

Abstract
Background
DNA methylation is one way to encode epigenetic information and plays a crucial role in regulating gene expression during embryonic development. DNA methylation marks are established by the DNA methyltransferases and, recently, a mechanism for active DNA demethylation has emerged involving the ten-eleven translocator proteins and thymine DNA glycosylase (TDG). However, so far it is not clear how these enzymes are recruited to, and regulate DNA methylation at, specific genomic loci. A number of studies imply that sequence-specific transcription factors are involved in targeting DNA methylation and demethylation processes. Oestrogen receptor beta (ERβ) is a ligand-inducible transcription factor regulating gene expression in response to the female sex hormone oestrogen. Previously, we found that ERβ deficiency results in changes in DNA methylation patterns at two gene promoters, implicating an involvement of ERβ in DNA methylation. In this study, we set out to explore this involvement on a genome-wide level, and to investigate the underlying mechanisms of this function.
Results
Using reduced representation bisulfite sequencing, we compared genome-wide DNA methylation in mouse embryonic fibroblasts derived from wildtype and ERβ knock-out mice, and identified around 8000 differentially methylated positions (DMPs). Validation and further characterisation of selected DMPs showed that differences in methylation correlated with changes in expression of the nearest gene. Additionally, re-introduction of ERβ into the knock-out cells could reverse hypermethylation and reactivate expression of some of the genes. We also show that ERβ is recruited to regions around hypermethylated DMPs. Finally, we demonstrate here that ERβ interacts with TDG and that TDG binds ERβ-dependently to hypermethylated DMPs.
Conclusion
We provide evidence that ERβ plays a role in regulating DNA methylation at specific genomic loci, likely as the result of its interaction with TDG at these regions. Our findings imply a novel function of ERβ, beyond direct transcriptional control, in regulating DNA methylation at target genes. Further, they shed light on the question how DNA methylation is regulated at specific genomic loci by supporting a concept in which sequence-specific transcription factors can target factors that regulate DNA methylation patterns.
Electronic supplementary material
The online version of this article (doi:10.1186/s13072-016-0055-7) contains supplementary material, which is available to authorised users.
Keywords: Oestrogen receptor β, Thymine DNA glycosylase, DNA methylation, Reduced representation bisulfite sequencing, Mouse embryonic fibroblasts, Mouse embryonic stem cells
Background
DNA methylation and histone modifications are ways to encode epigenetic information and play a crucial role in regulating gene expression during embryonic development [1, 2]. Aberrant epigenetic patterns are found in various human diseases, including cancer, obesity, and psychiatric disorders [3].
The most common epigenetic DNA modification is methylation at the fifth position of cytosine (5mC). The DNA methylation pattern is established and maintained by DNA methyltransferases (DNMTs) that transfer methyl groups from S-adenosyl methionine to cytosines, mainly at CpG dinucleotides [4]. DNA methylation has long been considered as fairly stable and only removable by passive mechanisms, i.e. by inhibition of DNMT activity during DNA replication. More recently, however, pathways of active DNA demethylation have been found to operate during embryonic development, primordial germ cell maturation [5], and cell differentiation [6]. Active demethylation is initiated by the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) by ten-eleven translocation (TET) proteins, a family of Fe(II)- and 2-oxoglutarate-dependent DNA dioxygenases [7]. Genome-wide mapping revealed that 5hmC is mostly found in pluripotent cells and neurons, in bodies of transcribed genes, and in gene regulatory regions (promoters and transcriptional enhancers) [8], often concomitant with the bivalent chromatin marks lysine 4 di- and tri-methylation and lysine 27 tri-methylation at histone H3 (H3K4m2/3 and H3K27m3, respectively) [9]. Such regions are poised for activation or permanent silencing during lineage commitment and terminal cell differentiation [10]. 5hmC can be further processed to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) by TET proteins. These modifications are recognised and excised by the thymine DNA glycosylase (TDG) [11] and replaced by an unmethylated cytosine by the base excision repair [12]. Evidence for active DNA demethylation by this mechanism stems from the findings that Tdg deficiency is embryonic lethal in mice [13, 14] and leads to changes in the distribution of cytosine modifications during stem cell differentiation [13, 15, 16], in particular in gene regulatory regions such as promoters and enhancers. Further, 5fC and 5caC accumulate in the absence of Tdg in embryonic stem cells (ESCs) at promoter and enhancer regions [15, 16].
An open question is how factors involved in regulation of DNA modifications are targeted to specific genomic loci. It has been suggested that transcription factor binding to their recognition sites leads to de novo methylation at proximal regions [17–19]. Further, non-coding RNAs are thought to guide DNMTs [20–22] or enzymes involved in active DNA demethylation [23] to specific regions, resulting in silencing or activation of these loci, respectively. Nevertheless, the exact mechanism of how DNA methylation is regulated at specific genomic regions is still not well understood.
Nuclear receptors (NRs) are inducible transcription factors that have been suggested to regulate epigenetic events, particularly histone modifications [24] but also DNA methylation [25–30]. Previously, we reported that the NR oestrogen receptor beta (ERβ) protects a single CpG in the promoter region of glucose transporter 4 (Glut4) from being hypermethylated [25]. Hypermethylation of this CpG in the absence of ERβ correlated with changes in expression and inducibility of Glut4. Thus, ERβ shows features of a transcription factor involved in the local regulation of DNA methylation.
ERβ is one of the two ER isoforms that mediate the physiological effects of oestrogens, the female sex hormones. It is involved in the development and functioning of the reproductive organs, but also of other tissues, e.g. the brain [31] and adipose tissue [32]. It is mostly found in the cell nucleus where it, upon activation, binds to regulatory elements [oestrogen response elements (EREs)] at target genes. There are a number of co-activators that enhance ER transcriptional activity, including chromatin-remodelling factors [33]. The ERs are not only activated by endogenous hormones, but also by pharmaceuticals and food-derived compounds such as phytoestrogens, plant protection products, and plasticisers. Exposure to a number of these compounds was shown to induce alterations of DNA methylation [34, 35].
Based on our previous results, we set out to analyse the effects of ERβ deficiency on DNA methylation at a genome-wide level. Using reduced representation bisulfite sequencing (RRBS) in mouse embryonic fibroblasts (MEFs) derived from wildtype (wt) and ERβ knock-out mice, we identified more than 8000 differentially methylated positions (DMPs). Differences in methylation correlated with changes in expression of the nearest genes and were reversible by re-introducing ERβ into knock-out MEFs. Further, we show here that ERβ interacts with TDG and that TDG is ERβ-dependently recruited to identified DMPs. Thus, we provide evidence that ERβ plays a role in regulating DNA methylation at specific genomic loci by targeting TDG to these regions.
