

Abstract
Stress and the major stress hormone corticosterone induce profound influences in the brain. Altered histone modification and transcriptional dysfunction have been implicated in stress-related mental disorders. We previously found that repeated stress caused an impairment of prefrontal cortex (PFC)-mediated cognitive functions by increasing the ubiquitination and degradation of AMPA-type glutamate receptors via a mechanism depending on the E3 ubiquitin ligase Nedd4. Here, we demonstrated that in PFC of repeatedly stressed rats, active glucocorticoid receptor had the increased binding to the glucocorticoid response element of histone deacetylase 2 (HDAC2) promoter, resulting in the upregulation of HDAC2. Inhibition or knock-down of HDAC2 blocked the stress-induced impairment of synaptic transmission, AMPAR expression, and recognition memory. Furthermore, we found that, in stressed animals, the HDAC2-dependent downregulation of histone methyltransferase Ehmt2 (G9a) led to the loss of repressive histone methylation at the Nedd4-1 promoter and the transcriptional activation of Nedd4. These results have provided an epigenetic mechanism and a potential treatment strategy for the detrimental effects of chronic stress.
SIGNIFICANCE STATEMENT Prolonged stress exposure can induce altered histone modification and transcriptional dysfunction, which may underlie the profound influence of stress in regulating brain functions. We report an important finding about the epigenetic mechanism controlling the detrimental effects of repeated stress on synaptic transmission and cognitive function. First, it has revealed the stress-induced alteration of key epigenetic regulators HDAC2 and Ehmt2, which determines the synaptic and behavioral effects of repeated stress. Second, it has uncovered the stress-induced histone modification of the target gene Nedd4, an E3 ligase that is critically involved in the ubiquitination and degradation of AMPA receptors and cognition. Third, it has provided the epigenetic approach, HDAC2 inhibition or knock-down, to rescue synaptic and cognitive functions in stressed animals.
Keywords: AMPA receptors, corticosterone, E3 ubiquitin ligase, histone deacetylase, histone methyltransferase, histone modification
Introduction
Stress has complex and profound impact on the prefrontal cortex (PFC), a key brain region involved in working memory and self-regulatory and goal-directed behaviors (Popoli et al., 2012; McEwen and Morrison, 2013). Chronic or severe stress could trigger or precipitate mental illnesses, including schizophrenia, posttraumatic stress disorder, and bipolar disorder (McEwen, 2002; de Kloet et al., 2005; Arnsten, 2009). Neuroimaging studies have often found PFC hypoactivity in patients with these psychiatric disorders (Callicott et al., 1998; Blumberg et al., 2003; Shin et al., 2005; Francati et al., 2007). Animal studies have discovered that the loss of glutamatergic transmission in PFC neurons induced by repeated stress and glucocorticoid receptor (GR) activation leads to the impairment of PFC-dependent cognitive processes (Yuen et al., 2012; Wei et al., 2014a).
To identify potential therapeutic targets for various stress-related disorders, we needed to find out what could regulate the GR signaling and stress response. Emerging evidence suggests that epigenetic regulation of gene transcription via chromatin remodeling plays a pivotal role in cognitive processes (Borrelli et al., 2008; Fischer et al., 2010; Gräff and Tsai, 2013). A key epigenetic process is the addition and removal of acetyl groups to histone proteins. Acetylation of histone proteins by histone acetyltransferases usually leads to a relaxing of chromatin architecture, which allows transcription factors to interact with specific gene promoters and activate gene transcription. Deacetylation of histone proteins by histone deacetylases (HDACs) typically leads to the opposite effect, condensing chromatin and silencing gene expression.
Altered histone acetylation and transcriptional dysfunction have been linked to a wide range of neurologic and psychiatric disorders, including depression and drug addiction (Tsankova et al., 2007; Abel and Zukin, 2008; Kazantsev and Thompson, 2008). Among the four major HDAC families (Carey and La Thangue, 2006; Chuang et al., 2009), particular attention has been paid to class I (HDAC1, HDAC2, HDAC3, and HDAC8) and II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10) HDACs. Loss of HDAC5 or HDAC2 is associated with the altered histone acetylation and global patterns of gene expression in the nucleus accumbens in animal models of drug addiction and depression, which is found to be important for the maladaptive behavioral changes after chronic exposure to cocaine or emotional stimuli (Renthal et al., 2007; Covington et al., 2009).
The promoter region of HDAC2 contains a well conserved recognition sequence for GR, the glucocorticoid response element (GRE) (Gräff et al., 2012), raising the possibility that GR activation may alter HDAC2 transcription, which affects the expression of neuroplasticity genes epigenetically. Our previous studies have found that GR mediates the stress-induced depression of glutamatergic transmission via the enhanced ubiquitin/proteasome-mediated degradation of GluR1 subunits, which is controlled by the E3 ubiquitin ligase Nedd4 (Yuen et al., 2012), which has been found to play a key role in homeostatic regulation of AMPAR trafficking (Schwarz et al., 2010, Hou et al., 2011). In this study, we sought to find out whether HDAC2 is a potential key regulator for the GR-induced upregulation of Nedd4 expression in PFC and if HDAC2 inhibition could block the detrimental effects of repeated stress.
Materials and Methods
Repeated stress paradigm.
All experiments were performed with the approval of the Institutional Animal Care and Use Committee of the State University of New York at Buffalo. As described in our previous studies on stress (Yuen et al., 2012), Sprague Dawley male rats at the prepubertal age (3–5 weeks, juvenile adolescent), a highly plastic period (Sturman and Moghaddam, 2011), were used. Animals were group housed in cages with a light (6:00 A.M.–6:00 P.M.)/dark (6:00 P.M.–6:00 A.M.) cycle. For repeated restraint stress, rats were placed in air-accessible cylinders for 2 h daily (10:00 A.M.–12:00 P.M.) for 7 d starting at postnatal days 21–23 (p21–p23). The container size was similar to the animal size, which made the animal almost immobile in the container.
Experiments were performed 24–48 h after the termination of repeated stress (restraint, 7 d). For acute stress (forced-swim stress), rats were placed in a cylindrical glass tank (24.5 cm high × 18.5 cm diameter) filled with water to a depth of 20 cm. Rats were forced to swim in warm water (23–25°C) for 20 min and killed 1 h after acute stress (Yuen et al., 2009). Animals were anesthetized by inhaling halothane for a few minutes before decapitation. Controls versus stressed animals were handled in the same way in parallel for all the biochemical and molecular experiments, as well as electrophysiological measurements, which excludes the possibility that the differences between these animals were due to anesthesia.
Primary neuronal culture.
Rat PFC cultures were prepared by modification of methods described previously (Wang et al., 2003; Wei et al., 2014b). Briefly, the PFC was dissected from 18-d-old rat embryos and cells were dissociated using trypsin and titrated through a Pasteur pipette. Neurons were plated on coverslips coated with poly-l-lysine in DMEM with 10% fetal calf serum at a density of 1 × 105 cells/cm2. When neurons attached to the coverslip within 24 h, the medium was changed to Neurobasal medium with a B27 supplement. Two to 3 d after plating, 5 μm AraC was added to inhibit glial growth. Subsequently, half of the medium was changed to a conditional medium once a week. Neurons were maintained for 2–3 weeks before being used for recordings.
Quantitative real-time RT-PCR.
To compare the mRNA levels, quantitative RT-PCR was used. Total RNA was isolated from rat PFC punches using TRIzol reagent (Invitrogen) and treated with DNase I (Invitrogen) to remove genomic DNA. Then, SuperScript III first-strand synthesis system for RT-PCR (Invitrogen) was used to obtain cDNA from the tissue mRNA, followed by treatment with RNase H (2 U/ l) for 20 min at 37°C. Quantitative real-time RT-PCR was performed using the iCycler iQ Real-Time PCR Detection System and iQ Supermix (Bio-Rad) according to the manufacturer's instructions. In brief, GAPDH was used as the housekeeping gene for quantitation of the expression of target genes (HDACs) in samples from control versus stressed rats. Fold changes in the target genes by stress were determined by the following equaton: 2−δ(δCT), where δCT = CT(target) − CT(GAPDH), and δ(δCT) = δCT(stressed) − δCT(control). CT (threshold cycle) is defined as the fractional cycle number at which the fluorescence reaches 10× the SD of the baseline. A total reaction mixture of 25 μl was amplified in a 96-well thin-wall PCR plate (Bio-Rad) using the following PCR cycling parameters: 95°C for 5 min followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 60 s. Primers for rat HDAC1 are as follows: 5′-TTACTACTACGACGGGGATG-3′ (forward); 5′-ACCATAGTTGAGCAGCAAAT-3′ (reverse). Primers for rat HDAC2 are as follows: 5′-ATGGCGTACAGTCAAGGAG-3′ (forward); 5′-CCATTTTTCGGTATAAACCAT-3′ (reverse). Primers for rat HDAC4 are as follows: 5′-CTCCCAAAGCCATCCAGATG-3′ (forward); 5′-GAGAACTGGTGGTCCAAGCG-3′ (reverse). Primers for rat HDAC5 are as follows: 5′-TGGCCTTGGATGGGCATTAG-3′ (forward); 5′-CCGACTCGTTGGGAGAGTTC-3′ (reverse). Primers for rat HDAC6 are as follows: 5′-TGCAGAAGCCAAGCGTTCC-3′ (forward); 5′-GGGTACAGCACCCTTCTTCC-3′ (reverse). Primers for rat Nedd4-1 are as follows: 5′-GGAGCTGGACCTGAGATTTATC-3′ (forward); 5′-TATGTTCCCTCCAGTCGTTCAC-3′ (reverse). Primers for rat Ehmt1 are as follows: 5′-CATAGCAAAAGCAGACACGA-3′ (forward); 5′-ACTTTCCAAGGTTTCCTTTC-3′ (reverse). Primers for rat Ehmt2 are as follows: 5′-TCCAGCATTTCCGCATGAGTGA-3′ (forward); 5′-CAACTGTTCAGCTAGAGCTTCG-3′ (reverse). Primers for rat GAPDH are as follows: 5′-GACAACTCCCTCAAGATTGTCAG-3′ (forward); 5′-ATGGCATGGACTGTGGTCATGAG-3′ (reverse). Quantitative real-time RT-PCR was performed in triple reactions.
