first confirmed that systemic administration of ANA12 inhibits TrkB signaling in the retina. Mice were injected intraperitoneally with ANA12 or vehicle and, 3.5 h later, retinas were challenged by intravitreal BDNF or BSA for a further 30 min. Retinas were dissected and processed to analyze p[Y516]TrkB. Consistent with previous findings (Cazorla et al., 2011b), ANA12 selectively inhibited BDNF-stimulated TrkB receptor activation in the WT retina (Fig. 4A), without affecting NGF-stimulated phosphorylation of TrkA (Fig. 4B). Using pERG, we therefore examined whether inhibition of TrkB by ANA12 treatment for 4 h affected RGC activity in WT mice. ANA12 administration significantly decreased by 30% the peak amplitude of the P1 wave (Fig. 4C). Despite a trend toward increasing P1 peak latency, ANA12 did not significantly change P1 peak latency (Fig. 4D). To verify the block of TrkB signaling, after ERG recording, we analyzed phosphorylated ERKs (pERKs) in the retinas of treated mice. In mice treated with ANA12, pERK levels were significantly reduced (Fig. 4E), suggesting that TrkB inhibition prevents light-induced ERK activation. To further explore the role of ERKs in visual response, we injected the ERK inhibitor PD0325901 or vehicle in the vitreous of WT mice and analyzed RGC activity by pERG. Vehicle injection alters neither peak amplitude (5.0 ± 0.9 μV) nor peak latency (88.2 ± 14.1 ms) of P1 compared with noninjected mice (P1 peak amplitude, 5.4 ± 0.4 μV; p = 0.46; P1 peak latency, 91.5 ± 6.1 ms, p = 0.54, n = 9). In eyes injected with PD0325901, RGC response was significantly altered. PD0325901 did not affect P1 peak latency (Fig. 4G), but significantly reduced the peak amplitude of P1 (Fig. 4F). We also analyzed levels of pERKs by Western blot. Retinal levels of pERKs were significantly reduced by PD0325901 injection (Fig. 4H), confirming the inhibition of ERKs.
Figure 4.
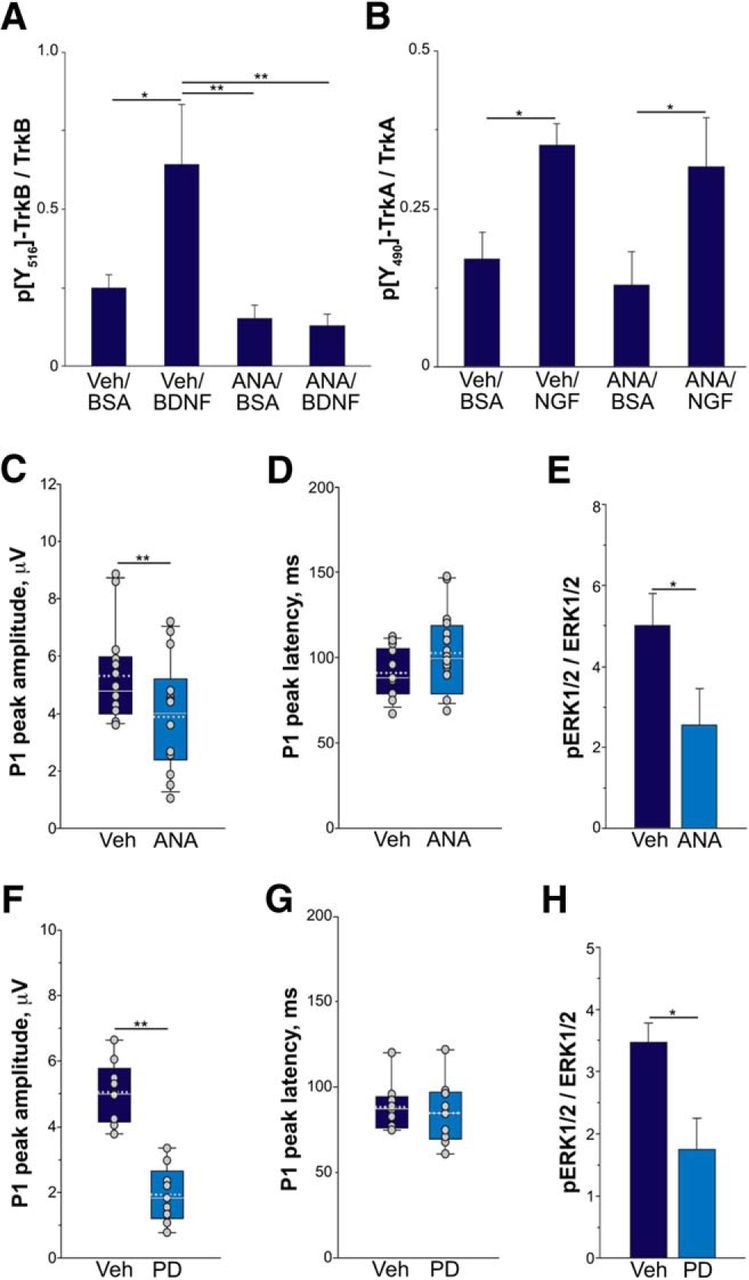
Inhibition of TrkB and ERK reduces RGC activity. A, B, WT mice were injected intraperitoneally with 1 mg/kg ANA12 (ANA) or vehicle (VEH). BDNF, NGF, or BSA were injected in the vitreous 3.5 h later. After 30 min, retinas were dissected and p[Y516]TrkB or p[Y490]TrkA levels were analyzed by Western blot (A) or ELISA (B) as described in Materials and Methods and normalized to total TrkB or TrkA expression. Bars represent the average ratio ± SEM. *p < 0.05, **p < 0.01, two-way ANOVA followed by Holm–Sidak multiple-comparison test. C, D, RGC activity of WT mice after inhibition of TrkB with ANA12. WT mice were injected intraperitoneally with 1 mg/kg ANA12 or vehicle and 4 h later were anesthetized and subjected to pERG. Box plots of peak amplitude (C) or latency (D) of P1 wave in WT mice treated with VEH (n = 14) or ANA12 (n = 16) are shown. Average values of individual mice are overlaid on each plot (gray circles). In each box, median (white line) and mean (dotted gray line) values are indicated. **p < 0.01, Mann–Whitney rank-sum test. E, pERK levels after ANA12 injection. pERKs were analyzed by Western blot and normalized to total ERK expression. Bars represent the average ratio ± SEM. *p < 0.05, Student's t test. F, G, RGC activity of WT mice after inhibition of ERKs. WT mice were injected with 20 nm PD0325901 (PD) or VEH in the vitreous and 1 h later were anesthetized and subjected to pERG. F, G, Box plots of peak amplitude (F) or latency (G) of P1 wave in WT mice treated with VEH or PD are shown. Average values of individual mice are overlaid on each plot (gray circles). In each box, median (white line) and mean (dotted gray line) values are indicated. **p < 0.01, Student's t test. n = 9 per group. H, pERK levels after PD injection. pERKs were analyzed by Western blot and normalized to total ERK expression. Bars represent the average ratio ± SEM. *p < 0.05, Student's t test.