Results
ERβ deficiency leads to methylation changes in developmental genes
To identify genomic loci that show DNA methylation changes in the absence of ERβ [GenBank: NM_207707, Swiss-Prot: O08537], we conducted reduced representation bisulfite sequencing (RRBS [36]) with MEFs derived from ERβ+/+ (wt) and ERβ−/− (βerko) mice [25]. Sequencing resulted in roughly 40 million reads of which 40 % were unambiguously mapped to the mouse genome. Around 3 × 105 CpGs were covered by the screen, which corresponds to 2.5 % of all CpGs in the mouse genome. In both cell lines, around 52 % of the covered CpGs were unmethylated (<20 % methylation), around 33 % fully methylated (>80 % methylation), and around 15 % displayed methylation between 20 and 80 % (Fig. 1a). The majority of positions were fully methylated or unmethylated in both cell types (Fig. 1b). We chose to focus on CpGs which were covered by more than four reads in both cell types and were either unmethylated (<20 % reads indicating methylation) in wt and methylated (>80 % methylation) in βerko cells or vice versa. These criteria identified 8071 DMPs, 6016 of which were highly methylated in wt and less methylated in βerko MEFs (hereafter referred to as hypomethylated DMPs) and 2055 were highly methylated in βerko and less methylated in wt MEFs (hereafter referred to as hypermethylated DMPs). Two thousand nine hundred and fifteen hypo- and 133 hypermethylated CpGs were found in clusters, forming 466 and 32 clusters, respectively. Annotation of DMPs showed the expected enrichment for promoter and intragenic regions compared to the whole genome (Fig. 1c, d; Table 1). However, gene-associated regions were more often found in hypo- than in hypermethylated loci (Table 1). Gene ontology (GO) analysis of genes containing DMPs showed enrichment for pathways involved in developmental processes for both hypo- and hypermethylated genes (Table 2).
Fig. 1.
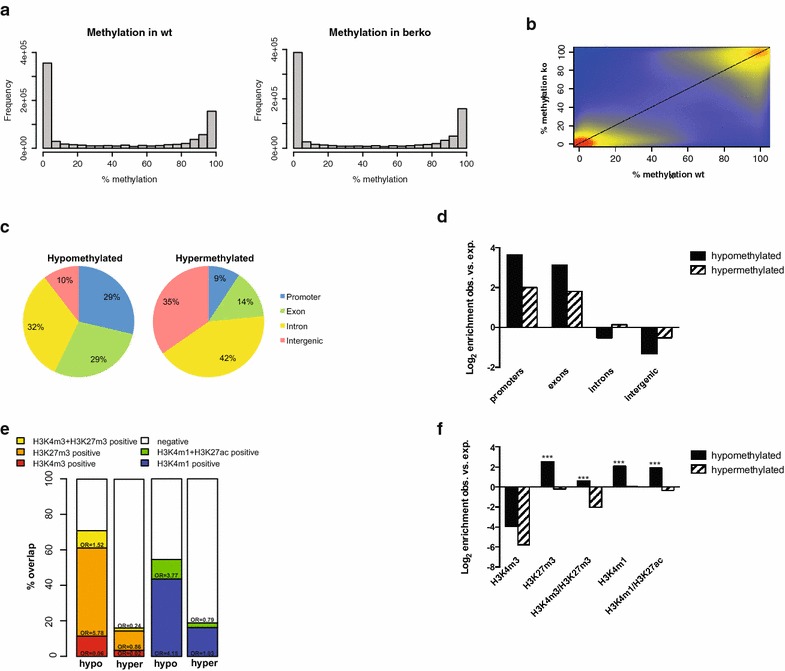
ERβ deficiency leads to altered DNA methylation patterns. a Histogram showing the distribution of methylation at the sequenced cytosines in wt and βerko MEFs. b Scatterplot of percentage (%) methylation in wt vs. βerko MEFs at cytosines covered in both cell types. c Pie chart presenting the genomic distribution of hypo- and hypermethylated positions. A position was considered hypermethylated if more than 80 % of the reads indicated methylation and hypomethylated if less than 20 % indicated methylation in βerko MEFs. d Enrichment (log2 ratios of observed over random) of hypo- and hypermethylated positions at different genomic features. e Comparison of regions identified by RRBS with datasets for histone modifications in MEFs [36] using GenomeInspector (Genomatix). Bar plots indicate percentages of hypomethylated (hypo) and hypermethylated (hyper) CpGs either marked by H3K4m3 (red), H3K27me3 (orange), both marks (yellow), or none of them (negative, white), or H3K4m1 (blue), H3K4m1 plus H3K27ac (green), or none of them (negative, white). Odds ratios (ORs) and p values according to Fisher exact test. f Enrichment (log2 ratios of observed over random) of histone modifications at hypo- and hypermethylated positions
Table 1.
Genomic distribution of hyper- and hypomethylated differentially methylated positions (DMPs)
| % in genome | % in hypermethylated | Enrichment in hypermethylated | % in hypomethylated | Enrichment in hypomethylated | |
|---|---|---|---|---|---|
| Exonic | 5.5 | 19.1 | 3.5 | 48 | 8.7 |
| Intronic | 37.4 | 38.2 | 1 | 27 | 0.7 |
| Intergenic | 57.1 | 42.6 | 0.7 | 25 | 0.4 |
| Promoter | 2.6 | 10 | 3.8 | 30.8 | 11.8 |
Table 2.
Gene ontology (GO) enrichment analysis of genes whose promoter is either hyper- or hypomethylated
| GO term | p value | # Genes (63 in total) |
|---|---|---|
| Hypermethylated genes | ||
| Embryonic morphogenesis | 4.91E−04 | 5 |
| Embryonic appendage morphogenesis | 6.39E−04 | 3 |
| Embryonic limb morphogenesis | 6.39E−04 | 3 |
| Regulation of transcription, DNA dependent | 7.62E−04 | 8 |
| Regulation of RNA metabolic process | 8.50E−04 | 8 |
| Transcription, DNA dependent | 1.01E−03 | 8 |
| RNA biosynthetic process | 1.03E−03 | 8 |
| Appendage morphogenesis | 1.12E−03 | 3 |
| Limb morphogenesis | 1.12E−03 | 3 |
| Embryonic development | 1.19E−03 | 6 |
| GO term | p value | # Genes (223 in total) |
|---|---|---|
| Hypomethylated genes | ||
| Developmental process | 5.29E−10 | 52 |
| Multicellular organismal development | 2.70E−09 | 48 |
| Anatomical structure development | 4.73E−09 | 44 |
| System development | 1.93E−08 | 41 |
| Regulation of transcription from RNA polymerase II promoter | 6.70E−08 | 20 |
| Regulation of transcription, DNA dependent | 7.84E−08 | 27 |
| Regulation of RNA metabolic process | 1.10E−07 | 27 |
| Positive regulation of cellular biosynthetic process | 1.42E−07 | 20 |
| Transcription from RNA polymerase II promoter | 1.66E−07 | 20 |
| Transcription, DNA dependent | 1.88E−07 | 27 |
Included genes show more than 30 % difference in methylation between wt and βerko MEFs, and at least 30 % of all promoter CpGs are covered by more than four reads in both cell types
Hypomethylated regions overlap with silenced transcriptional regulators
To further characterise the genomic regions containing DMPs, we compared our data obtained by RRBS with published datasets for enrichment of different chromatin marks. We found the largest significant overlaps for hypomethylated DMPs with histone 3 lysine 27 tri-methylation (H3K27m3) and lysine 4 mono-methylation (H3K4m1) in MEFs [36] (Fig. 1e, f). These marks are indicative for repressed promoter regions and poised transcriptional enhancers, respectively. Smaller, significant overlaps were found for histone 3 lysine 4 tri-methylation (H3K4m3) in combination with H3K27m3 (bivalent chromatin), and for H3K4m1 in combination with histone 3 lysine 27 acetylation (H3K27ac) (active enhancer regions). No significant overlap was found with hypermethylated positions. These results suggest that many of the hypomethylated DMPs lie in regions involved in transcriptional regulation, which are however inactive in wt MEFs.