Western blotting of nuclear proteins.
Nuclear extracts from rat brains were prepared according to the manufacturer's instructions (Life Technologies) with modifications. Briefly, 10 PFC punches (diameter is 1.5 mm) from fresh rat slices (600 μm) per animal were collected and then homogenized with 500 μl of homogenization buffer (20 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 0.5% NP-40, 1 mm PMSF, with cocktail protease inhibitor). The homogenate was incubated on ice for 15 min and followed by centrifugation at 3000 rpm at 4°C for 10 min. The nuclear pellet was resuspended in 50 μl of nuclear extract buffer (100 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, 10% glycerol, 1 mm PMSF, with cocktail protease inhibitor) and incubated on ice for 30 min with vortexed periodically to resuspend the pellet. After centrifugation, the supernatant for nuclear fractions was collected, boiled in 2 × SDS loading buffer for 5 min, and then separated on 10% SDS-polyacrylamide gels. Western blotting experiments for nuclear proteins were performed with anti-HDAC1 (1:500, sc-7872; Santa Cruz Biotechnology), anti-HDAC2 (1:1000, sc-7899; Santa Cruz Biotechnology), pGR (1:1000, 4161; Cell Signaling Technology), and H3 (1:1000, 4499; Cell Signaling Technology). The level of histone acetylation of was measured by blotting the PFC slices (in 1% SDS lysis buffer) with antibodies against pan-acetylated H3 (1:1000, 06–599; Millipore), H3 (1:1000, 4499; Cell Signaling Technology), and actin (1:1000, sc-1616-R; Santa Cruz Biotechnology).
ChIP.
Briefly, 10 PFC punches (diameter is 1.5 mm) from fresh rat slices (600 μm) per animal were collected, cross-linked with 1% formaldehyde for 12 min, and quenched by the addition of glycine at a final concentration of 0.125 m for 5 min before freezing at −80°C. The 1% SDS lysis buffer containing PMSF and cocktail proteinase inhibitors (200 μl) was added before sample sonication. A Fisher Scientific Sonic Dismembrator Model 300 was used, with sonication at 28% of power with microtip for 10 15 s pulse (30 s pause between pulses) at 4°C while the samples were immersed in an ice-water bath. This procedure resulted in DNA fragment sizes of 0.2–1 Kb. After centrifugation, ∼20% of the supernatant was saved to serve as an input control. To reduce nonspecific background, the supernatant was diluted in ChIP dilution buffer and precleared with 80 μl of salmon sperm DNA/protein A agarose-50% slurry (16–157; Millipore) for 30 min at 4°C with agitation. The precleared supernatant was incubated with antibodies against S211pGR (4161, 1:100 per reaction, corresponding to Ser232 of rat GR; Cell Signaling Technology), H3K9me2 (ab1220, 5 μg per reaction; Abcam), or pan-acetylated H3 (06–599, 5 μg per reaction; Millipore) overnight at 4°C under constant rotation, following by incubation with 60 μl of salmon sperm DNA/Protein A agarose-50% slurry for 1 h at 4°C. Rabbit or mouse IgG was used as a nonspecific control for immunoprecipitation assays. After five washes, bound complex was eluted from the beads by incubating with 250 μl of elution buffer twice at room temperature. After reversing crosslinks in 65°C for 4 h, immunoprecipitated DNA, and input DNA were purified by phenol/chloroform extraction. Purified DNA was subjected to qPCRs with primers against rat HDAC2 promoter GRE region (forward, +200 bp ∼+219 bp relative to transcriptional start site (TSS), 5′-CATGGCGTACAGTCAAGGAG-3′; reverse, +378 bp ∼+395 bp relative to TSS, 5′-GGACCGGAGAGGAGCGAA-3′), Nedd4-1 promoter (Primers at −500 bp relative to TSS: forward, −512 bp ∼−531 bp, 5′-TTGAGGCTCAGCTATGCAGG-3′; reverse, −395 bp ∼−414 bp, 5′-GAAAGCGTGCTCTTGAGGGT-3′; primers at −1500 bp relative to TSS: forward, −1585 bp ∼−1604 bp, 5′-CCAGGCCCATCAATGCTATA-3′; reverse, −1442 bp ∼−1461 bp, 5′-CTAGTGAGAAGACCTGTCTC-3′) or Ehmt2 promoter (primers at −700 bp relative to TSS: forward, −761 bp ∼−780 bp, 5′-AATCTGGCGGCTCAGAACCA-3′; reverse, −602 bp ∼−621 bp, 5′-GGTCCAGAACTGGGGTTGTA-3′; primers at −1500 bp relative to TSS: forward, −1506 bp ∼−1525 bp, 5′-CCATAGATTCTCCCAGCGCT-3′; reverse, −1346 bp ∼−1365 bp, 5′-TGTGGTCCATGGCTTGCACT-3′).
Electrophysiological recordings in slices and cultures.
Standard whole-cell voltage-clamp recordings was used to measure synaptic currents in rat layer V medial PFC pyramidal neurons in brain slices as we described previously (Yuen et al., 2011, 2012; Wei et al., 2014a, 2014b). Rats were anesthetized with halothane (Sigma-Aldrich) inhalation. Brains were immediately removed, iced, and cut into 300 μm slices with a vibratome (VP1000S; Leica). Slices were then incubated in artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 26 NaHCO3, 3 KCl, 2 MgCl2, 1.25 NaH2PO4, 1 CaCl2, 10 glucose, pH 7.4, 300 mOsm for 1–6 h at room temperature (20–22°C) bubbling with 95% O2, 5% CO2. PFC-containing slices were positioned in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated ACSF. Bicuculline (10 μm) and CNQX (25 μm) were added in NMDAR-EPSC recordings. Bicuculline and D-APV (25 μm) were added in AMPAR-EPSC recordings. Patch electrodes were filled with internal solution containing the following (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2.2 QX-314, 12 phosphocreatine, 5 MgATP, 0.2 Na2GTP, and 0.1 leupeptin, pH 7.2–7.3, 265–270 mOsm. Cells were visualized with a 40× water-immersion lens and illuminated with near infrared (IR) light and the image was detected with an IR-sensitive CCD camera. A Multiclamp 700A amplifier was used for these recordings. Tight seals (2–10 GΩ) from visualized neurons were obtained by applying negative pressure. With additional suction, the membrane was disrupted into the whole-cell configuration. Evoked EPSCs were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Astro Med). A bipolar stimulating electrode (FHC) was placed ∼100 μm from the recorded neuron (layer V PrL) in a deeper layer (layer VI). To obtain the input/output responses, EPSC was elicited by a series of stimulation intensities with the same duration of pulses. Membrane potential was maintained at −70 mV for AMPAR-EPSC recordings. For NMDAR-EPSC, the cell (clamped at −70 mV) was depolarized to +60 mV for 3 s before stimulation to fully relieve the voltage-dependent Mg2+ block. Data analyses were performed with Clampfit (Axon), Kaleidagraph (Albeck Software), and GraphPad Prism 6 software.
Miniature EPSCs (mEPSCs) in cultured PFC neurons were recorded with the same internal solution used for recording evoked AMPAR- or NMDAR-EPSCs in slices. The external solution contained the following (in mm): 127 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 12 glucose, 10 HEPES, and 0.001 TTX, pH 7.3–7.4, 300–305 mOsm. Bicuculline (10 μm) and D-APV (25 μm) were added to block GABAAR and NMDAR activation. The membrane potential was held at −70 mV. Synaptic currents were analyzed with the Mini Analysis Program (Synaptosoft).
ShRNA lentiviral knock-down.