Together, these results indicate that both TrkB and ERKs are required for full RGC activity, suggesting that reduced TrkB/ERK signaling likely contributes to retinal dysfunction in P301S mice.
RGC activity and TrkB expression are preserved in young P301S mice
To investigate the relationship of retinal activity and TrkB expression with tau inclusions, we next examined how these vary with the progression of tauopathy. In P301S retinas, occasional tau inclusions positively stained by the Congo red analog FSB were detected as early as 7–8 weeks of age (Gasparini et al., 2011; Schön et al., 2012). The number of inclusions increases in a stepwise manner, reaching steady-state values at 3 months of age (number of RGCs with FSB-positive tau inclusions/retina: 2.5 months, 10.4 ± 1.8; 3 months, 33.0 ± 1.7; 5 months, 36.7 ± 1.1). To examine how the progression of tau pathology affects retinal function, we assessed pERG response in 1-month-old, 3-month-old, and 5-month-old WT and P301S mice. In P301S mice, the peak amplitude of both P1 and N2 waves were reduced compared with WT only at 5 months of age, while they were preserved at younger ages (Fig. 5A,B). Similarly, the latency of both P1 and N2 was reduced in 5-month-old P301S mice (Fig. 1), while it was unchanged at age 1 month (average ± SD: P1: WT, 92.8 ± 12.4 ms; P301S, 104.8 ± 10.9 ms; p = 0.09, Student's t test. N2: WT, 250.8 ± 14.3 ms; P301S, 250.4 ± 24.8 ms; p = 0.97, Student's t test) and 3 months (average ± SD: P1: WT, 82.2 ± 6.9 ms; P301S, 92.5 ± 16.8 ms; p = 0.15, Student's t test. N2: WT, 242.9 ± 17.1 ms; P301S, 241.4 ± 13.9 ms; p = 0.87, Student's t test). We next examined TrkB levels in relation to tauopathy progression. Similar to retinal function, changes in TrkB were detected only in 5-month-old transgenic mice: TrkB was upregulated in 5-month-old P301S mice and was normal at 1 month and 3 months of age (Fig. 5C,D). These results indicate that retinal dysfunction and changes in TrkB levels coincide in time and occur after a steady state of tau inclusions has already developed.
Figure 5.

RGC activity and TrkB expression are preserved in the early stages of tauopathy. RGC activity and TrkB expression were analyzed in P301S and WT mice at preinclusion (1 month of age), onset (3 months of age), and full-blown stages (5 months of age) of retinal tauopathy as described in Materials and Methods. A, B, Peak amplitude of P1 (A) and N2 (B) waves in WT and P301S mice as detected by pERG. Bars represent the average amplitude ± SEM. *p < 0.05, Student's t test. C, Western blot analysis of the TrkB in retinas of WT and P301S mice at 1, 3, and 5 months of age. Tubulin was analyzed as loading control. The asterisk indicates the band of truncated TrkB. Each lane corresponds to retinal lysate from one mouse. D, TrkB protein expression in the retina of 1-month-old, 3-month-old, and 5-month-old P301S and WT mice. TrkB was analyzed by Western blot and normalized to tubulin. Bars represent the average ratio ± SEM. **p < 0.01, Student's t test. In all panels, numbers in brackets indicate the number of animals used per group.
Tauopathy levels drive TrkB upregulation
Recent findings indicate that tau pathology spreads in a hierarchical manner among anatomically connected areas (Kfoury et al., 2012; de Calignon et al., 2012; Ahmed et al., 2014; Hyman, 2014). Here, we took advantage of this self-propagating behavior to accelerate retinal tauopathy in young P301S transgenic mice and investigate how tauopathy specifically relates to the changes of retinal activity and TrkB levels. To enhance tau pathology, we injected brain extract from 5-month-old P301S mice (P301SBE) in the superior colliculus (SC) of P301S mice at 1.5 months of age (Fig. 6A,B). Six weeks later, we examined tau pathology in the retina, ON, and brain of injected and not-injected (NI) mice. Changes in the content of total tau and phospho-tau were detected only in anatomically connected areas. Specifically, in P301SBE-injected transgenic mice, levels of total tau protein were significantly increased in the ON (Fig. 6F,G). In the SC and retina, although there was a trend toward an increase (+38 and +50%, respectively), the difference in total tau was not statistically significant (Figs. 6C,D, 7A,B). In contrast, tau phosphorylation was significantly enhanced in all three connected areas in P301SBE-injected transgenic mice. In fact, levels of AT8-positive phospho-tau were increased by >50-fold in the SC (Fig. 6C,E), 45-fold in the ON (Fig. 6F,H), and sixfold in the retina (Fig. 7C). Immunohistochemistry also showed increased phospho-tau in the SC and visual cortex of mice injected with P301SBE (Fig. 6J,K). Levels of tau and phospho-tau did not vary in the cerebellum (Fig. 6I), which is not anatomically connected with the SC. These results indicate that 6 weeks after P301SBE injection in the SC, tau pathology worsens in anatomically connected areas of the visual system of 3-month-old P301S mice. This method can therefore be used to experimentally manipulate levels of tau pathology to study tauopathy-dependent molecular changes.