Re-expression of ERβ complements hyper- but not hypomethylation
We selected ten hypo- and ten hypermethylated DMPs with different genomic position and histone methylation patterns (Table 3). First, we analysed DNA methylation at DMPs by methylation-dependent restriction digest followed by real-time PCR in wt and βerko MEFs as well as in βerko MEFs complemented with an ectopically expressed ERβ cDNA (βerkohERβ) (Fig. 2a). We confirmed hypomethylation for all ten hypomethylated DMPs in Table 3 (Fig. 2a, left panel) and hypermethylation for eight of the ten hypermethylated DMPs (Fig. 2a, right panel). Re-introduction of ERβ restored wt methylation at a subset (4 of 8) of hypermethylated DMPs (Fig. 2a, right panel, DMP hyper1–4) but not at hypomethylated DMPs.
Table 3.
Features of validated differentially methylated positions (DMPs)
| DMP | Genomic location | CGI | Promoter MEFs | Promoter ESCs | Enhancer MEFs | Enhancer ESCs |
|---|---|---|---|---|---|---|
| Hypo1 | Intergenic | No | H3K4m3/H3K27m3 | H3K4m3 | H3K4m1 | |
| Hypo2 (Dyx1c1) | Promoter region | Yes | H3K4m3 | H3K4m3 | ||
| Hypo3 | Intragenic (intron) | No | H3K4m1 | |||
| Hypo4 | Intragenic (intron) | No | H3K4m1 | |||
| Hypo5 | Intragenic (intron) | No | H3K4m3 | H3K4m1/H3K27ac | H3K4m1 | |
| Hypo6 | Promoter region | No | H3K27m3 | H3K27m3 | H3K4m1 | H3K4m1/H3K27ac |
| Hypo7 | Intragenic (intron) | Yes | H3K4m3/H3K27m3 | H3K27m3 | H3K4m1 | |
| Hypo8 (HoxD9) | Promoter region | Yes | H3K4m3/H3K27m3 | H3K4m3/H3K27m3 | H3K4m1 | |
| Hypo9 | Promoter region | Yes | H3K4m3/H3K27m3 | H3K4m3 | H3K4m1/H3K27ac | |
| Hypo10 | Promoter region | Yes | H3K4m3/H3K27m3 | H3K4m3/H3K27m3 | H3K4m1/H3K27ac | H3K4m1 |
| Hyper1 (HoxA9) | Promoter region | No | H3K4m3 | H3K4m3/H3K27m3 | ||
| Hyper2 (HoxA10) | Promoter region | No | H3K4m3/H3K27m3 | H3K27m3 | H3K4m1 | |
| Hyper3 | Promoter region | No | H3K4m3/H3K27m3 | H3K27m3 | H3K4m1 | |
| Hyper4 (Tnfaip2) | Promoter region | Yes | H3K4m3 | H3K4m3 | H3K4m1 | H3K4m1 |
| Hyper5 | Intragenic (intron) | No | ||||
| Hyper6 (Pitx1) | Promoter region | Yes | H3K4m3/H3K27m3 | H3K4m3/H3K27m3 | H3K4m1 | |
| Hyper7 | Intragenic (intron) | No | ||||
| Hyper8 | Intergenic | Yes | H3K4m3 | H3K4m3 | H3K4m1 |
Genomic location, presence of a CpG island (CGI), and comparison with datasets for histone modifications enriched at promoter or enhancer regions in MEFs and embryonic stem cells (ESCs) using GenomeInspector (Genomatix). Hypo and hyper refer to hypo- and hypermethylation, respectively
Fig. 2.
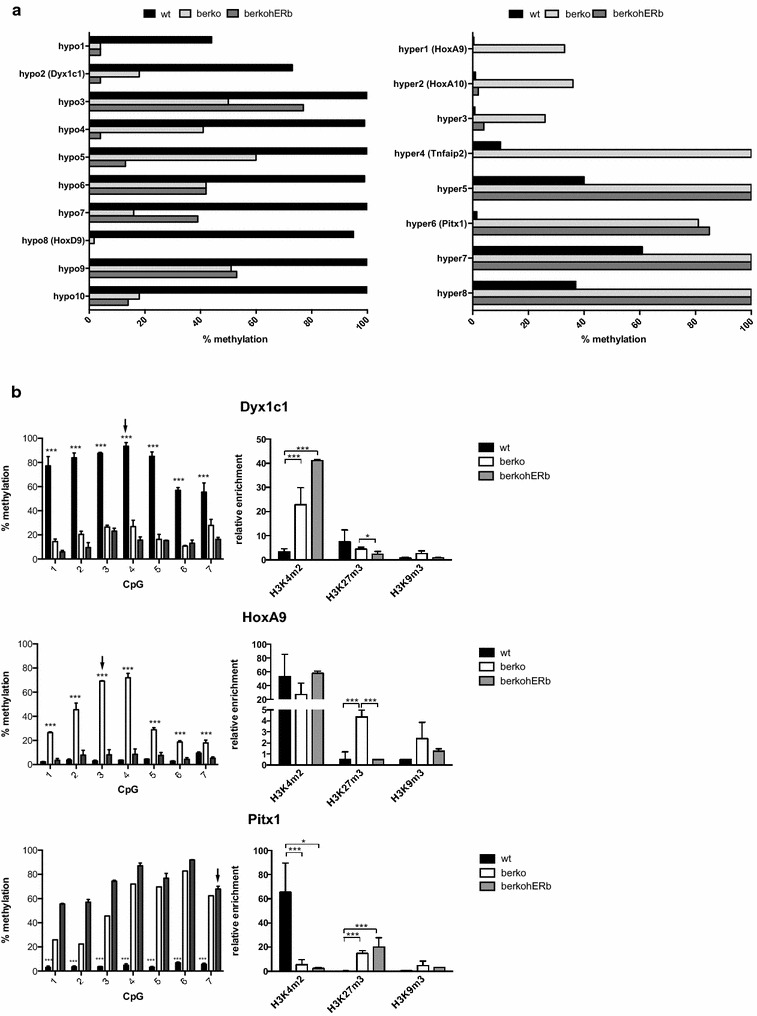
Hyper- but not hypomethylation in βerko MEFs is reversible by re-introduction of ERβ into βerko MEFs. a DNA methylation analysis of ten hypo- and eight hypermethylated positions. DNA methylation was assessed by methylation-specific enzymatic digest followed by qPCR. Positions with gene names in brackets were chosen for further analysis. b DNA methylation (left panel) and histone modifications (right panel) of differentially methylated genes in wt, βerko, and βerkohERβ MEFs. DNA methylation was assessed by pyrosequencing of bisulfite-treated DNA; black arrows mark DMPs identified by RRBS. Triple Asterisk indicates significant differences (p < 0.005) for wt vs. βerko and βerkohERβ (Dyx1c1 and Pitx1) or βerko vs. wt and βerkohERβ (HoxA9). Histone modifications were analysed using ChIP followed by qPCR and normalised to HPRT (H3K4m2 and H3K27m3) or GAPDH promoter (H3K9m3) (means + SD; n ≥ 3)
Next, we analysed DNA methylation at regions surrounding DMPs (within 200–400 bp) and the effect of the ERβ agonist DPN on the methylation by pyrosequencing. Further, chromatin marks histone 3 lysine 4 dimethylation (H3K4m2) lysine 27 tri-methylation (H3K27m3) and lysine 9 tri-methylation (H3K9m3) around the DMPs was assessed by chromatin immunoprecipitation (ChIP). We found that the DNA methylation patterns at adjacent CpGs were similar to the ones at the identified DMPs, with one exception (shown in Fig. 2b and Additional file 1). DPN treatment, even for 4 days, had no effect on the DNA methylation pattern (Additional file 1). Histone modifications in the analysed regions matched the DNA methylation patterns: hypomethylated genes, exemplified by Dyx1c1 [GenBank: NM_026314] in Fig. 2b, showed enrichment compared to a control region of both H3K4m2 and H3K27m3, reflecting a bivalent chromatin state, whereas hypermethylated genes, HoxA9 [GenBank: NM_010456] and Pitx1 [GenBank: NM_011097], displayed only enrichment for H3K4m2 in wt MEFs. This pattern was inverted in βerko MEFs, and complemented in βerkohERβ MEFs for HoxA9 for which DNA methylation was complementable (Fig. 2b).