The shRNA oligonucleotide targeting rat HDAC2 sequence (CCCAATGAGTTGCCATATAAT; Open Biosystems) was inserted to the lentiviral vector pLKO.3G (Addgene), which contains an eGFP marker. For the production of lentiviral particles, a mixture containing the pLKO.3G shRNA plasmid (against HDAC2), psPAX2 packaging plasmid, and pMD2.G envelope plasmid (Addgene) was transfected to HEK-293FT cells using Lipofectamine 2000. The transfection reagent was removed 12–15 h later, and cells were incubated in fresh DMEM (containing 10% FBS + penicillin/streptomycin) for 24 h. The medium harvested from the cells, which contained lentiviral particles, was concentrated by centrifugation (2000 × g, 20 min) with Amicon Ultra Centrifugal Filter with Ultracel-100 membrane (Millipore). The concentrated virus was stored at −80°C. To test the specific knock-down effect, HEK-293 cells or rat cortical cultures (15–17 d in vitro) were transfected or infected with HDAC2 shRNA plasmid or lentivirus, followed by immunoblotting or immunocytochemistry with anti-HDAC2 (1:1000, sc-7899; Santa Cruz Biotechnology), anti-HDAC6 (1:1000,, ab1440; Abcam), anti-HDAC1 (1:500, sc-7872; Santa Cruz Biotechnology), or anti-MAP2 (1:500, sc-80013; Santa Cruz Biotechnology). The intensity of HDAC2 in infected neurons was analyzed with ImageJ software and normalized with the intensity of MAP2. The HDAC6 shRNA lentivirus was produced in the same way as we described previously (AAAGCAAAGACAGCTAAGGCA; Lee et al., 2012). In vivo delivery of the viral suspension (2 μl) was achieved by stereotaxic injection bilaterally into PFC with a Hamilton syringe (needle gauge 31) as we described previously (Yuen et al., 2012; Wei et al., 2014a, 2014b). Rats (∼3 weeks old) were used for lentivirus injection and the 7 d repeated stress exposure began 2 d after viral injection. Electrophysiological experiments and behavioral testing were performed 1–2 d after stress cessation.
Biochemical measurement of surface and total proteins.
To examine the surface expression of AMPA receptors, surface biotinylation was performed as described previously (Yuen et al., 2009, 2012; Wei et al., 2014a, 2014b). In brief, PFC slices were incubated with ACSF containing 1 mg/ml sulfo-N-hydroxysuccinimide- LC-Biotin (Pierce) for 20 min on ice. The slices were then rinsed 3 times in Tris-buffered saline to quench the biotin reaction, followed by homogenization in modified radioimmunoprecipitation assay buffer (1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, 50 mm NaPO4, 150 mm NaCl, 2 mm EDTA, 50 mm NaF, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, and 1 mg/ml leupeptin). The homogenates were centrifuged at 14,000 × g for 15 min at 4°C. Protein (15 μg) was removed to measure total protein. For surface protein, 150 μg of protein incubated with 100 μl of 50% neutravidin agarose (Pierce) for 2 h at 4°C and bound proteins were resuspended in SDS sample buffer and boiled. Quantitative Western blots were performed on both total and biotinylated (surface) proteins using antibodies against GluR1 (1:1000, AB1504; Millipore), GluR2 (1:500, 75-002; NeuroMab), and actin (1:1000, sc-1616-R; Santa Cruz Biotechnology). The intensity of Western blot bands was analyzed with ImageJ software and normalized with the intensity of actin.
Immunoprecipitation.
To examine GluR1 ubiquitination in PFC slices from control versus stressed rats with the injection of various HDAC inhibitors, the immunoprecipitation was performed as described previously (Yuen et al., 2012). PFC slices were collected and homogenized in lysis buffer containing the following (in mm): 50 NaCl, 30 sodium pyrophosphate, 50 NaF, 10 Tris, 5 EDTA, 0.1 Na3VO4, and 1 PMSF, with 1% Triton X-100 and protease inhibitor tablet. Lysates were ultracentrifuged (200,000 × g) at 4°C for 1 h. Supernatant fractions were incubated with polyclonal antibody against GluR1 (AB1504, 1:100 per reaction; Millipore) overnight at 4°C, followed by incubation with 50 μl of protein A/G plus agarose (Santa Cruz Biotechnology) for 1 h at 4°C. Immunoprecipitates were washed 3 times with lysis buffer and then boiled in 2× SDS loading buffer for 5 min and separated on 7.5% SDS-polyacrylamide gels. Western blotting experiments were performed with anti-ubiquitin (1:1000, sc-8017; Santa Cruz Biotechnology).
Behavioral testing.
Temporal order recognition (TOR) memory, a PFC-mediated explicit memory process requiring judgments of the prior occurrence of stimuli based on the relative familiarity, special, or recency information (Barker et al., 2007), was used as we described previously (Yuen et al., 2012; Wei et al., 2014a). This TOR memory task comprised two sample phases and one test trial. In each sample phase, the animals were allowed to explore two identical objects for a total of 3 min. The animal was then removed from the platform to its home cage. Different objects were used for sample phases I and II, with a 1 h delay between the sample phases. The test trial (3 min) was given 3 h after sample phase II. During the test trial, an object from sample phase I and an object from sample phase II were used. If temporal order memory is intact, the animals will spend more time exploring the object from sample I (i.e., the novel object presented less recently) compared with the object from sample II (i.e., the familiar object presented more recently). We calculated a discrimination ratio, the proportion of time spent exploring the novel (less recent) object (i.e., the difference in time spent exploring the novel and familiar objects divided by the total time spent exploring both objects) during the test trial. This measure takes into account individual differences in the total amount of exploration time.
Statistics.
All data are expressed as the mean ± SEM. Experiments with two groups were analyzed statistically using unpaired Student's t tests. Experiments with more than two groups were subjected to one-way or two-way ANOVA, followed by post hoc Tukey tests.
Results
HDAC2 is upregulated by GR activation in the PFC of animals exposed to repeated stress
To examine the potential involvement of histone modification in the action of stress, we first compared the expression of HDACs in PFC from control versus repeatedly stressed rats (2 h restraint/d for 7 d). Slice punches from prelimbic cortex and dorsal anterior cingulate cortex were used for the comparison. Quantitative RT-PCR analyses indicated that the level of HDAC2 mRNA was significantly higher in PFC lysates from stressed animals (Fig. 1A, 1.62 ± 0.21-fold of control, n = 6 pairs, p < 0.05), whereas the mRNA level of other HDAC family members was largely unchanged by stress (HDAC1: 1.16 ± 0.24-fold of control; HDAC4: 0.82 ± 0.14-fold of control; HDAC5: 0.92 ± 0.10-fold of control; HDAC6: 0.90 ± 0.13-fold of control, n = 8 pairs, p > 0.05). Western blot analyses also showed that the protein level of HDAC2, but not HDAC1, in the nucleus fraction of PFC was significantly higher in animals exposed to repeated stress (Fig. 1B, HDAC2: 1.83 ± 0.16-fold of control, n = 3 pairs, p < 0.05; HDAC1: 1.20 ± 0.19-fold of control, n = 4 pairs, p > 0.05). In contrast, in animals exposed to acute stress (20 min forced swim), no significant increase of nuclear HDAC2 was observed in PFC (Fig. 1B, 0.98 ± 0.10-fold of control, n = 4 pairs, p > 0.05), suggesting that prolonged stress exposure is required for the upregulation of HDAC2.
Figure 1.

HDAC2 is selectively upregulated by GR activation in PFC of stressed animals. A, Quantitative real-time RT-PCR data on the mRNA level of HDAC family members in PFC from control (con) groups versus rats exposed to 7 d restraint stress (RS). *p < 0.05, t test. B, Immunoblots and quantification analysis of the protein level of HDAC1 and HDAC2 in the nuclear fraction of PFC pyramidal neurons from control (con) versus rats exposed to repeated stress (RS) or acute stress (AS). *p < 0.05, t test. C, Immunoblots and quantification analysis of the protein level of active GR (S211pGR) in the nuclear fraction of PFC neurons from control (con) versus repeatedly stressed (RS) rats. *p < 0.05, t test. D, ChIP assay data on the binding of pGR to the HDAC2 promoter GRE region in PFC from control versus stressed rats. *p < 0.05, t test. Top, Diagram showing the location of GRE and primers (F, forward; R, reverse). E, Quantitative real-time RT-PCR data on the HDAC2 mRNA level in PFC from control versus repeatedly stressed rats with intraperitoneal injection of the GR antagonist RU486 or vehicle. *p < 0.05, ANOVA.