Figure 7.

Increased levels of tau induces upregulation of TrkB in the retina. WTBE or P301SBE were injected in the SC (2.92 mm behind bregma) of P301S mice at 1.5 months of age and retinas were analyzed 6 weeks later. A, Western blot analysis of TrkB, tau, and phospho-tau protein in retinas of 3-month-old mice injected with WTBE, with P301SBE, or not injected (NI). Total tau and phospho-tau were detected using the Tau-5 and AT8 monoclonal antibodies, respectively. Actin was analyzed as a loading control. B, C, Quantitative analysis of total tau (B) and phospho-tau (C, pTau) in the retinas of 3-month-old P301S mice injected with WTBE, with P301SBE, or NI. Values were normalized to actin (B) or total tau (C). Data are expressed as a percentage of NI and bars represent the average ± SEM. *p < 0.05, Kruskal–Wallis one-way ANOVA on ranks followed by the pairwise multiple-comparison procedure according to Dunn's method. D, Quantitative analysis of TrkB in the retinas of 3-month-old P301S mice injected with WTBE, with P301SBE, or NI. E, F, Regression analysis of levels of TrkB and total tau (E; R2 = 0.64; adjusted R2 = 0.62; p < 0.01) and TrkB and phospho-tau detected by AT8 antibody (F; R2 = 0.73; adjusted R2 = 0.71; p < 0.01) in retinas from mice injected with P301SBE. In B–F: WTBE, n = 10; P301SBE, n = 16; NI, n = 12. G, pERG response in transgenic mice injected with WTBE, with P301SBE, or NI. Curves represent the respective average response ± SD. H, P1 peak latency. I, P1 peak amplitude. J, Response amplitude measured at the median P1 peak latency of NI mice. Graphs in H–J represent average ± SD. In G–J, WTBE, n = 7; P301SBE, n = 9; NI, n = 8. *p < 0.05, **p < 0.01, Kruskal–Wallis ANOVA on ranks followed by pairwise Student–Newman–Keuls multiple-comparison method.
We next examined the retinal expression of TrkB as a function of tau content. In transgenic mice injected with P301SBE, retinal levels of TrkB were increased by 50% (Fig. 7A,D). To investigate the relationship between levels of TrkB and tau, we performed regression analyses. We found that retinal levels of TrkB significantly varied in direct proportion with those of total tau (Fig. 7E) and of AT8-positive phospho-tau (Fig. 7F), indicating that the TrkB upregulation is likely dependent on tau levels.
To test whether increasing the levels of WT murine tau is sufficient to enhance tauopathy and TrkB expression in the retina, we injected 1.5-month-old P301S transgenic mice with brain extract from WT mice (WTBE). Six weeks later, levels of total tau were unchanged in SC (Fig. 6C,D), ON (Fig. 6F,G), and retina (Fig. 7A,B) of WTBE-injected transgenic mice. While levels of total tau and phospho-tau were not significantly changed in the SC or ON (Fig. 6C–H), there was a significant increase in phospho-tau in the retina of WTBE-injected transgenic mice (Fig. 7C), similar to that achieved in the retina of sham-injected transgenic mice (AT8/tau ratio: sham, 11.5 ± 1.8; WTBE, 11.2 ± 2.0). However, levels of TrkB in the retina were not significantly affected in WTBE-injected (Fig. 7D) and sham-injected (TrkB/actin: NI, 0.9 ± 0.1; sham, 0.8 ± 0.1) transgenic mice compared with NI, suggesting that altered phosphorylation of murine tau alone does not affect TrkB levels.
Tauopathy levels drive RGC activity dysfunction
To examine the effects of accelerating tau pathology on RGC function, we analyzed pERG response in transgenic mice injected with either WTBE or P301SBE and in NI P301S transgenic mice. We found that the pERG response was significantly slower in P301SBE-injected transgenic mice compared with WTBE-injected or NI transgenic mice (Fig. 7G,H) and both P1 (Fig. 7G,H) and N2 (latency ± SD: WTBE, 213.0 ± 16.0 ms; P301SBE, 290.3 ± 46.2 ms; p < 0.05, Mann–Whitney rank-sum test) waves peaked at increased latency. Consistently, the mean P1 amplitude of the pERG response measured at the median latency of NI (i.e., 93.5 ms) was significantly reduced in P301SBE-injected transgenic mice compared with both NI and WTBE-injected mice (Fig. 7J). Despite a trend toward reduction, the peak amplitude of P1 was not significantly different from those of NI and WTBE-injected mice (Fig. 7I). These results suggest that levels of tau pathology exceeding a certain threshold are required to significantly alter RGC activity in the P301S mouse, mainly affecting the response rising time.
TrkB–fyn interaction is altered in the P301S retina
We next examined the mechanisms by which tau pathology promotes TrkB upregulation and impairs signaling. Through binding microtubules, tau regulates both anterograde and retrograde axonal transport in vitro (Dixit et al., 2008). It has been also shown that the N terminus of tau binds to dynactin p150, a subunit of the dynein retrograde motor complex (Magnani et al., 2007). This motor protein is involved in the trafficking of neurotrophin receptors, including TrkB (Deinhardt et al., 2006), and accumulates at tau-rich sites in axons of retinal explants from adult P301S mice (Magnani et al., 2007). We therefore hypothesized that the tau-dependent uncoupling of TrkB from its downstream signaling pathway could be due to receptor mislocalization.