ERβ regulates transcription of differentially methylated targets
To investigate if differential methylation is associated with transcriptional changes, we compared gene expression in wt, βerko, and βerkohERβ MEFs using the Affymetrix® Mouse Gene 1.1. ST platform. In total, we identified 4949 unique genes that showed a change in expression between wt and βerko MEFs (listed in Additional file 2). By re-introducing ERβ, 2051 genes showed differential gene expression compared to βerko MEFs (listed in Additional file 3). Two thousand five hundred and six genes were up-regulated, i.e. showed higher expression in βerko than in wt, and 2523 genes were down-regulated, i.e. showed lower expression in βerko than in wt. The genes nearest to DMPs (1494 unique genes for hypermethylated DMPs and 2475 for hypomethylated DMPs) were than compared to the differentially expressed genes. Twenty-nine percent (17 % up- and 12 % down-regulated) of the genes closest to hypermethylated DMPs and 36 % (20 % up- and 16 % down-regulated) of the ones closest to hypomethylated DMPs showed differential expression in βerko compared to wt MEFs (Fig. 3a). For both the hyper- and the hypomethylated genes, the overlap was bigger with up- than with down-regulated genes (17 vs. 12 % and 20 vs. 16 %, respectively). The expression of around 27 and 25 % of the genes overlapping with hyper- and hypomethylated genes, respectively, was rescued by re-introducing ERβ into the βerko MEFs (Fig. 3a).
Fig. 3.
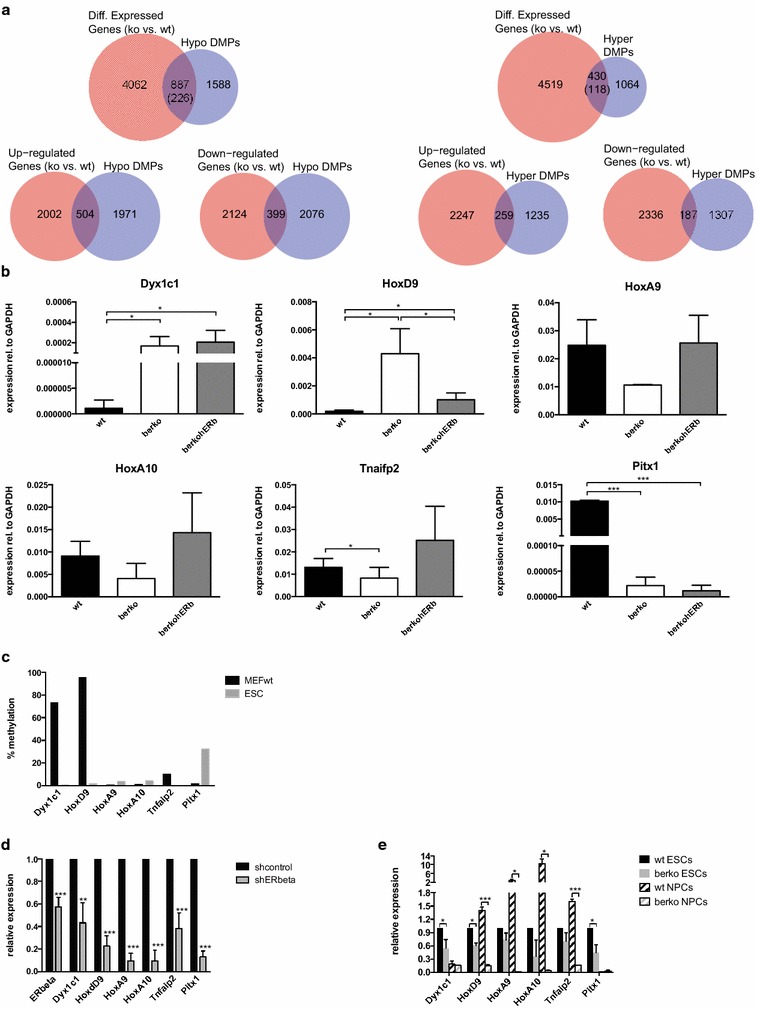
ERβ-dependent transcription of differentially methylated genes in MEFs and ESCs. a Venn diagram visualising overlaps between differentially methylated (identified by RRBS) and differentially expressed (identified by microarray expression analysis) genes in wt and βerko cells. b Gene expression analysis of hypomethylated (Dyx1c1, HoxD9), hypermethylated complementable (HoxA9, HoxA10, and Tnfaip2), and hypermethylated non-complementable genes in wt, βerko, and βerkohERβ MEFs. Gene expression was analysed by RT-qPCR (mean + SD; n ≥ 3). c DNA methylation of differentially methylated genes in wt MEFs and ESCs, assessed by methylation-specific enzymatic digest followed by qPCR. d ERβ-dependent expression of differentially methylated genes in ESCs. Gene expression was assessed by qRT-PCR 4 days after transfection with plasmid encoding for shRNA against ERβ or non-targeting control (means + SD; n ≥ 3). All the genes showed significantly decreased expression compared to shcontrol (** p < 0.01, *** p < 0.005). e ERβ-dependent expression of differentially methylated genes in wt and berko ESCs and NPCs derived thereof. Gene expression was assessed by qRT-PCR (means + SD; n ≥ 3; * p < 0.05, *** p < 0.005)
For validation and further analysis of the relationship between methylation and transcription, we chose six genes that were identified as differentially expressed between wt and βerko MEFs and validated as differentially methylated in Fig. 2a: Dyx1c1, HoxD9 [GenBank: NM_013555], HoxA9, HoxA10 [GenBank: L08757], Tnfaip2 [GenBank: NM_009396], and Pitx1. Transcription levels measured by qPCR were inversely correlated with the DNA methylation in MEFs; Dyx1c1 and HoxD9 showed higher expression in βerko and βerkohERβ cells, and HoxA9, HoxA10, Tnfaip2, and Pitx1 higher expression in wt cells (Fig. 3b). As observed for DNA methylation, wt gene expression was restored by re-introduction of ERβ for HoxA9, HoxA10, and Tnfaip2 (Fig. 3b). No effect of the ERβ ligand DPN was found on the expression of the tested genes (Additional file 1).
We also wanted to know if these genes are transcriptionally regulated by ERβ independently of their methylation status. Thus, we turned to mouse ESCs where DNA methylation is generally low. Indeed, DNA methylation was low at the investigated DMPs in ESCs (Fig. 3c). To test whether ERβ is involved in the transcriptional regulation of DMP-associated genes in ESCs, we assessed their expression in the absence and presence of ERβ in ESCs using small hairpin (sh)RNA mediated knocked-down of ERβ. As shown in Fig. 3d, knock-down of ERβ resulted in decreased expression of all of the tested genes. These results were in large parts confirmed in ESCs derived from βerko mice (Fig. 3e). Further, when these cells were differentiated towards neuronal precursor cells (NPCs), the effect of ERβ deficiency became larger, but only in those targets whose expression increased in NPCs (Fig. 3e). Genes that became silenced upon differentiation lost their ERβ dependency. Thus, all investigated targets identified as differentially methylated between wt and βerko MEFs are regulated by ERβ, but this is dependent on the cell type.