To determine how HDAC2 transcription is increased in repeatedly stressed animals, we examined the involvement of the GR because the proximal promoter region of HDAC2 has a well conserved recognition element for GR (Gräff et al., 2012). GR phosphorylation at Ser211 has been robustly associated with the activated form of GR (Kino et al., 2007). We found that the nuclear level of S211pGR was significantly higher in PFC of animals exposed to repeated stress (Fig. 1C, 1.75 ± 0.22-fold of control, n = 4 pairs, p < 0.05), indicating the stress-induced activation of GR. Next, we examined the binding of S211pGR to HDAC2 promoter. A primer pair against the HDAC2 promoter GRE region was used in ChIP assays. As shown in Figure 1D, S211pGR binding to HDAC2-GRE was significantly increased in PFC lysates from stressed animals (1.53 ± 0.16-fold of control, n = 7 pairs, p < 0.05). In addition, we tested whether the upregulation of HDAC2 mRNA level in stressed animals could be prevented by GR antagonism. As shown in Figure 1E, the GR antagonist RU486 (10 mg/kg, i.p. injected daily 30 min before stress) blocked the stress-induced increase of HDAC2 mRNA (1.31 ± 0.18-fold of control, n = 11 pairs, p > 0.05). These data suggest that GR is responsible for the transcriptional activation of HDAC2 by repeated stress.
HDAC2 inhibition or knock-down blocks the depression of PFC glutamatergic transmission by repeated stress or prolonged corticosterone (CORT) treatment
To test the involvement of HDAC family members in the synaptic effects of repeated stress in vivo, we injected animals with various HDAC inhibitors. Because the effects of repeated stress (restraint, 7 d) on glutamate receptors started to appear at day 5 (Yuen et al., 2012), we chose day 4 as our start point for the administration of HDAC inhibitors (injected intraperitoneally 1 h before restraint stress, beginning on day 4 until the end of the stress protocol). Because the cognitive impairment induced by repeated stress persisted for 2–3 d after stress cessation (Yuen et al., 2012), all of the electrophysiological, biochemical, and behavioral measurements in this study were performed 1–2 d after stress. As shown in Figure 2A, repeated stress caused a substantial reduction of the input/output curves of AMPAR-EPSC induced by a series of stimulus intensities (55–70% decrease, n = 8, p < 0.01, ANOVA), whereas in animals injected with trichostatin A (TSA, 0.5 mg/kg), a pan HDAC inhibitor permeant to the blood–brain barrier (Chuang et al., 2009), the reducing effect of repeated stress was largely blocked (<5% decrease, n = 7, p > 0.05, ANOVA).
Figure 2.

HDAC inhibitors block the reducing effect of repeated stress or CORT on AMPAR-EPSC. A, Input/output curves of AMPAR-EPSC evoked by a series of stimulus intensities in PFC pyramidal neurons from control (con) versus repeatedly stressed (RS) rats with the intraperitoneal injection of TSA (0.5 mg/kg, a pan-HDAC inhibitor) or vehicle. **p < 0.01, ANOVA. B, Dot plots showing the amplitude of AMPAR-EPSC evoked by the same stimulus in PFC pyramidal neurons from control versus stressed animals with intraperitoneal injections of TSA, SB (0.4 g/kg, a pan-HDAC inhibitor except for HDAC6), MGCD0103 (15 μg/kg, a selective inhibitor for HDAC1 and HDAC2), or MS-275 (15 μg/kg, a selective inhibitor for HDAC1). Inset, Representative AMPAR-EPSC traces. Scale bars, 50 pA, 20 ms. C, Immunoblots and quantification analysis of the level of acetylated H3 and total H3 in cortical slices either from rats injected intraperitoneally with various agents (vehicle, 15 μg/kg MGCD0103, or 15 μg/kg or 5 mg/kg MS-275) or treated with vehicle versus MS-275 (10 μm, 4 h in vitro). **p < 0.01, *p < 0.05, t test. D, Bar graphs showing the mEPSC amplitude and frequency in cultured cortical neurons treated with CORT (100 nm, 7 d) in the absence or presence of TSA (1 μm), SB (500 μm), tubastatin A (10 μm, an inhibitor for HDAC6), MGCD0103 (500 nm), or MS-275 (600 nm). **p < 0.01, *p < 0.05, ANOVA. Inset, Representative mEPSC traces. Scale bar, 20 pA, 5 s.
Next, we injected different HDAC inhibitors to stressed animals and compared AMPAR-EPSC evoked at a single intensity (60 μA; Fig. 2B). Similar to the blockade seen with TSA (control: 138.6 ± 11.5 pA, n = 16, stress: 61.5 ± 10.1 pA, n = 11, p < 0.001, ANOVA; TSA: 131.9 ± 10.3 pA, n = 9, TSA + stress: 154.2 ± 14.1 pA, n = 9, p > 0.05, ANOVA), injection of sodium butyrate (SB, 0.4 g/kg), a pan HDAC inhibitor except for HDAC6 (Ying et al., 2006; Schroeder et al., 2007), also blocked the reduction of AMPAR-EPSC in repeatedly stressed animals (SB: 148.4 ± 16.8 pA, n = 8, SB + stress: 144.1 ± 26.4 pA, n = 7, p > 0.05, ANOVA). MGCD0103 (15 μg/kg), a selective inhibitor for HDAC1 and HDAC2 (Ferguson et al., 2013), abolished the reducing effect of stress (MGCD0103: 147.9 ± 3.1 pA, n = 9, MGCD0103 + stress: 149.9 ± 12.7 pA, n = 9, p > 0.05, ANOVA), whereas MS-275 (15 μg/kg), a selective inhibitor for HDAC1 (Eyüpoglu et al., 2006; Simonini et al., 2006), failed to do so (MS-275: 146.9 ± 13.3 pA, n = 9, MS-275 + stress: 70.2 ± 8.5 pA, n = 10, p < 0.001, ANOVA). The brain permeability of MGCD0103 and MS-275 injections (injected intraperitoneally once daily for 4 d) was shown by the increased histone acetylation in frontal cortex (Fig. 2C, MGCD0103, 15 μg/kg: 1.77 ± 0.27-fold of control, n = 8 pairs; MS-275, 15 μg/kg: 1.27 ± 0.10-fold of control, n = 8 pairs; MS-275, 5 mg/kg: 1.37 ± 0.12-fold of control, n = 5 pairs, p < 0.05). In vitro treatment (MS-275, 10 μm, 4 h) in PFC slices induced a similar increase in H3 acetylation (MS-275, 1.26 ± 0.04-fold of control, n = 6 pairs, p < 0.01). These data suggest the potential involvement of HDAC2 in the synaptic action of repeated stress.
The effect of repeated behavioral stress can be mimicked by the prolonged treatment with the stress hormone CORT (Yuen et al., 2012), so we examined the role of HDAC in CORT-induced changes in glutamatergic responses in vitro. Cortical cultures (10–12 d in vitro) were exposed to CORT (100 nm) for 5–7 d in the presence of various HDAC inhibitors (added on the third day). mEPSC, a synaptic response resulting from quantal release of single glutamate vesicles, was recorded after washing off these agents. As shown in Figure 2D, a significant main effect was found on mEPSC amplitude and frequency after long-term CORT treatment (F(11,79) = 6.9 for amp.; F(11,79) = 3.8 for freq., p < 0.001, ANOVA, control: 23.8 ± 1.0 pA, 7.9 ± 1.9 Hz, n = 12; CORT: 16.9 ± 0.5 pA, 4.4 ± 0.8 Hz, n = 13, p < 0,01), which was blocked by TSA (1 μm, TSA: 21.6 ± 1.4 pA, 6.8 ± 1.1 Hz, n = 6; TSA+CORT: 20.4 ± 0.7 pA, 4.7 ± 0.8 Hz, n = 6, p > 0.05) or SB (500 μm, Marinova et al., 2011, SB: 22.7 ± 1.5 pA, 5.8 ± 0.9 Hz, n = 4; SB + CORT: 18.3 ± 1.6 pA, 5.1 ± 1.0 Hz, n = 5, p > 0.05). In addition, MGCD0103 (500 nm) blocked the effect of CORT (MGCD0103: 22.1 ± 2.1 pA, 6.6 ± 2.1 Hz, n = 7; MGCD0103 + CORT: 25.2 ± 1.2 pA, 8.8 ± 1.8 Hz, n = 7, p > 0.05), but MS-275 (600 nm) was ineffective (MS-275: 22.3 ± 0.9 pA, 7.1 ± 1.0 Hz, n = 5; MS-275 + CORT: 16.5 ± 0.8 pA, 1.8 ± 0.4 Hz, n = 4, p < 0.01). Tubastatin A (10 μm), an inhibitor for HDAC6 (Butler et al., 2010), also failed to blocked the CORT-induced reduction of mEPSC (tubastatin: 23.6 ± 1.2 pA, 6.6 ± 0.7 Hz, n = 6; tubastatin + CORT: 15.4 ± 0.6 pA, 2.9 ± 0.8 Hz, n = 5, p < 0.01). These in vitro data further suggest that HDAC2 is required for synaptic action of corticorsteroid stress hormones.