To test this hypothesis, we first performed immunofluorescence analysis of TrkB and phospho-tau on retinal sections from 5-month-old WT and P301S mice. In P301S retinas, phospho-tau immunoreactivity is detected in all RGCs, but the signal intensity varies among cells: the brightest signal is indeed detected in the subset of RGCs that bears filamentous inclusions of phospho-tau with β-sheet secondary structure (Gasparini et al., 2011). In WT retinas, TrkB immunoreactivity was evenly distributed in the inner plexiform layer, where RGC dendrites are located, in the RGC layer, which contain RGC cell bodies, and in the nerve fiber layer that is formed by RGC axons (Fig. 8A). Consistent with biochemical findings, TrkB immunoreactivity was more intense in RGCs of P301S than those of WT mice. Specifically, in P301S retinas, the TrkB signal was enhanced in the RGC soma, where it colocalized with phospho-tau aggregates (Fig. 8Bb). Further, in RGCs bearing the highest amounts of phospho-tau, TrkB accumulated both in soma and dystrophic dendrites (Fig. 8Bb′). To investigate whether TrkB accumulates in tau inclusions, we isolated insoluble tau filaments from the brains and retinas of 5-month-old WT and P301S mice by sarkosyl extraction. Similarly to transgenic retinas, TrkB levels were increased in total lysates of P301S brains (Fig. 8C,D). In the sarkosyl-insoluble fraction, TrkB was present in similar amounts in WT and P301S transgenic brains (Fig. 8C,D) or retinas (data not shown), arguing against cosegregation of the receptor with filamentous tau aggregates.
Figure 8.

TrkB–fyn interaction is altered in P301S retina. A, B, Representative maximal projections of z-stack confocal images of WT (A) and P301S (B) retinas immunostained with antibodies against TrkB (A, B; green), human tau (A, Bb), and phospho-tau (AT8; Bb′, red). Nuclei were counterstained with Hoechst-33342 (blue). Scale bars, 25 μm. Arrows and arrowheads indicate RGC soma and axons, respectively. Open triangles indicate dystrophic RGC dendrites containing phospho-tau aggregates. Bb, Human tau and TrkB colocalization in the soma of P301S RGCs. Bb′, Accumulation of TrkB in a typical P301S RGC containing mature filamentous aggregates of phospho-tau. C, Western blot analysis of TrkB and phospho-tau in the total brain lysate (BL) and sarkosyl-insoluble (SI) fraction of brain from WT and P301S mice at 5 months of age. Phospho-tau was detected using the PHF-1 monoclonal antibody. The asterisk indicates the band of truncated TrkB. D, Quantitative analysis of TrkB in total BL and SI fraction of WT and P301S brain. Levels of TrkB were analyzed by Western blot (C). For BLs, TrkB levels are normalized over actin (P301S, n = 6; WT, n = 7). For the SI fraction, densitometric values are normalized over levels in WT and bars represent average ± SEM. P301S, n = 12; WT, n = 12. A.U., Arbitrary units. E, Western blot and quantitative analyses of fyn in total retina lysate of WT and P301S mice at 5 months of age. Fyn levels were normalized over tubulin. n = 12 per group. F, Coimmunoprecipitation of tau with fyn. Lysates (input) were immunoprecipitated (IP) using anti-fyn antibody or incubated with protein G agarose (PAG) beads alone as control. Samples were analyzed by Western blot for fyn and tau. Two different exposures of the same Western blot for tau are shown. Densitometry of fyn and tau Western blot from two independent coimmunoprecipitations is shown in the graph. G, Western blot analysis of pY18-tau, total tau, and actin. Total tau was detected using the Tau-5 monoclonal antibody. H, Quantitative analysis of pY18-tau in the retinas of 5-month-old P301S mice. pY18-tau values were normalized to total tau. Bars represent the average ratio ± SEM. **p < 0.01, Mann–Whitney rank-sum test. WT, n = 11; P301S, n = 10. I, J, Representative maximal projections of z-stack confocal images of WT (I) and P301S (J) retinas immunostained with antibodies against pY18-tau (red) and fyn (green). Nuclei are counterstained with DAPI (blue). Scale bars, 25 μm. Jj, pY18-tau and fyn colocalization in P301S RGCs. Jj′, Typical degenerating P301S RGC filled up with filamentous aggregates of phospho-tau. Arrows and arrowheads indicate RGC soma and axons, respectively. Open triangles indicate pY18-tau/fyn colocalization in dystrophic RGC dendrites. In A, B, I, and J: NFL, Nerve fiber layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
TrkB localization and signaling cascade depends on the interaction with the SH2 domain of fyn (Iwasaki et al., 1998; Pereira and Chao, 2007; Zonta and Minichiello, 2013), which also binds phosphorylated and mutant tau (Bhaskar et al., 2005; Usardi et al., 2011). We therefore investigated whether tau–fyn interaction was altered in P301S retinas. In agreement with previous findings (Lee et al., 2004; Ittner et al., 2010), tau coimmunoprecipitates with fyn in both WT and P301S retinas. The total levels of fyn were similar in total retinal lysates of both WT and P301S mice (Fig. 8E). However, the amount of tau associated with fyn was greater in P301S than in WT retinas (Fig. 8F). Consistent with an increased interaction of the two proteins, we found that in P301S retinas, tau was phosphorylated on tyrosine Y18 (pY18-tau; Fig. 8G,H), a residue phosphorylated by fyn upon binding (Lee et al., 2004). Fyn localization was also altered in transgenic retinas. While in WT retinas fyn mainly localized in RGC axons (Fig. 8I), in P301S retinas fyn also localized in RGC soma and partially colocalized with pY18-tau immunoreactivity (Fig. 8Jj,Jj′).
Overall, these results indicate that in P301S retinas, tau pathology is associated with TrkB accumulation in RGC soma and dystrophic dendrites, enhanced tau–fyn interaction, and fyn-mediated tau phosphorylation.