ERβ binds to regions around DMPs
To investigate direct involvement of ERβ in regulating these genes, we assessed the association of ERβ with regions around DMPs by ChIP in MEFs as well as in ESCs (Fig. 4a). ERβ was enriched in wt and βerkohERβ MEFs compared to βerko MEFs and an unrelated control region at these hypermethylated loci where re-introduction of ERβ was capable of reversing hypermethylation and increase expression of the associated gene (Fig. 4a, left panel). No enrichment was measured at hypomethylated loci in MEFs, but was evident in ESCs. This corroborates the findings that ERβ deficiency in ESCs leads to decreased transcription of these genes.
Fig. 4.
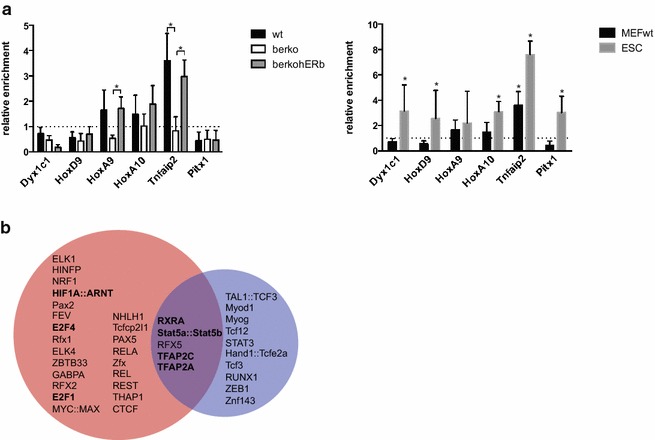
ERβ binds to regions around DMPs. a ERβ recruitment to differentially methylated genes in MEFs (left panel) and ESCs (right panel). HA-tagged ERβ was precipitated and differentially methylated regions were analysed by qRT-PCR. Asterisk indicates significant (p < 0.05) differences between wt/βerkohERβ and βerko MEFs or significant (p < 0.05) enrichment compared to binding at an unrelated, heterochromatic region on chromosome 2 [13] (means + SD; n ≥ 3). b Venn diagram visualising the enriched transcription factor binding motifs around hypo- or hypermethylated DMPs or both. Motifs that have been seen enriched in ERβ-ChIP-seq studies or that are bound by transcription factors known to interact with ERs are depicted in bold
We next investigated if there are classical ER binding sites, EREs situated around the identified DMPs. Somewhat surprisingly, we could not identify any EREs in the regions we found ERβ-enrichment using ChIP. However, the hypermethylated loci where re-introduction of ERβ was capable of reversing hypermethylation had two motifs in common: Myog and activator protein 2 (AP2). The latter has been found enriched at ERβ binding sites in a breast cancer cell line using ChIP-seq [37]. The hypomethylated loci shared a binding motif for the regulatory factor for X-box 1 (Rfx1). No interaction between this factor and ERs has been described; however, interestingly, the family of Rfx transcription factors is involved in regulation and reading of DNA methylation marks [38–40].
Motif enrichment analysis of regions around all DMPs identified 636 potential EREs (genomic positions are listed in Additional file 4); however, no enrichment for ERE motifs was found compared to the reference genome. On the other hand, significant enrichment for binding sites of other transcription factors was identified (Fig. 4b, links to haystack motif enrichment analyses are provided in Additional files 5 and 6). Among these motifs, AP2, E2F, NRF1, and CTCF recognition sites had been shown to be enriched at ERβ binding sites [37]. Further, motifs were enriched for transcription factors known to interact with ERβ: Stat5 [41], RXRα [42], and ARNT [43]. Hyper- and hypomethylated loci showed enrichment for different motifs but shared binding sites for RXRα heterodimers, Stat5a::Stat5b heterodimers, RFX5, and AP2 (Fig. 4b).
Together, these results suggest that ERβ binds to the analysed differentially methylated regions not via classical EREs but via other binding sites or transcription factors.
ERβ interacts with thymine DNA glycosylase
Next, we investigated how ERβ can regulate DNA methylation at these specific sites. We hypothesised that ERβ may target factors regulating DNA methylation to these loci. ERα was previously shown to interact with TDG [44], an essential component of active DNA demethylation [13, 15, 16, 45]. We thus examined if ERβ can interact with TDG [GenBank: NM_172552, Swiss-Prot: P56581] as well. To this end, we conducted GST-pulldown assays using ERβ coupled to glutathion-S-transferase (GST) that was immobilised to Glutathione Sepharose and incubated with recombinant TDG. GST alone was used as a negative control. As shown in Fig. 5a, more TDG was recovered from GST-ERβ bound Sepharose compared to GST only. Quantification of four experiments showed that this increase in TDG recovery was statistically significant (Fig. 5a, right panel). However, there was still a considerable amount of unspecific binding of TDG to the matrix. Thus, we complemented these experiments with far-western blot analyses, immobilising the GST-tagged ERβ to nitrocellulose membrane and probing this membrane with recombinant TDG. Subsequent probing with a specific antibody against TDG highlighted a protein co-migrating with GST-ERβ but not with the GST-tag alone. GST-tagged SUMO-1, which has been shown to interact with TDG [46], was used as a positive control. As only unspecific bands were detected on a membrane that had not been probed with TDG (Fig. 5b, right panel), these results indicated a physical interaction of ERβ with TDG.
Fig. 5.