Although a relatively low dose of MS275 and MGCD0103 (15 μg/kg) was used in our in vivo study, an interaction with their less affine targets cannot be ruled out. To further confirm the specific involvement of HDAC2 in regulating repeated stress-induced reduction of glutamatergic responses, we performed RNA interference-mediated knock-down of HDAC2 in vitro or in vivo. As illustrated in Figure 3A, HDAC2 shRNA caused a specific and effective suppression of the expression of HDAC2, but not other HDAC family members, in HEK293 cells. Immunocytochemical analyses (Fig. 3B) of cortical cultures infected with HDAC2 shRNA lentivirus for 7 d also demonstrated the strong reduction of HDAC2 expression in neuronal nuclei (control shRNA: 1.0 ± 0.016, n = 20; HDAC2 shRNA: 0.62 ± 0.017, n = 38, p < 0.001). We then examined the effect of prolonged CORT treatment (100 nm, 7 d) on mEPSC in cortical cultures transfected with HDAC2 shRNA. GFP alone or a HDAC6 shRNA, which has been shown to induce a substantial knock-down of HDAC6 expression in our previous study (Lee et al., 2012), was used as a control. As shown in Figure 3C, a significant main effect was found on mEPSC in CORT-treated neurons with different transfections (amplitude, F(5,21) = 12.9, p < 0.001, ANOVA; frequency, F(5,21) = 10.1, p < 0.001, ANOVA). Post hoc analysis indicated that the potent reduction of mEPSC by CORT in GFP-transfected neurons (GFP: 21.2 ± 0.7 pA, 4.2 ± 0.2 Hz, n = 3; GFP + CORT: 15.2 ± 0.9 pA, 1.3 ± 0.2 Hz, n = 4, p < 0.01) was lost in neurons with HDAC2 knock-down (HDAC2 shRNA: 22.7 ± 1.1 pA, 5.0 ± 1.1 Hz, n = 4; HDAC2 shRNA + CORT: 21.5 ± 2.1 pA, 4.8 ± 0.4 Hz, n = 4, p > 0.05), but not in those with HDAC6 knock-down (HDAC6 shRNA: 26.0 ± 0.6 pA, 4.7 ± 0.3 Hz, n = 3; HDAC6 shRNA + CORT: 15.1 ± 0.4 pA, 1.6 ± 0.2 Hz, n = 4, p < 0.01).
Figure 3.
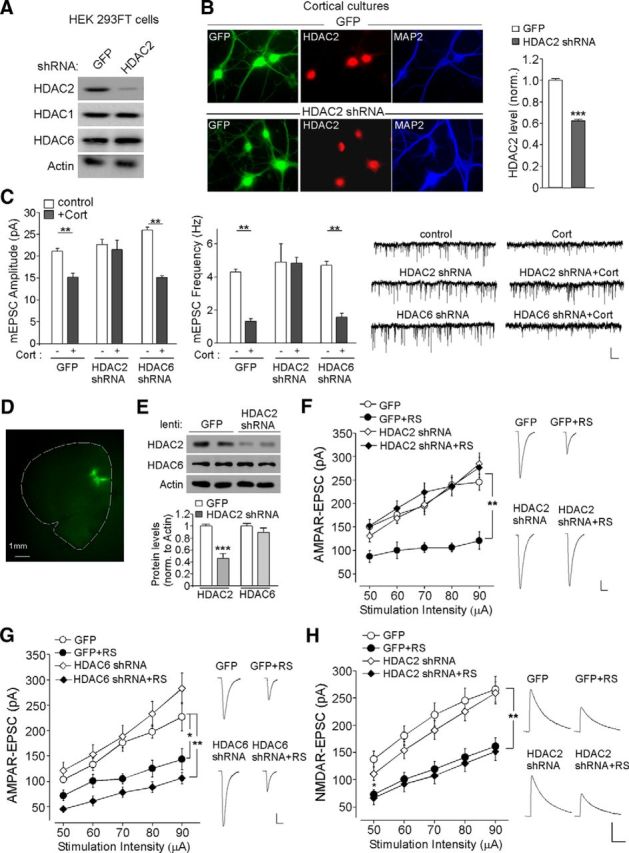
HDAC2 knock-down blocks the effect of prolonged CORT or repeated stress on AMPAR-mediated synaptic responses. A, Western blots showing the expression of HDAC2, HDAC1, and HDAC6 in HEK293 cells transfected with HDAC2 shRNA or a GFP control shRNA. Actin was used as a loading control. B, Immunocytochemical staining and quantification analysis of HDAC2 in cultured PFC neurons infected with HDAC2 shRNA lentivirus or a GFP lentivirus. ***p < 0.001, t test. C, Bar graphs and representative mEPSC traces showing the effect of CORT treatment (100 nm, 7 d) on mEPSC amplitude and frequency in cortical cultures transfected with GFP, HDAC2 shRNA, or HDAC6 shRNA. Scale bar, 20 pA, 5 s. **p < 0.01, ANOVA. D, Image showing the spread of HDAC2 shRNA lentivirus stereotaxically injected to the prelimbic region of PFC. E, Immunoblots and quantification analysis of HDAC2 and HDAC6 expression in rat PFC infected with HDAC2 shRNA lentivirus or GFP lentivirus. ***p < 0.001, t test. F, G, Summarized input/output curves of AMPAR-EPSC in control (con) versus repeatedly stressed (RS) rats with the PFC injection of GFP lentivirus, HDAC2 shRNA lentivirus (F), or HDAC6 shRNA lentivirus (G). Inset, Representative AMPAR-EPSC traces. Scale bars, 50 pA, 20 ms. **p < 0.01, *p < 0.05, ANOVA. H, Summarized input/output curves of NMDAR-EPSC in control (con) versus repeatedly stressed (RS) rats with the PFC injection of GFP or HDAC2 shRNA lentivirus. Inset, Representative NMDAR-EPSC traces. Scale bars, 100 pA, 200 ms. **p < 0.01, ANOVA.
Next, we performed the stereotaxic injection of HDAC2 shRNA lentivirus into the medial PFC and further validated HDAC2 knock-down in vivo. HDAC2 shRNA lentivirus was injected into the PrL region of medial PFC (Fig. 3D). GFP+ areas were punched out from PFC slices for protein detection at day 6 after injection. As shown in Figure 3E, HDAC2 shRNA lentivirus infection significantly and specifically diminished the expression of HDAC2 (HDAC2: 0.46 ± 0.08-fold of control, n = 6 pairs, p < 0.001; HDAC6: 0.90 ± 0.07-fold of control, n = 6 pairs, p > 0.05). We next investigated the involvement of HDAC2 in regulating the electrophysiological effects of repeated stress. GFP or HDAC6 shRNA lentivirus (Lee et al., 2012) was also used as a control. A significant main effect was found in stressed animals with different viral infections (Fig. 3F,G, n = 5–13 per group, p < 0.01, ANOVA). Post hoc analysis showed that repeated stress caused a substantial downregulation of the AMPAR-EPSC amplitude in GFP-injected animals (42–56% decrease, GFP: n = 5; GFP + stress: n = 5, p < 0.01), but not in HDAC2 shRNA-injected animals (1–16% decrease, HDAC2 shRNA: n = 13; HDAC2 shRNA + stress: n = 11, p > 0.05). In contrast, the reducing effect of repeated stress was intact in HDAC6 shRNA-injected animals (58–63% decrease, HDAC6 shRNA: n = 8; HDAC6 shRNA + stress: n = 8, p < 0.01). Together, these electrophysiological results have identified HDAC2 as the key factor required for the downregulation of AMPAR responses by repeated stress.
Because NMDARs are also lost in PFC pyramidal neurons from repeatedly stressed rats (Yuen et al., 2012), we examined the impact of HDAC2 on stress-induced changes in NMDARs. A significant main effect was found in repeatedly stressed animals with different viral infections (Fig. 3H, n = 12–15 per group, p < 0.01, ANOVA). Post hoc analysis showed that repeated stress caused a substantial decrease of NMDAR-EPSC amplitudes in GFP-injected animals (39–47% decrease, GFP: n = 13; GFP + stress: n = 15, p < 0.01) and the reducing effect of repeated stress was intact in HDAC2 shRNA-injected animals (40–44% decrease, HDAC2 shRNA: n = 12; HDAC2 shRNA + stress: n = 13, p < 0.01), indicating that HDAC2 knock-down failed to block the downregulation of NMDAR responses by repeated stress.