Discussion
Our findings demonstrate that in the retina of 5-month-old P301S mice, functional changes in neuronal activity are accompanied and possibly mediated by tau-induced BDNF signaling impairment. The activity of RGCs and retinal acuity are significantly reduced in P301S mice, while the overall function of the outer retina is preserved. Moreover, in P301S mice, the activity-dependent secretion of BDNF in the vitreous is significantly decreased, likely due to diminished RGC activity. In P301S retinas, despite being selectively upregulated, TrkB receptors are uncoupled from their downstream signaling through ERK1/2. Finally, we show that impairment of both retinal function and TrkB upregulation stem from tau pathology and that tau-dependent TrkB signaling deficit possibly mediates tau-induced neuronal dysfunction.
At 5 months of age, P301S tau transgenic mice accumulate hyperphosphorylated tau and develop tau filamentous inclusions in a small subset of RGCs. These changes neither alter the overall anatomy of the retina—inner or outer—nor induce loss of RGCs (Gasparini et al., 2011), recapitulating pathological alterations of early-stage tauopathy. Here, we find that this mild pathological phenotype is sufficient to impair retinal activity. Indeed, consistent with the anatomical distribution of retinal tau pathology in the P301S mouse, the activity of RGCs is selectively reduced, while the outer retina is fully functional. This demonstrates that PR function and synaptic transmission to bipolar cells are not compromised in transgenic mice and the reduced pERG response is not a secondary effect due to visual signaling loss in the outer retina.
The impaired RGC activity seems directly related to reaching a certain threshold of tau pathology and consequent TrkB dysfunction in RGCs. In P301S mice, retinal activity is normal at 1 and 3 months of age and declines at 5 months. Similarly, TrkB levels are upregulated only at 5 months of age. Accelerating tau pathology by injection of P301SBE, but not WTBE, in the SC of 1.5-month-old transgenic mice significantly impairs retinal activity at 3 months of age, slowing RGC response to pattern light stimuli. Again, retinal functional changes induced by P301SBE are accompanied by upregulation of TrkB. Thus, in P301S mice, tau-induced changes of TrkB and retinal function are concurrent, suggesting a tight link between defective TrkB signaling and RGC dysfunction. This view is also supported by results obtained using the TrkB inhibitor ANA12. In WT mice, ANA12 significantly reduces RGC response to pattern light stimuli, reproducing to some extent the retinal dysfunction of P301S mice, and suggesting that TrkB signaling contributes to mouse visual response. However, despite previous (Cazorla et al., 2011b) and current results indicating that ANA12 is selective for TrkB, we cannot exclude the possibility that some ANA12 actions may be mediated by non-Trk-mediated mechanisms because an extensive receptor interaction screening for ANA12 is still missing.
Compelling evidence gathered in the past two decades demonstrates that BDNF/TrkB signaling acts as modulator of neuronal activity and synaptic plasticity in the brain (Park and Poo, 2013) through presynaptic and postsynaptic molecular changes, including enhanced neurotransmitter release, expression and activity of neurotransmitter receptors, modulation of ion channels, and, in the long term, transcriptional effects and structural remodeling of axons, dendrites, and dendritic spines (Lai et al., 2012; Mariga et al., 2015). Consistently, our results indicate that TrkB contributes to the maintenance of physiological retinal activity and suggest that reduced TrkB signaling impairs the visual response in the mouse, likely through an ERK-mediated mechanism.
There is evidence that ERKs, in addition to controlling proliferation and differentiation during development, play a key role in modulating synaptic plasticity in mature neurons (Thomas and Huganir, 2004). For example, in the visual system, ERK activation is required for visual cortical plasticity (Di Cristo et al., 2001), experience-dependent plasticity, and visual recognition memory (Boggio et al., 2007; Silingardi et al., 2011). Further, ERKs regulate several synaptic targets in the adult brain (Thomas and Huganir, 2004), including NMDA and AMPA receptors that have been demonstrated to be also altered by soluble species and phosphorylated forms of tau (Hoover et al., 2010; Ittner et al., 2010). ERKs also mediate several of the neuronal and synaptic mechanisms of BDNF/TrkB signaling, including enhancement of neurotransmitter release (Jovanovic et al., 2000), modulation of Nav and VGCC ion channels (Lesser and Lo, 1995; Woodall et al., 2008), gene expression (Gaiddon et al., 1996), and structural synaptic plasticity (Alonso et al., 2004; Takei et al., 2004). Within this context, we now report that ERKs are also required for the physiological response of mouse RGCs. In WT mice, reducing ERK activation by TrkB inhibition with ANA12 or blocking ERKs by the specific ERK inhibitor PD0325901 significantly decreases light-induced RGC activity. Further, in the P301S mouse, defective ERK signaling is associated with impaired visual response. These results indicate that ERK signaling is required for proper visual response in the mouse. Future investigation is warranted to dissect the molecular, transcriptional, or structural mechanisms affected in the early stage of tauopathy and their link with impaired neurotrophin signaling through ERKs.
Neuropathological studies in human brain samples indicate that BDNF signaling is altered in AD and non-AD tauopathies, both at the ligand and receptor levels. BDNF mRNA and protein levels are decreased in some tauopathies, including Pick's disease, corticobasal degeneration (Belrose et al., 2014), and AD (Ferrer et al., 1999). In the AD brain, TrkB receptors' immunoreactivity is also significantly altered in tangle-bearing neurons (Ferrer et al., 1999). Within this context, our work has yielded key insights into the early effects of tauopathy on this signaling pathway in relation to neuronal function. We show that in P301S mice, despite normal retinal expression and synthesis, vitreal levels of BDNF are strongly reduced. This finding apparently diverges from previous work showing that BDNF is not downregulated in the hippocampus of the THY-Tau22 transgenic mouse (Burnouf et al., 2012). Technical and experimental design differences among various studies may account for the discrepancy. First, by taking advantage of the natural compartmentalization of the retina, our study, unlike other studies (Burnouf et al., 2012), distinguishes between different pools of BDNF, i.e., retinal versus secreted, which reveals a selective reduction of secreted BDNF. Further, different stages of tauopathy or the presence of multiple mutations in tau (Schindowski et al., 2006) may affect BDNF expression differently in the brain of the THY-Tau22 transgenic model.