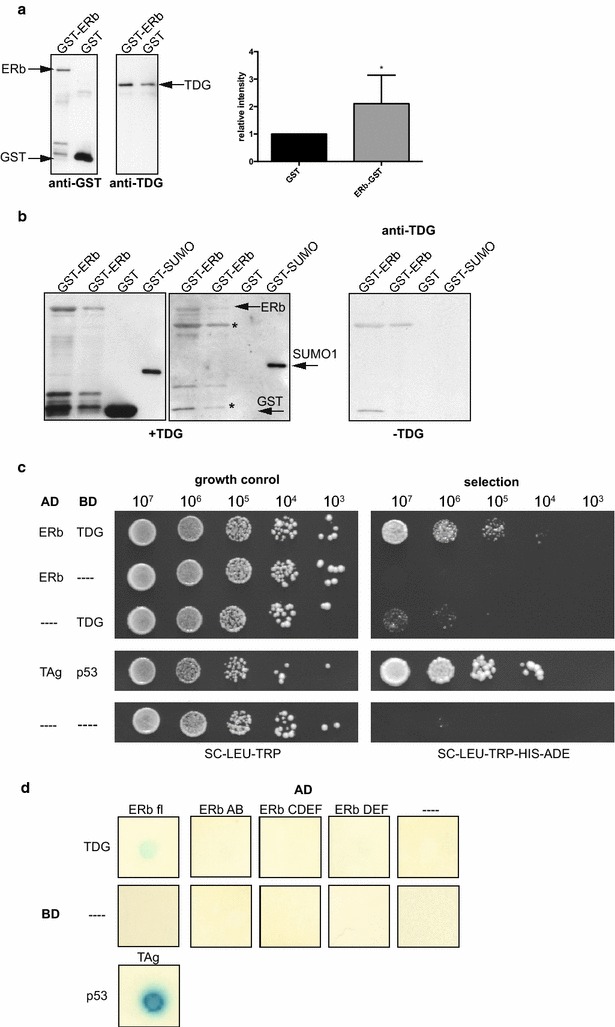
ERβ interacts directly with TDG. a Interaction of ERβ and TDG in GST-pulldown assays. GST-tagged ERβ or GST alone was immobilised on Glutathione Sepharose and incubated with recombinant TDG. The left panel shows representative western blots of the eluates using an anti-GST and an anti-TDG antibody. Quantification of four independent experiments is shown the right panel. Asterisk indicates significantly (p < 0.05) increased intensity of the band corresponding to TDG in the presence of ERβ. b Interaction of ERβ and TDG on far-western blots. GST-tagged ERβ (lanes 1 + 2), GST (lane 3), and GST-tagged SUMO-1 as a positive control (lane 4) were immobilised on a membrane and probed with recombinant TDG. Proteins were detected using antibody against GST (left panel) or TDG (middle panel). The right panel shows a membrane not probed with recombinant TDG. Asterisk marks unspecific bands. c Interaction between ERβ and TDG in yeast two-hybrid assays. ERβ fused to the AD and TDG fused to the BD of GAL4 were expressed in the yeast strain AH109. Serial dilutions of cells were spotted on control (SC-LEU-TRP, left panel) and selective medium (SC-LEU-TRP-HIS-ADE, right panel) to monitor activity of the reporter genes ADE2 and HIS3. As a positive control, murine p53 fused to GAL4 BD was used in combination with SV40 large T-antigen fused to GAL4 AD. The Gal4 BD and/or GAL4 AD alone served as negative controls (—). d Domain mapping for ERβ using yeast two-hybrid assays. Activity was tested in the yeast strain Y187 using lacZ as a reporter gene. Activity of the lacZ-encoded β-galactosidase leads to cleavage of X-gal and concomitant accumulation of a blue product (5,5′-dibromo-4,4′-dichloro-indigo). In addition to constructs as in B, individual ERβ domains (AB, CDEF, DEF) were fused to the GAL4 AD and used for transformation of Y187 cells. 106 cells were dropped onto SC plates lacking leucine and tryptophan. After 24 h of growth, cells were lysed and incubated with X-Gal for up to 17 h to monitor appearance of blue colour
To corroborate these biochemical assays, we performed yeast two-hybrid assays in the Saccharomyces cerevisiae strain AH109. This strain harbours the two Gal4-inducible reporter genes HIS3 and ADE2 and protein interactions can be assessed by growth on selective medium lacking adenine and histidine. As expression of TDG fused to the GAL4 activation domain (AD) induced some auto-activation of the reporter genes, as previously observed (data not shown), we addressed potential interactions by expressing ERβ fused to the GAL4 AD and TDG fused to the GAL4 DNA binding domain (BD). As shown in Fig. 5c, co-expression of ERβ and TDG enabled growth on selective medium. In contrast, little or no growth was detected when either factor was combined with the corresponding vector control, indicative of specific interactions between ERβ and TDG. This result was confirmed using another yeast strain (Y187) in which the lacZ gene, coding for the β-galactosidase, serves as the reporter gene to detect protein interactions (Fig. 5d). In addition to full-length ERβ, isolated domains of the receptor were tested to delineate the domain(s) responsible for interaction with TDG. We tested the AB, DEF, and CDEF domains of ERβ; however, no reporter activity above background could be detected for the interaction of TDG with any of these constructs (Fig. 5d). Together, these data provide strong evidence for a physical interaction between ERβ and TDG that requires the interaction of several domains of the receptor.
TDG is a transcriptional co-activator of ERβ and is recruited to DMPs in an ERβ-dependent manner
To test if the interaction with TDG has an effect on ERβ function, we conducted reporter gene assays, measuring ERβ transcriptional activity. To this end, Tdg−/− MEFs were co-transfected with plasmids encoding ERβ, a luciferase reporter gene driven by 3 EREs [47], and TDG. Four hours after transfection, cells were stimulated with E2 and harvested the next day for measurement of luciferase activity. Transfection of ERβ plasmid enhanced transcription of the reporter gene, and E2 treatment led to a further increase of luciferase activity (Fig. 6a). Co-transfection with TDG vector enhanced transcriptional activity of ERβ additionally. This increase was observed both in the absence (maximal 2-fold) as well as in the presence (maximal 2.5-fold) of ligand (Fig. 6a).
Fig. 6.
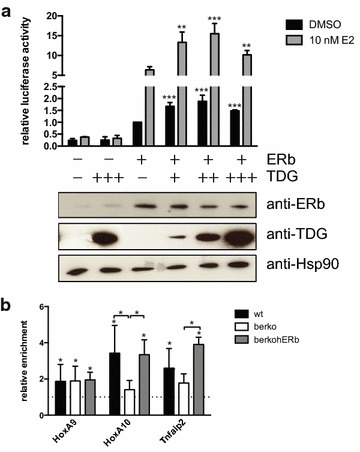
ERβ–TDG interaction affects gene regulation. a TDG increases ERβ transcriptional activity in reporter assays. Tdg−/− MEFs were transfected with plasmids encoding for the reporter gene 3× ERE-luc, TK-renilla, ERβ, and TDG in different concentrations (50, 150, and 300 ng transfected plasmid), and treated with 10 nM E2. After 16 h, firefly luciferase activity was measured and normalised against renilla luciferase activity (means + SD; n ≥ 3). TDG co-expression increased luciferase activity significantly (** p < 0.01, *** p < 0.005). Expression of ERβ and TDG was confirmed by western blot analysis. b TDG is recruited to ERβ-regulated genes in MEFs. TDG recruitment to indicated genes in wt, βerko, and βerkohERβ MEFs was analysed by ChIP-qPCR. Asterisk indicates significant (p < 0.05) enrichment compared to binding at an unrelated, heterochromatic region on chromosome 2 [13] (means + SD; n ≥ 3)
We then asked if TDG is recruited to the differentially methylated loci at which ERβ is enriched in wt MEFs. Using ChIP assays, we found TDG recruitment to these genes in wt and βerkohERβ MEFs (Fig. 6b). Notably, TDG enrichment at HoxA10 and Tnfaip2 appeared to be dependent on ERβ as it was not detectable in βerko MEFs, indicating that ERβ recruits TDG to these loci.
TDG regulates genes associated with DMPs in ESCs
The proposed role for TDG in DNA demethylation is to process 5caC and 5fC [11]. Indeed, TDG-deficient ESCs show accumulation of 5fC and 5caC at gene regulatory elements [15, 16]. We therefore reasoned that if ERβ recruits TDG to certain genomic regions to regulate DNA methylation, the loss of ERβ would result in less TDG recruitment and, hence, accumulation of 5fC and 5caC at these loci. Comparison of genome-wide 5fC data [16] with our RRBS data revealed an overlap of hyper- and hypomethylated DMPs with 5fC in wt ESCs of 10 and 12.5 %, respectively. However, in TDG-deficient cells, this overlap increased to 32 and 46 % (Fig. 7a), which corresponds to a 1.6-fold and 2.9-fold of observed vs. expected enrichment, respectively (Fig. 7b). This indicates that nearly half of the hypomethylated DMPs identified in our screen overlapped with regions where TDG processes 5fC. Thus, we tested if TDG also transcriptionally regulates the differentially methylated targets in ESCs. Gene expression was analysed in TDG-deficient ESCs and cells complemented with TDG. A clear down-regulation was observed in ESCs lacking TDG (Fig. 7c), demonstrating that they are regulated by TDG.
Fig. 7.