Inhibiting HDAC2 blocks the loss of AMPAR expression in animals exposed to repeated stress
Our previous study found that the suppression of glutamatergic transmission by repeated stress results from the reduced number of AMPA receptors (Yuen et al., 2012). Therefore, here, we performed Western blotting and surface biotinylation experiments to determine whether HDAC inhibitors could restore the total and surface level of AMPAR subunits in PFC slices from stressed rats. HDAC inhibitors were injected intraperitoneally daily (1 h before restraint stress, beginning on day 4 until the end of the stress protocol). As shown in Figure 4, A and B, in vehicle-injected animals exposed to repeated stress, the number of AMPAR subunits in the surface pool was significantly decreased (surface GluR1: 60% decrease, surface GluR2: 43% decrease, n = 9 pairs, p < 0.01, ANOVA) and a significant decrease in the number of total GluR1 subunits, but not GluR2 subunits, was also observed (41% decrease, n = 8 pairs, p < 0.01, ANOVA), consistent with our previous findings (Yuen et al., 2012). Injecting TSA (0.5 mg/kg, a pan-HDAC inhibitor) or MGCD0103 (15 μg/kg, a selective inhibitor for HDAC1 and HDAC2) abolished the decreasing effects of repeated stress on surface GluR1/2 (TSA + RS: <10% decrease, n = 4 pairs, p > 0.05; MGCD0103 + RS: <5% decrease, n = 5 pairs, p > 0.05) and total GluR1 (TSA + RS: <10% decrease, n = 4 pairs, p > 0.05; MGCD0103 + RS: <10% decrease, n = 4 pairs, p > 0.05), whereas injecting MS-275 (15 μg/kg, a selective inhibitor for HDAC1) failed to do so (surface GluR1/2, MS-275 + RS: 54–64% decrease, n = 4 pairs, p < 0.05; total GluR1, MS-275 + RS: 63% decrease, n = 5 pairs, p < 0.01). This suggests that HDAC2 is involved in the downregulation of AMPAR expression by repeated stress.
Figure 4.
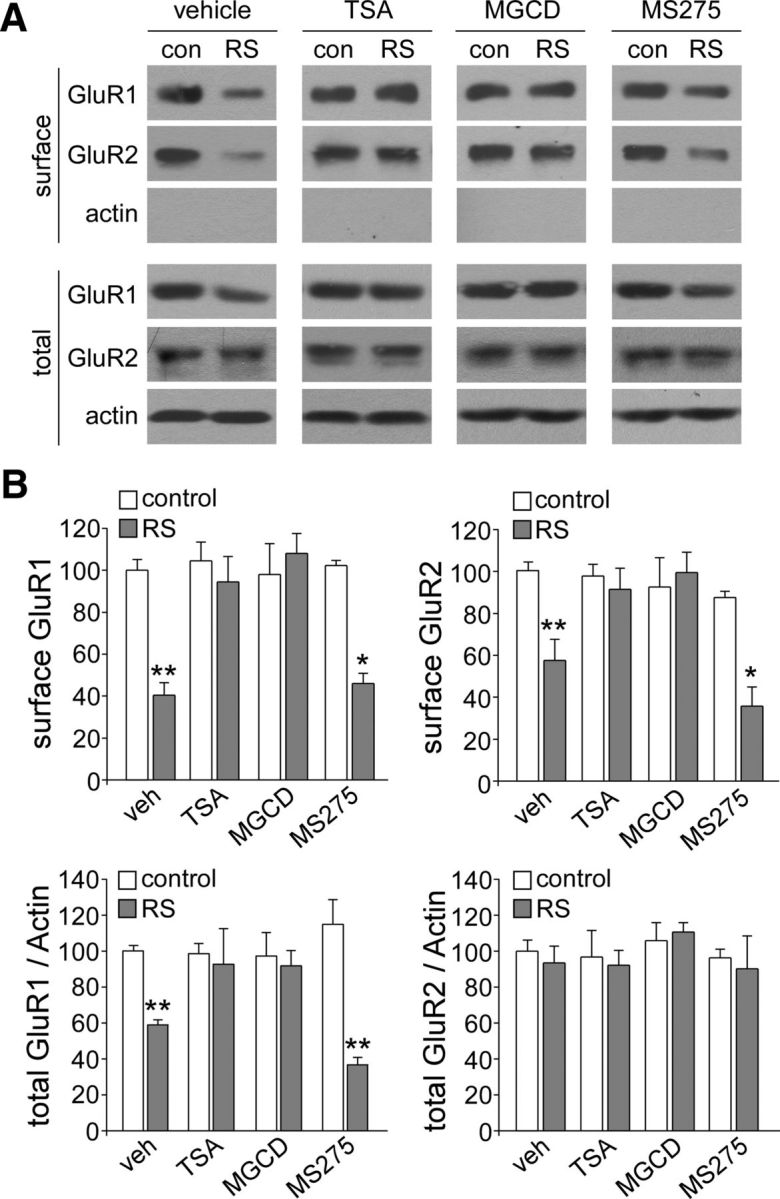
HDAC2 inhibitors block the reducing effect of repeated stress on the total and surface levels of AMPAR subunits in PFC. A, B, Immunoblots (A) and quantification analysis (B) of the total and surface AMPAR subunits in PFC from control (con) versus repeatedly stressed (RS) rats with the intraperitoneal injection of vehicle, TSA, MGCD0103, or MS-275. **p < 0.01, *p < 0.05, ANOVA.
HDAC2 inhibition or knock-down ameliorates the cognitive impairment induced by repeated stress
To determine the involvement of HDAC in behavioral deficits induced by repeated stress, we examined TOR memory, a cognitive process controlled by prefrontal cortex (Barker et al., 2007) and impaired by repeated stress (Yuen et al., 2012). Rats were intraperitoneally injected with various HDAC inhibitors during the 7 d restraint stress (beginning on day 4) and tested 1–2 d after the last stress exposure. The discrimination ratio (DR), an index of the object recognition memory, showed a significant main effect (Fig. 5A, F(5,39) = 5.59, p < 0.001, ANOVA). Post hoc analysis indicated a profound impairment of TOR memory by repeated stress in vehicle-injected animals (DR in vehicle: 55.9 ± 3.0%, n = 6; DR in vehicle + stress: −1.8 ± 19.0%, n = 6, p < 0.05). TOR memory was rescued in the stressed animals injected with MGCD0103 (15 μg/kg, i.p., 4 d), an HDAC1 and 2 inhibitor (DR in MGCD0103: 57.1 ± 11.5%, n = 6; DR in MGCD0103 + stress: 52.7 ± 8.5%, n = 7, p > 0.05). However, injection of MS-275 (15 μg/kg, i.p., 4 d), an HDAC1 inhibitor, was ineffective in altering the cognitive impairment by stress (DR in MS-275: 51.8 ± 11.0%, n = 7; DR in MS-275 + stress: 3.1 ± 12.3%, n = 8, p < 0.05). As shown in Figure 5B, the unstressed control group spent more time exploring the novel (less recent) object in the test trial (novel object: 24.9 ± 1.5 s, familiar object: 7.0 ± 0.7 s, n = 6, p < 0.001), whereas the stressed rats lost the preference for the novel object (novel object: 16.7 ± 4.9 s, familiar object: 15.3 ± 3.5 s, n = 6, p > 0.05). The preference for the novel object in stressed animals was recovered by MGCD0103 injection (novel object: 26.7 ± 5.3 s, familiar object: 7.1 ± 1.3 s, n = 7, p < 0.01), but not MS-275 injection (novel object: 19.6 ± 3.9 s, familiar object: 17.8 ± 2.4 s, n = 8, p > 0.05).
Figure 5.

HDAC2 inhibition or knock-down blocks the effect of repeated stress on TOR memory. A, Bar graphs showing the DR of TOR tasks in control (con) versus repeatedly stressed (RS) rats with the intraperitoneal injection of vehicle, MGCD0103 (15 μg/kg), or MS-275 (15 μg/kg). *p < 0.05, ANOVA. B, Bar graphs showing the exploration time for novel versus familiar objects in the test trial of TOR tasks in control (con) versus repeatedly stressed animals with the intraperitoneal injection of vehicle, MGCD0103 (15 μg/kg), or MS-275 (15 μg/kg). ***p < 0.001, **p < 0.01, *p < 0.05, t test. C, Bar graphs showing the DR of TOR tasks in control versus repeatedly stressed animals injected with GFP or HDAC2 shRNA lentivirus. **p < 0.01, *p < 0.05, ANOVA. D, Bar graphs showing the exploration time for novel versus familiar objects in the test trial of TOR tasks in control (con) versus repeatedly stressed animals with PFC injection of GFP or HDAC2 shRNA lentivirus. *p < 0.05, t test.
Next, we performed the stereotaxic injection of HDAC2 shRNA lentivirus into the PFC and examined TOR memory in animals exposed to repeated stress. A significant main effect was found on the discrimination ratio (Fig. 5C, F(3,23) = 7.59, p < 0.01, ANOVA). Post hoc analysis indicated a significant impairment of TOR memory by repeated stress in animals injected with GFP lentivirus (DR in GFP: 48.0 ± 10.1%, n = 6; DR in GFP + stress: −1.6 ± 6.8%, n = 6, p < 0.01). TOR memory was rescued in stressed animals injected with HDAC2 shRNA lentivirus (31.7 ± 5.2%, n = 6). HDAC2 knock-down did not affect the TOR memory in unstressed control animals (42.8 ± 9.4%, n = 6). As shown in Figure 5D, stressed rats injected with GFP lentivirus lost the preference of the novel over the familiar object in the test trial (novel object: 15.5 ± 1.7 s, familiar object: 16.2 ± 2.0 s, n = 6, p > 0.05), whereas stressed rats injected with HDAC2 shRNA lentivirus showed the reinstatement of the preference for the novel object (novel object: 21.5 ± 2.7 s, familiar object: 11.8 ± 2.7 s, n = 6, p < 0.05). Together, these data indicate that selective inhibition or knock-down of HDAC2 is able to block stress-induced behavioral deficits.