We also show that the reduction of vitreal BDNF likely stems from impaired RGC activity in P301S retina. Accordingly, we find that in WT mice, inhibiting retinal activity by TTX significantly reduces BDNF levels in the vitreous, reproducing the transgenic phenotype. This confirms and extends findings in cultured neurons, which have demonstrated that BDNF secretion depends on neuronal activity (Matsumoto et al., 2008); in the retina in vivo, the secretion of BDNF is also modulated by RGC activity.
Further, we gain some insight on the mechanisms by which initial tauopathy disrupts BDNF signaling at the receptor level. In 5-month-old P301S tau mice, despite being upregulated, retinal TrkB receptors fail to signal through the ERK pathways. In P301S retinas, TrkB receptors accumulate in RGC soma and dystrophic dendrites. TrkB signaling depends on the interaction of the receptor with the tyrosine kinase fyn (Iwasaki et al., 1998; Pereira and Chao, 2007), which also interacts with tau (Lee et al., 2004; Pooler et al., 2012). The tau–fyn interaction is enhanced when tau is phosphorylated (Usardi et al., 2011) or bears specific mutations, including the P301L mutation (Bhaskar et al., 2005). Even though the expression of fyn in WT retinas is similar to that in P301S retinas, the tau–fyn interaction is increased in transgenic retinas and fyn partially colocalizes with phospho-tau. This suggests that excessive tau–fyn binding may prevent the stabilization of other protein complexes, including TrkB, and induce abnormal tau phosphorylation. Consistently, in P301S transgenic retinas, there is robust phosphorylation of tau on Y18, which is typically phosphorylated by fyn and accumulates in paired helical filaments extracted from the human AD brain (Lee et al., 2004).
In conclusion, our results support a model in which early tau pathology impairs TrkB signaling through ERKs. This would result in neuronal dysfunction and reduction of activity-dependent BDNF secretion, starting a deleterious vicious cycle that possibly leads to neurodegeneration at later stages. These findings are clinically relevant from diagnostic and therapeutic perspectives. From a diagnostic viewpoint, the results suggest that retinal examination may be useful for diagnosing AD, even though the eye is not the main area targeted by AD pathology. In fact, the retinas of some, though not all (Ho et al., 2014), AD patients develop amyloid plaques (Koronyo-Hamaoui et al., 2011; Tsai et al., 2014), inclusions of phosphorylated tau (Schön et al., 2012), RGC degeneration (Blanks et al., 1989, 1991, 1996a,b), and retinal dysfunction (Parisi, 2003; Iseri et al., 2006). Interestingly, in a subset of cases, AD manifests with early visual symptoms (Lee and Martin, 2004). Such symptoms are believed to be of cortical origin and posterior cortical atrophy has been proposed as a new clinical entity (Crutch et al., 2012). However, a systematic analysis of AD patients with visual symptoms and their retinal function is still lacking. Further investigation should explore the potential use of noninvasive, clinically available electrophysiological techniques, such as ERG, to detect early pathophysiological changes in this subset of AD cases.
From the therapeutic perspective, clinical trials using BDNF have been conducted in neurodegenerative disorders, such as amyotrophic lateral sclerosis and diabetic neuropathy, with inconclusive results (Ochs et al., 2000; Thoenen and Sendtner, 2002). While BDNF bioavailability might have been an issue in the failure of such trials, other possible reasons for failure include pharmacodynamic aspects, such as possible lack of target engagement. Rather, our data suggest that pathological dysfunction of the TrkB/BDNF pathway occurs at an early stage of tauopathy, preventing activation of the pathway by exogenous BDNF and leading to neuronal dysfunction. This raises concerns about the benefit of BDNF/TrkB-targeting drugs (e.g., 7, 8-dihydroxyflavone; Marongiu et al., 2013) for tau-induced neurodegeneration. Overall, our results underscore the need for a better understanding of the molecular changes associated with early tauopathy stages to support informed therapeutic decisions.
Footnotes
This work was supported by a grant from Compagnia di San Paolo to L.G. and by Istituto Italiano di Tecnologia intramural funds to L.G. We thank Dr. P. Davies, Albert Einstein College of Medicine (New York, NY) for kindly providing MC1 and PHF-1 anti-tau antibodies. We are grateful to Ms. Odetta Antico for technical assistance in immunofluorescence studies.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, Bose S, Clavaguera F, Tolnay M, Lavenir I, Goedert M, Hutton ML, O'Neill MJ. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathologica. 2014;127:667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia S, Silhol M, Moulière F, Meffre J, Höllinger I, Maurice T, Tapia-Arancibia L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–326. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/WNL.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. New Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrose JC, Masoudi R, Michalski B, Fahnestock M. Increased pro-nerve growth factor and decreased brain-derived neurotrophic factor in non-Alzheimer's disease tauopathies. Neurobiol Aging. 2014;35:926–933. doi: 10.1016/j.neurobiolaging.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer's disease. Brain Res. 1989;501:364–372. doi: 10.1016/0006-8993(89)90653-7. [DOI] [PubMed] [Google Scholar]
- Blanks JC, Torigoe Y, Hinton DR, Blanks RH. Retinal degeneration in the macula of patients with Alzheimer's disease. Ann NY Acad Sci. 1991;640:44–46. doi: 10.1111/j.1749-6632.1991.tb00188.x. [DOI] [PubMed] [Google Scholar]
- Blanks JC, Torigoe Y, Hinton DR, Blanks RH. Retinal pathology in Alzheimer's disease. I. Ganglion cell loss in foveal/parafoveal retina. Neurobiol Aging. 1996a;17:377–384. doi: 10.1016/0197-4580(96)00010-3. [DOI] [PubMed] [Google Scholar]