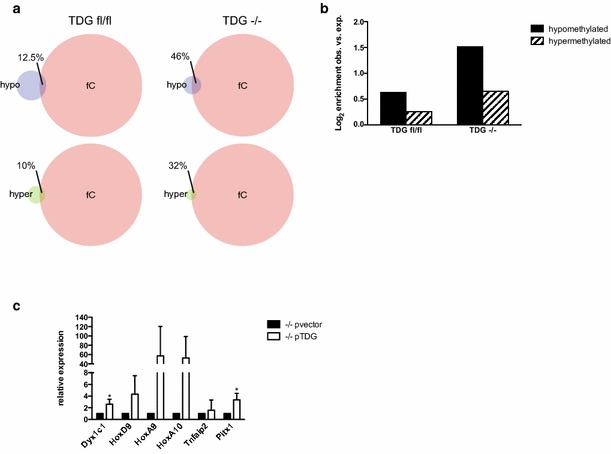
TDG regulates genes associated with DMPs in ESCs. a Venn diagram showing the overlap of hypo- and hypermethylated DMPs in βerko MEFs identified by RRBS with datasets for 5fC enrichment in deficient (Tdg −/−) or proficient (Tdg fl/fl) ESCs [16] using GenomeInspector (Genomatix). Hypo and hyper refer to the hypo- and hypermethylated regions identified in this study, respectively. b Enrichment (log2 ratios of observed over random) of 5fC in deficient (Tdg −/−) or proficient (Tdg fl/fl) ESCs at differently methylated positions. c TDG dependency of differentially methylated genes in ESCs. Gene expression in TDG-deficient (Tdg −/− pvector) and TDG-complemented (Tdg −/− pTDG) cells was assessed by qRT-PCR (means + SD; n = 3). Asterisk indicates significant (p < 0.05) increase in TDG-proficient vs. TDG-deficient cells
Discussion
Several studies suggest that NRs including the ERs are directly involved in regulating DNA methylation [25–30]. In this study, we systematically addressed the role of ERβ in regulating DNA methylation at specific loci. To this end, we carried out RRBS comparing ERβ-proficient and ERβ-deficient MEFs. Using this method, we limited the screen to CpG-rich regions and did not discriminate between 5mC and 5hmC modifications. In the analysis, we only considered DMPs that were fully methylated (80–100 %) in one cell type and unmethylated (0–20 %) in the other. Despite these constraints, we could identify more than 8000 DMPs, one-third of which was hyper- and two-thirds were hypomethylated. Around 30 % of the genes closest to hypo- or hypermethylated DMPs were differentially expressed between ERβ-proficient and ERβ-deficient cells, and expression of about one-third of them was rescued by re-expressing ERβ. Correlation between DNA methylation and expression changes was also demonstrated for selected genes associated with DMPs. Further, we found that they are bound and transcriptionally regulated by ERβ when they were actively transcribed. Further, we demonstrated that ERβ interacts physically and functionally with TDG, and that TDG associates with DMPs, in some cases ERβ-dependently, and is involved in the regulation of associated genes.
Based on our results, we propose that ERβ binds to regulatory regions of target genes and recruits TDG to these places. This interaction enhances gene expression on one hand and prevents DNA methylation on the other hand (illustrated in Additional file 7). The latter can be envisaged as the result of an interplay between TDG and the TET proteins that oxidise methylated CpGs to 5fC and 5caC, which in turn can be processed by TDG and the base excision repair to unmethylated C. Lack of ERβ results in less TDG recruitment and, hence, changes in gene transcription and DNA methylation, both hyper- and hypomethylation depending on the activity state of the respective gene. Hypermethylated DMPs were associated with genes actively transcribed in MEFs whose expression and methylation patterns could be restored upon re-introduction of ERβ into knock-out cells. This implicates an active demethylation mechanism involving TDG, as described previously for other genes in somatic cells [27, 28, 48, 49]. Hypomethylated genes, on the other hand, were inactivated in MEFs but transcribed and regulated by ERβ and TDG in ESCs, and hypomethylated DMPs overlapped remarkably with loci where 5fC accumulated in TDG-deficient ESCs [16]. As this mark is not recognised by the maintenance DNMT, 5fC accumulation leads to passive demethylation during cell division. Thus, at genes that become silenced during differentiation from ESCs to MEFs, lack of ERβ, and hence diminished TDG recruitment, could lead to erroneous passive demethylation, resulting in hypomethylation in the differentiated cells. This is corroborated by the findings that hypomethylated DMPs overlap with repressive chromatin marks in MEFs (Fig. 1e, f), that associated genes are involved in embryonic development (Table 2), and that genes that become repressed in NPCs are not regulated by ERβ anymore (Fig. 3e). At this point, we do not have an explanation for the occurrence of hypermethylated positions whose methylation pattern is not revertible by re-expression of ERβ other than clonal differences between wt and βerko cells.
Notably, as in our previous study [50], we could not find any effect of oestrogen on ERβ’s function at the loci investigated, neither on its transcriptional activity nor on its effects on DNA methylation and interaction with TDG. In contrast, TDG was shown to interact with ERα and enhance its transcriptional activity, in the presence of ligand [44]. The interaction between ERα and TDG is mediated by SRC-1, and overexpression of this co-factor in COS1 cells resulted in enhancement of ERα’s ligand-independent transcriptional activity by TDG [51]. Thus, availability of other factors involved in ER–TDG interaction in the different cell systems could explain the difference between the two receptor isoforms. On the other hand, ligand-independent function of ERβ has been shown previously in different contexts, e.g. when modulating ERα-induced gene expression in breast cancer cells [52]. Additionally, we have found that ERβ is tightly bound to the chromatin in extracts of different cell types even in the absence of ligand (our unpublished observation).
We could neither identify any classical EREs in the regions that ERβ bound to, nor were EREs found enriched around differentially methylated sites. This suggests that ERβ binds to these regions in complex with other factors. Trans-recruitment of big transcription factor complexes has been shown to be important for functional enhancer regions [53]. Indeed, we could find enriched motifs for TF that are known to interact with ERs, such as Stat5, RXR, and ARNT. Further, we found motifs that have been shown to be enriched in an ERβ-ChIP-seq study [37]. These included AP2 motifs that we found on all the investigated hypermethylated targets whose methylation and expression was restored by re-expressing ERβ but not in the non-complementable ones. The selected hypomethylated DMPs had Rfx1 sites in common, and motifs for Rfx factors were enriched on a genome-wide level. To our knowledge, no interaction between Rfx and ERβ has been described but, interestingly, these transcription factors, as well as CTCF that is enriched at hypomethylated DMPs, are implicated in regulation and reading of DNA methylation marks [38–40, 54]. Additionally, Our results imply that the function ERβ exerts at the identified targets differs from the classical ligand-induced ER signalling pathway. How ERβ binds to these loci and if and how the identified factors interact with ERβ has to be determined in future studies.
Although the factors that regulate changes in DNA methylation patterns during cell differentiation are identified, how they are recruited to and regulated at specific genomic loci is still unclear. R loop formation at CpG islands has been shown to exclude DNMT3a and DNMT3b, hence preventing methylation of these structures [55]. On the other hand, it has been suggested that locus-specific de novo methylation is induced by recruitment of DNMTs by non-coding RNAs [20–22] or by proximal sequence elements providing for specific transcription factor binding [17]. The involvement of transcription factors in regulating DNA methylation patterns has also been shown at distal regulatory regions with low methylation (LMRs) [18, 19]. The data presented here further support the notion that transcription factors can target DNA methylation and demethylation events, and provide a mechanism underlying this role. We suggest that interaction with factors regulating DNA methylation patterns is not limited to ERβ but could be general principle applying for many sequence-specific transcription factors. Indeed, several studies report an association between NR-induced transcription and DNA methylation changes [27, 28, 49].