Repeated stress upregulates Nedd4, the E3 ligase for GluR1 ubiquitination, via an HDAC2-dependent mechanism involving histone methylation
We have found previously that the loss of AMPARs in repeatedly stressed animals is due to the increased ubiquitination and degradation of GluR1 subunits by the E3 ubiquitin ligase Nedd4-1 (Yuen et al., 2012). Therefore, we examined the role of HDAC2 in Nedd4-1 regulation. First, we examined Nedd4-1 expression in control versus stressed rats with the injection of various HDAC inhibitors. As shown in Figure 6A, the level of Nedd4-1 protein was significantly higher in PFC lysates from stressed animals (1.40 ± 0.08-fold of control, n = 5 pairs, p < 0.05, ANOVA). Injections of MGCD0103 blocked the increasing effect of stress on Nedd4-1 expression (1.07 ± 0.13-fold of control, n = 4 pairs, p > 0.05). We also measured Nedd4-1 protein levels in PFC punches from control versus stressed animals injected with GFP or HDAC2 lentivirus. As shown in Figure 6B, repeated stress caused a substantial upregulation of Nedd4-1 in GFP-injected animals (1.47 ± 0.11-fold of control, n = 7 pairs, p < 0.01, ANOVA), but not in HDAC2 shRNA-injected animals (1.0 ± 0.07-fold of control, n = 8 pairs, p > 0.05). It suggests that HDAC2 is required for the stress-induced increase of Nedd4 expression. Quantitative PCR analyses further indicated that the level of Nedd4-1 mRNA was significantly elevated in PFC from stressed animals (Fig. 6C, 1.70 ± 0.13-fold of control, n = 8 pairs, p < 0.01), suggesting that transcriptional activation of Nedd4-1 occurs after repeated stress.
Figure 6.

HDAC2-dependent downregulation of histone methyltransferase Ehmt2 (G9a) mediates the loss of repressive histone methylation at the Nedd4-1 promoter in stressed animals. A, B, Immunoblots and quantification analysis of the protein level of Nedd4-1 in PFC punches from control versus stressed rats with the intraperitoneal injection of MGCD0103 versus vehicle control (A) or the local injection of GFP versus HDAC2 shRNA lentivirus (B). **p < 0.01, *p < 0.05, ANOVA. C, Quantitative real-time RT-PCR data on the mRNA level of Nedd4-1 in PFC from control versus repeatedly stressed rats. **p < 0.01, t test. D, Representative blots and quantification showing the ubiquitination of GluR1 subunits in control (con) versus repeatedly stressed (RS) rats with the intraperitoneal injection of vehicle, MGCD0103, or MS-275. Lysates of PFC slices were immunoprecipitated with anti-GluR1 and then blotted with an ubiquitin antibody. **p < 0.01, ANOVA. E, ChIP assay data showing the H3K9Me2 level at rat Nedd4-1 promoter regions in PFC lysates from control versus repeatedly stressed (RS) rats. *p < 0.05, t test. Top, Diagram showing the location of primers. F, Quantitative real-time RT-PCR data on the mRNA level of Ehmt1 and Ehmt2 in PFC from control versus repeatedly stressed rats without or with the intraperitoneal injection of MGCD0103. *p < 0.05, ANOVA. G, Quantitative real-time RT-PCR data on the mRNA level of Ehmt2 in PFC from control versus repeatedly stressed rats with the local injection of GFP or HDAC2 shRNA lentivirus. **p < 0.01, ANOVA. H, ChIP assay data showing the acetylated histone H3 level at rat Ehmt2 promoter regions in PFC lysates from control versus repeatedly stressed (RS) rats. *p < 0.05, t test. Top, Diagram showing the location of primers.
Next, we measured GluR1 ubiquitination in PFC slices from control versus stressed rats with the injection of various HDAC inhibitors. As shown in Figure 6D, the level of ubiquitinated GluR1 showed a significant main effect (F(5,30) = 35.4, p < 0.001, ANOVA). Post hoc analysis indicated that the stress-induced increase in GluR1 ubiquitination could be detected in vehicle-injected animals (2.50 ± 0.15-fold of control, n = 5 pairs, p < 0.01) or MS-275 (15 μg/kg)-injected animals (2.14 ± 0.06-fold of control, n = 6 pairs, p < 0.01), but not those injected with MGCD0103 (15 μg/kg, 0.90 ± 0.12-fold of control, n = 9 pairs, p > 0.05). This suggests that HDAC2 is required for Nedd4-mediated GluR1 ubiquitination in response to repeated stress.
A puzzling question is why increased HDAC2 leads to increased Nedd4-1 gene transcription in response to repeated stress, because histone deacetylation compacts chromatin structure and represses gene transcription (Cheung et al., 2000; Lee and Workman, 2007). One possibility is that Nedd4-1 upregulation is caused by changes in histone methylation resulting from increased HDAC2. We therefore examined the potential alteration of histone methylation at Nedd4-1 promoter by repeated stress. A ChIP assay was used to examine H3K9 dimethylation (H3K9me2, linked to gene silencing) at the Nedd4-1 promoter. As shown in Figure 6E, stressed animals had significantly reduced H3K9 dimethylation at the proximal Nedd4-1 promoter region (500 bp from TSS, 0.43 ± 0.10-fold of control, n = 10 pairs, p < 0.05), but not the immediate 1500 bp upstream region, suggesting that the elevated Nedd4-1 gene expression may be caused by the loss of repressive histone methylation in stressed animals.
To identify the potential histone methyltransferase mediating the histone modification of Nedd4-1, we used quantitative RT-PCR to examine the potential changes in the expression of Ehmt1 (GLP) and Ehmt2 (G9a), which specifically catalyze H3K9 dimethylation, in stressed animals. As shown in Figure 6F, compared with unstressed control rats, the level of Ehmt2 mRNA was significantly reduced in rats exposed to repeated stress (0.53 ± 0.14-fold of control, n = 12 pairs, p < 0.05, ANOVA); however, such a reduction was not observed in stressed rats with the injection of HDAC2 inhibitor MGCD0103 (1.0 ± 0.11-fold of control, n = 5, p > 0.05). In contrast, the mRNA level of Ehmt1 was not significantly changed. We also measured Ehmt2 mRNA level in PFC punches from control versus stressed animals injected with GFP or HDAC2 lentivirus. As shown in Figure 6G, in GFP-injected rats, repeated stress induced a substantial downregulation of the Ehmt2 mRNA level (0.63 ± 0.06-fold of control, n = 7 pairs, p < 0.01, ANOVA); however, such an effect was not observed in stressed rats injected with HDAC2 shRNA (0.99 ± 0.07-fold of control, n = 6 pairs, p > 0.05). These data suggest that repeated stress induces a HDAC2-dependent, selective downregulation of Ehmt2, which could be responsible for the loss of repressive H3K9 dimethylation at the Nedd4-1 promoter.
To further determine whether the stress-induced downregulation of Ehmt2 transcription might be due to the upregulated HDAC2, we performed ChIP assays to examine the impact of repeated stress on the histone acetylation of Ehmt2 promoter. As shown in Figure 6H, stressed animals had significantly reduced Histone H3 acetylation at the proximal Ehmt2 promoter region (700 bp from TSS, 0.35 ± 0.08-fold of control, n = 10 pairs, p < 0.05) and the immediate 1500 bp upstream region (0.49 ± 0.07-fold of control, n = 9 pairs, p < 0.05). This suggests that the decreased Ehmt2 gene expression in stressed animals may result from the loss of histone acetylation due to elevated HDAC2.
Discussion
Histone-modification-based epigenetic control of gene transcription in mature neurons provides a key molecular mechanism to guide dynamic plasticity in response to various stimuli (Borrelli et al., 2008; Maze et al., 2013). Our previous studies have shown that repeated stress induces the increased ubiquitination and degradation of AMPA receptors and impaired cognitive functions (Yuen et al., 2012). Here, we have revealed an epigenetic mechanism involved in the stress-induced transcriptional activation of Nedd4 (Fig. 7), an E3 ubiquitin ligase critical for the regulation of AMPAR ubiquitination/degradation (Schwarz et al., 2010; Hou et al., 2011) and PFC-mediated cognition (Yuen et al., 2012). In animals exposed to repeated stress, HDAC2 is increased by activated GR, resulting in an elevation of Nedd4 expression. Inhibition or knock-down of HDAC2 blocks the stress-induced impairment of synaptic transmission, AMPAR expression, and recognition memory, suggesting that HDAC2-selective inhibitors may have therapeutic potentials for stress-related mental disorders.
Figure 7.