- Blanks JC, Schmidt SY, Torigoe Y, Porrello KV, Hinton DR, Blanks RH. Retinal pathology in Alzheimer's disease. II. Regional neuron loss and glial changes in GCL. Neurobiol Aging. 1996b;17:385–395. doi: 10.1016/0197-4580(96)00009-7. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggio EM, Putignano E, Sassoè-Pognetto M, Pizzorusso T, Giustetto M. Visual stimulation activates ERK in synaptic and somatic compartments of rat cortical neurons with parallel kinetics. PloS ONE. 2007;2:e604. doi: 10.1371/journal.pone.0000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnouf S, Belarbi K, Troquier L, Derisbourg M, Demeyer D, Leboucher A, Laurent C, Hamdane M, Buee L, Blum D. Hippocampal BDNF expression in a tau transgenic mouse model. Curr Alzheimer Res. 2012;9:406–410. doi: 10.2174/156720512800492468. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Arrang JM, Prémont J. Pharmacological characterization of six trkB antibodies reveals a novel class of functional agents for the study of the BDNF receptor. Br J Pharmacol. 2011a;162:947–960. doi: 10.1111/j.1476-5381.2010.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011b;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Ryu SH, Suh PG. On/off-regulation of phospholipase C-gamma 1-mediated signal transduction. Adv Enzyme Regul. 2007;47:104–116. doi: 10.1016/j.advenzreg.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res. 1997;49:71–81. doi: 10.1016/S0169-328X(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–178. doi: 10.1016/S1474-4422(11)70289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Di Cristo G, Berardi N, Cancedda L, Pizzorusso T, Putignano E, Ratto GM, Maffei L. Requirement of ERK activation for visual cortical plasticity. Science. 2001;292:2337–2340. doi: 10.1126/science.1059075. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici L, Origlia N, Falsini B, Cerri E, Barloscio D, Fabiani C, Sansò M, Giovannini L. Rescue of retinal function by BDNF in a mouse model of glaucoma. PloS One. 2014;9:e115579. doi: 10.1371/journal.pone.0115579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera D, Mazzaro N, Canale C, Gasparini L. Resting microglia react to Abeta42 fibrils but do not detect oligomers or oligomer-induced neuronal damage. Neurobiol Aging. 2014;35:2444–2457. doi: 10.1016/j.neurobiolaging.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marín C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Martí E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. doi: 10.1097/00005072-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Gaiddon C, Loeffler JP, Larmet Y. Brain-derived neurotrophic factor stimulates AP-1 and cAMP-responsive element dependent transcriptional activity in central nervous system neurons. J Neurochem. 1996;66:2279–2286. doi: 10.1046/j.1471-4159.1996.66062279.x. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32:419–433. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Gezen-Ak D, Dursun E, Hanaðasý H, Bilgiç B, Lohman E, Araz ÖS, Atasoy IL, Alaylioðlu M, Önal B, Gürvit H, Yýlmazer S. BDNF, TNFα, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer's disease or mild cognitive impairment. J Alzheimers Dis. 2013;37:185–195. doi: 10.3233/JAD-130497. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Grishanin RN, Yang H, Liu X, Donohue-Rolfe K, Nune GC, Zang K, Xu B, Duncan JL, Lavail MM, Copenhagen DR, Reichardt LF. Retinal TrkB receptors regulate neural development in the inner, but not outer, retina. Mol Cell Neurosci. 2008;38:431–443. doi: 10.1016/j.mcn.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Webber DJ, Bilican B, Goedert M, Spillantini MG, Chandran S. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 2010;30:9973–9983. doi: 10.1523/JNEUROSCI.0834-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Ho CY, Troncoso JC, Knox D, Stark W, Eberhart CG. Beta-amyloid, phospho-tau and alpha-synuclein deposits similar to those in the brain are not identified in the eyes of Alzheimer's and Parkinson's disease patients. Brain Pathol. 2014;24:25–32. doi: 10.1111/bpa.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000;57:846–851. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res. 2000;76:347–354. doi: 10.1016/s0169-328x(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT. Tau propagation, different tau phenotypes, and prion-like properties of tau. Neuron. 2014;82:1189–1190. doi: 10.1016/j.neuron.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Iseri PK, Altina° O, Tokay T, Yüksel N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J Neuroophthalmol. 2006;26:18–24. doi: 10.1097/01.wno.0000204645.56873.26. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Gay B, Wada K, Koizumi S. Association of the Src family tyrosine kinase Fyn with TrkB. J Neurochem. 1998;71:106–111. doi: 10.1046/j.1471-4159.1998.71010106.x. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalin I, Alyautdin R, Kocherga G, Bakar MA. Targeted delivery of brain-derived neurotrophic factor for the treatment of blindness and deafness. Int J Nanomedicine. 2015;10:3245–3267. doi: 10.2147/IJN.S77480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54(Suppl 1):S204–S217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KO, Wong AS, Cheung MC, Xu P, Liang Z, Lok KC, Xie H, Palko ME, Yung WH, Tessarollo L, Cheung ZH, Ip NY. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15:1506–1515. doi: 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi S, Sale A, Berardi N, Viegi A, Maffei L, Cenni MC. Retinal functional development is sensitive to environmental enrichment: a role for BDNF. FASEB J. 2007;21:130–139. doi: 10.1096/fj.06-6083com. [DOI] [PubMed] [Google Scholar]