A working model showing the epigenetic mechanism of repeated stress. In stressed animals, activated GR binds to GRE of HDAC2 promoter, resulting in HDAC2 upregulation. Consequently, Ehmt2 is suppressed, leading to the loss of repressive histone methylation at the Nedd4 promoter and increased transcription of Nedd4. Nedd4 induces GluR1 ubiquitination and degradation, causing the loss of glutamatergic transmission and PFC-mediated cognitive function. Inhibiting HDAC2 blocks the signaling cascade and detrimental effects of repeated stress.
HDAC2 is enriched at the promoters of genes regulated by neuronal activity or genes related to synaptic plasticity (Guan et al., 2009), implicating the importance of HDAC2 in regulating gene expression involved in basal and activity-induced neuronal functions. HDAC2, which is increased by Alzheimer's disease-related neurotoxic insults (Gräff et al., 2012), negatively regulates memory formation and synaptic plasticity (Guan et al., 2009). Treatment with HDAC inhibitor or knock-down of HDAC2 ameliorates the memory impairment in mice with HDAC2 overexpression (Guan et al., 2009; Gräff et al., 2012). Moreover, changes in HDAC expression have been found in the hippocampus and nucleus accumbens of depression models and depressed humans and infusion of HDAC inhibitors reverses the altered global patterns of gene expression and exerts robust antidepressant-like effects (Tsankova et al., 2006; Schroeder et al., 2007; Covington et al., 2009; Golden et al., 2013). HDAC inhibitors also augment the therapeutic effects of atypical antipsychotics by preventing the repressive histone modifications at the metabotropic glutamate 2 promoter (Kurita et al., 2012). Our findings on the stress/GR-induced HDAC2 upregulation and the amelioration of detrimental effects of repeated stress by HDAC2 inhibition have uncovered a key role of HDAC2 in the action of behavioral stress.
Because histone hypoacetylation correlates closely with gene silencing (Lee and Workman, 2007), how does the increase of HDAC2 result in the transcriptional activation of Nedd4 after repeated stress? One possibility is through altering histone methylation, which correlates with either transcriptional activation (e.g., H3K4) or repression (e.g., H3K9, H3K27). Our biochemical evidence suggests that the HDAC2-mediated suppression of histone methyltransferase Ehmt2 and loss of repressive H3K9 dimethylation at Nedd4 promoter may be responsible for the upregulation of Nedd4 in stressed animals. Interestingly, downregulation of Ehmt2 (G9a) and alteration of histone methylation at various target genes have also been found to play a crucial role in cocaine-induced behavioral plasticity (Maze et al., 2010; Covington et al., 2011; Kennedy et al., 2013).
Although HDAC1-mediated chromatin remodeling in NAc has often been implied in cocaine addiction (Kennedy et al., 2013) and depression (Golden et al., 2013), our pharmacological and RNA interference data point to the role of HDAC2 in controlling the effect of glucocorticoid stress hormones in mPFC. We have provided evidence showing that GR binds to GRE on HDAC2 promoter and increases HDAC2 transcription in stressed animals, which is accompanied by decreased histone acetylation at the Ehmt2 promoter and decreased Ehmt2 transcription. Therefore, the repressive H3K9Me2 at the Nedd4 promoter is decreased and Nedd4 transcription is increased in stressed animals. It awaits to be tested whether GR can bind directly to the Ehmt2 (and even the Nedd4) promoter region and recruit HDAC2 to regulate their transcription. Furthermore, it is likely that the stress/GR-induced alteration of histone modification enzymes will lead to changes in the expression of many other genes involved in synaptic transmission and plasticity, which will be explored in future studies.
HDAC2 knock-down did not block the reduction of NMDAR-EPSC in repeatedly stressed rats. A potential reason is that HDAC2 inhibition does not affect the stress-induced NR1 ubiquitination and degradation, which is mediated by a different E3 ligase, Fbx2 (Yuen et al., 2012). This suggests that specific epigenetic mechanisms are involved in the regulation of different glutamate receptors.
In summary, we demonstrate here that the Nedd4 gene, which mediates the loss of AMPA receptors and PFC functions in response to repeated stress (Yuen et al., 2012), is a molecular target of HDAC2-Ehmt2. The epigenetic blockade of synaptic and behavioral deficits in stressed animals offers a novel avenue to counteract the negative impact of chronic stress.
Footnotes
This work was supported by the National Institutes of Health Grants MH108842 and DA037618 and Department of Veteran Affairs Grant 1I01BX001633 to Z.Y. We thank Xiaoqing Chen for excellent technical support.
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Bird F, Alexander V, Warburton EC. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci. 2007;27:2948–2957. doi: 10.1523/JNEUROSCI.5289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg HP, Leung HC, Skudlarski P, Lacadie CM, Fredericks CA, Harris BC, Charney DS, Gore JC, Krystal JH, Peterson BS. A functional magnetic resonance imaging study of bipolar disorder: state- and trait-related dysfunction in ventral prefrontal cortices. Arch Gen Psychiatry. 2003;60:601–609. doi: 10.1001/archpsyc.60.6.601. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Ramsey NF, Tallent K, Bertolino A, Knable MB, Coppola R, Goldberg T, van Gelderen P, Mattay VS, Frank JA, Moonen CT, Weinberger DR. Functional magnetic resonance imaging brain mapping in psychiatry: methodological issues illustrated in a study of working memory in schizophrenia. Neuropsychopharmacology. 1998;18:186–196. doi: 10.1016/S0893-133X(97)00096-1. [DOI] [PubMed] [Google Scholar]
- Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr Opin Pharmacol. 2006;6:369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/S0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, Dietz DM, Lobo MK, Ghose S, Mouzon E, Neve RL, Tamminga CA, Nestler EJ. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Eyüpoglu IY, Hahnen E, Tränkle C, Savaskan NE, Siebzehnrübl FA, Buslei R, Lemke D, Wick W, Fahlbusch R, Blümcke I. Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS 275. Mol Cancer Ther. 2006;5:1248–1255. doi: 10.1158/1535-7163.MCT-05-0533. [DOI] [PubMed] [Google Scholar]
- Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, Holson EB, McKinsey TA. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2013;110:9806–9811. doi: 10.1073/pnas.1301509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24:202–218. doi: 10.1002/da.20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, Cahill ME, Dias C, Ribeiro E, Ables JL, Kennedy PJ, Robison AJ, Gonzalez-Maeso J, Neve RL, Turecki G, Ghose S, Tamminga CA, Russo SJ. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med. 2013;19:337–344. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J, Tsai LH. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci. 2013;14:97–111. doi: 10.1038/nrn3427. [DOI] [PubMed] [Google Scholar]
- Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 2011;72:806–818. doi: 10.1016/j.neuron.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, Bassel-Duby R, Olson EN, Nestler EJ. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci. 2013;16:434–440. doi: 10.1038/nn.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, Kawasaki E, Pant HC, Chrousos GP. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568. doi: 10.1210/me.2006-0345. [DOI] [PubMed] [Google Scholar]
- Kurita M, Holloway T, García-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, Dietz DM, Mocci G, Gabilondo AM, Hanks J, Umali A, Callado LF, Gallitano AL, Neve RL, Shen L, Buxbaum JD, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JB, Wei J, Liu W, Cheng J, Feng J, Yan Z. Histone Deacetylase 6 gates the synaptic action of acute stress in prefrontal cortex. J Physiol. 2012;590:1535–1546. doi: 10.1113/jphysiol.2011.224907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Marinova Z, Leng Y, Leeds P, Chuang DM. Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology. 2011;60:1109–1115. doi: 10.1016/j.neuropharm.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Noh KM, Allis CD. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology. 2013;38:3–22. doi: 10.1038/npp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging. 2002;23:921–939. doi: 10.1016/S0197-4580(02)00027-1. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/ncb2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, Orr SP, Pitman RK, Whalen PJ, Rauch SL. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, Guidotti A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A. 2006;103:1587–1592. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman DA, Moghaddam B. Reduced neuronal inhibition and coordination of adolescent prefrontal cortex during motivated behavior. J Neurosci. 2011;31:1471–1478. doi: 10.1523/JNEUROSCI.4210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Gu Z, Yan Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci. 2003;23:9852–9861. doi: 10.1523/JNEUROSCI.23-30-09852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Yuen EY, Liu W, Li X, Zhong P, Karatsoreos IN, McEwen BS, Yan Z. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol Psychiatry. 2014a;19:588–598. doi: 10.1038/mp.2013.83. [DOI] [PubMed] [Google Scholar]
- Wei J, Graziane NM, Wang H, Zhong P, Wang Q, Liu W, Hayashi-Takagi A, Korth C, Sawa A, Brandon NJ, Yan Z. Regulation of N-methyl-D-aspartate receptors by disrupted-in-schizophrenia-1. Biol Psychiatry. 2014b;75:414–424. doi: 10.1016/j.biopsych.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying M, Xu R, Wu X, Zhu H, Zhuang Y, Han M, Xu T. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. 2006;281:12580–12586. doi: 10.1074/jbc.M511677200. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A. 2009;106:14075–14079. doi: 10.1073/pnas.0906791106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–170. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–977. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]