- Landi S, Ciucci F, Maffei L, Berardi N, Cenni MC. Setting the pace for retinal development: environmental enrichment acts through insulin-like growth factor 1 and brain-derived neurotrophic factor. J Neurosci. 2009;29:10809–10819. doi: 10.1523/JNEUROSCI.1857-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AG, Martin CO. Neuro-ophthalmic findings in the visual variant of Alzheimer's disease. Ophthalmology. 2004;111:376–380. doi: 10.1016/S0161-6420(03)00732-2. discussion 380–381. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser SS, Lo DC. Regulation of voltage-gated ion channels by NGF and ciliary neurotrophic factor in SK-N-SH neuroblastoma cells. J Neurosci. 1995;15:253–261. doi: 10.1523/JNEUROSCI.15-01-00253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Grishanin RN, Tolwani RJ, Rentería RC, Xu B, Reichardt LF, Copenhagen DR. Brain-derived neurotrophic factor and TrkB modulate visual experience-dependent refinement of neuronal pathways in retina. J Neurosci. 2007;27:7256–7267. doi: 10.1523/JNEUROSCI.0779-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariga A, Zavadil J, Ginsberg SD, Chao MV. Withdrawal of BDNF from hippocampal cultures leads to changes in genes involved in synaptic function. Dev Neurobiol. 2015;75:173–192. doi: 10.1002/dneu.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu D, Imbrosci B, Mittmann T. Modulatory effects of the novel TrkB receptor agonist 7,8-dihydroxyflavone on synaptic transmission and intrinsic neuronal excitability in mouse visual cortex in vitro. Eur J Pharmacol. 2013;709:64–71. doi: 10.1016/j.ejphar.2013.03.044. [DOI] [PubMed] [Google Scholar]
- Matrone C, Barbagallo AP, La Rosa LR, Florenzano F, Ciotti MT, Mercanti D, Chao MV, Calissano P, D'Adamio L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J Neurosci. 2011;31:11756–11761. doi: 10.1523/JNEUROSCI.1960-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Miura G, Wang MH, Ivers KM, Frishman LJ. Retinal pathway origins of the pattern ERG of the mouse. Exp Eye Res. 2009;89:49–62. doi: 10.1016/j.exer.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs G, Penn RD, York M, Giess R, Beck M, Tonn J, Haigh J, Malta E, Traub M, Sendtner M, Toyka KV. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:201–206. doi: 10.1080/14660820050515197. [DOI] [PubMed] [Google Scholar]
- Parisi V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer's disease. Semin Ophthalmol. 2003;18:50–57. doi: 10.1076/soph.18.2.50.15855. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Pereira DB, Chao MV. The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J Neurosci. 2007;27:4859–4869. doi: 10.1523/JNEUROSCI.4587-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- Pláteník J, Fišar Z, Buchal R, Jirák R, Kitzlerová E, Zvìøová M, Raboch J. GSK3beta, CREB, and BDNF in peripheral blood of patients with Alzheimer's disease and depression. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:83–93. doi: 10.1016/j.pnpbp.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging. 2012;33:431.e427–438. doi: 10.1016/j.neurobiolaging.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Porciatti V. The mouse pattern electroretinogram. Doc Ophthalmol. 2007;115:145–153. doi: 10.1007/s10633-007-9059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porciatti V, Saleh M, Nagaraju M. The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2007;48:745–751. doi: 10.1167/iovs.06-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex TS, Allocca M, Domenici L, Surace EM, Maguire AM, Lyubarsky A, Cellerino A, Bennett J, Auricchio A. Systemic but not intraocular Epo gene transfer protects the retina from light- and genetic-induced degeneration. Mol Ther. 2004;10:855–861. doi: 10.1016/j.ymthe.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Sato K, Higuchi M, Iwata N, Saido TC, Sasamoto K. Fluoro-substituted and 13C-labeled styrylbenzene derivatives for detecting brain amyloid plaques. Eur J Med Chem. 2004;39:573–578. doi: 10.1016/j.ejmech.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Scattoni ML, Gasparini L, Alleva E, Goedert M, Calamandrei G, Spillantini MG. Early behavioural markers of disease in P301S tau transgenic mice. Behav Brain Res. 2010;208:250–257. doi: 10.1016/j.bbr.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, Buée L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön C, Hoffmann NA, Ochs SM, Burgold S, Filser S, Steinbach S, Seeliger MW, Arzberger T, Goedert M, Kretzschmar HA, Schmidt B, Herms J. Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PloS One. 2012;7:e53547. doi: 10.1371/journal.pone.0053547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz N, Schohl A, Ruthazer ES. Activity-dependent transcription of BDNF enhances visual acuity during development. Neuron. 2011;70:455–467. doi: 10.1016/j.neuron.2011.02.055. [DOI] [PubMed] [Google Scholar]
- Silingardi D, Angelucci A, De Pasquale R, Borsotti M, Squitieri G, Brambilla R, Putignano E, Pizzorusso T, Berardi N. ERK pathway activation bidirectionally affects visual recognition memory and synaptic plasticity in the perirhinal cortex. Front Behav Neurosci. 2011;5:84. doi: 10.3389/fnbeh.2011.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JH, Yu JT, Tan L. Brain-derived neurotrophic factor in Alzheimer's disease: risk, mechanisms, and therapy. Mol Neurobiol. 2015;52:1477–1493. doi: 10.1007/s12035-014-8958-4. [DOI] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci. 2002;5(suppl):1046–1050. doi: 10.1038/nn938. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tsai Y, Lu B, Ljubimov AV, Girman S, Ross-Cisneros FN, Sadun AA, Svendsen CN, Cohen RM, Wang S. Ocular changes in TgF344-AD rat model of Alzheimer's disease. Invest Ophthalmol Vis Sci. 2014;55:523–534. doi: 10.1167/iovs.13-12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usardi A, Pooler AM, Seereeram A, Reynolds CH, Derkinderen P, Anderton B, Hanger DP, Noble W, Williamson R. Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J. 2011;278:2927–2937. doi: 10.1111/j.1742-4658.2011.08218.x. [DOI] [PubMed] [Google Scholar]
- Woodall AJ, Richards MA, Turner DJ, Fitzgerald EM. Growth factors differentially regulate neuronal Cav channels via ERK-dependent signalling. Cell Calcium. 2008;43:562–575. doi: 10.1016/j.ceca.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Zonta B, Minichiello L. Synaptic membrane rafts: traffic lights for local neurotrophin signaling? Front Synaptic Neurosci. 2013;5:9. doi: 10.3389/fnsyn.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]