Conclusion
The data presented here suggest that ERβ can modulate DNA methylation patterns at specific genomic loci by interaction with TDG. This implies an important regulatory function of ERβ during cell differentiation and could be part of a mechanism underlying epigenetic alterations observed after exposure to compounds disrupting ER function in early development. Further, it supports a general concept in which transcription factors regulate the DNA methylation state at specific regions in the genome.
Methods
Plasmids and antibodies
Mission shRNA against ERβ was obtained from Sigma–Aldrich. Expression plasmid for HA-tagged mERβ (pSSH25-mERβ) was constructed by inserting PCR-amplified mERβ cDNA into pSHH25 [56] using XhoI and BlgII restriction sites. pSSH25-TDG for mammalian expression of HA-fused TDG [56], pPRS220 for yeast expression of Gal4-AD-fused TDG [46], 3× ERE-luc [47], and pSG5hERβ [57] used in luciferase assays have been published. pRL-TK for normalisation of luciferase activity was purchased from Promega. GST-ERβ was constructed by cloning cDNA encoding for human ERβ into pGEX-6P-3 (GE Healthcare) using BamHI and XhoI restriction sites. pACT2-ERβ was obtained by cloning cDNA encoding for human ERβ into pACT2 (Clontech) using SmaI and XhoI restriction sites. Antibodies used were as follows: rat monoclonal anti-HA (3F10) from Roche Applied Science, rabbit monoclonal anti-ERβ (05-824), rabbit polyclonal anti-H3K4m2 (07-030), and rabbit polyclonal anti-H3K27m3 (07-449) from Millipore, rabbit polyclonal anti-H3K9m3 (060-050) from Diagenode, and mouse monoclonal anti-Hsp90 (F-8) from Santa Cruz Biotechnology, Inc.
Cell culture and transfections
MEFs from wt and βerko mice [25] and βerko MEFs complemented with ERβ (βerkohERβ [25]) as well as Tdg−/− MEFs [56] were kept in high-glucose DMEM supplemented with 10 % FCS, 1 mM l-glutamine, 1 mM sodium pyruvate, and 1× non-essential amino acids and 5 μg/ml blasticidin (βerkohERβ). For stimulation with ER agonists, cells were put for at least 2 days into DMEM with 5 % dextran-coated charcoal-treated serum and treated with 10 nM E2 or DPN.
ESCs [58] were thawed on feeder cells in ESC medium (high-glucose DMEM supplemented with 15 % heat-inactivated FCS, 1 mM sodium pyruvate, 1× non-essential amino acids, and 0.1 mM β-mercaptoethanol) containing 1 U/μl LIF (Millipore). Upon feeder removal, cells were maintained in 2i medium (serum-free N2B27 [59] supplemented with 2i inhibitors [60], CHIR99021 (3 μM), and PD0325901 (1 μM), obtained from the division of signal transduction therapy, University of Dundee) containing 1 U/μl LIF. TDG knock-out ESCs complemented with TDG (Tdg−/− pTDG) or empty vector (Tdg−/− pvector) were maintained in 2i medium containing 1 μg/ml puromycin.
NPCs were generated from ERβ+/+and ERβ−/− ESCs, kindly contributed by Prof. Jan-Åke Gustafsson, via Embryoid Bodies (EBs). To generate EBs, feeder-independent mESCs were grown in serum-free 2i media for 2–3 passages and further split at a density of 5.5 × 105 cells/ml in EB medium (15 % knoc-kout Serum Replacement, 2 mM l-Glutamine (200 mM), 10 mM HEPES (1 M), 0.1 mM non-essential amino acids MEM (10 mM), 0.1 mM β-mercaptoethanol (50 mM), 10 µg/ml Gentamicin (10 mg/ml) in knoc-kout DMEM [high glucose with sodium pyruvate) (Invitrogen)] in a 100-mm non-adherent dish (non-tissue culture treated). The medium was replaced daily for 3 days with taking care of not to disturb cell aggregates. EBs were grown for 3 days in suspension and collected in a 50-mL conical tube containing 10 mL fresh EB medium, supplemented with 15 % ES-qualified FBS. EBs were transferred to gelatin-coated tissue culture plates at a ratio of 1:10 by gentle pipetting and cultured for 24 h at 37 °C and 5 % CO2. Medium was replaced by ITS–Fibronectin medium (495 ml knock-out DMEM/F12 without HEPES, 775 mg glucose, 36.5 mg l-glutamine, 1.2 g NaHCO3 (Invitrogen), and 5 ml ITS media supplement 100×, R and D systems). Recombinant bovine fibronectin (R and D Systems) was added to this medium at 5 µg/ml concentration. Cells were cultured for 6–8 days at 37 °C and 5 % CO2. ITS–Fibronectin medium was changed every other day. During this period, a monolayer grew from the attached EBs. ITS/Fibronectin medium was removed from the cell culture and the attached cells were washed twice with 10 mL of sterile DPBS. Cells were dissociated with Accutase solution and collected in a 15-mL tube containing ITS–Fibronectin medium by gentle pipetting. Cell clumps were removed (remnants of EB) by allowing the tube to stand just long enough to allow the cell clumps to settle to the bottom (about 5 min). Suspended cells were transferred to a new 15-mL tube by gentle pipetting and centrifuged for 5 min at 1500 rpm to pellet the cells. Cell pellets were resuspended in MN3FL medium (495 ml knock-out DMEM/F12 without HEPES, 775 mg glucose, 36.5 mg l-glutamine, 845 mg NaHCO3 (Invitrogen), 5 ml N2-Max media supplement 100×, 10 ng/ml FGF basic (R and D systems), and 1 µg/ml Laminin, Novus Biologicals). Cells were plated on poly-l-ornithine/laminin-coated dishes at a seeding density of 60,000 cells/cm2. Cells were fed MN3FL medium every day for 4–6 days until cell confluency reached close to 100 %. MN3FL medium was removed from the cell culture and attached cells were washed with sterile DPBS and dissociated with Accutase solution and passaged on poly-l-ornithine/laminin-coated dishes. Selected NPCs were grown further up to 6–10 passages.
For transfection with HA-ERβ, MEFs or ESCs were seeded onto 15-cm plates and transfected the following day using JetPRIME reagent (Polyplus transfection) according to the manufacturer’s protocol. One day after transfection, cells were harvested for ChIP.
For transfection with shRNA constructs, ESCs were seeded into 6-well dishes and transfected the following day using JetPRIME reagent according to the manufacturer’s protocol. Twenty-four hours after transfection, 1 μg/ml puromycin was added to the medium. Transfected cells were selected for 4 days changing medium daily, and harvested for RNA extraction.
Transfection for luciferase assays was performed using JetPEI (Polyplus transfection) in 24-well plates with 50 ng pRL-TK, 50 ng pSG5-hERβ, 100 ng 3× ERE-luc, and varying concentration 50, 150, and 300 ng) of pSHH25-TDG per well, according to the manufacturer’s protocol.
RRBS
Library preparation for RRBS was carried out as described [61]. In brief, genomic DNA derived from wt and βerko MEFs was isolated using the QIAamp DNA mini kit (Qiagen). Three batches of genomic DNA of each cell type were pooled, and 1 μg was digested with 20 U MspI overnight. The reaction was stopped by addition of 1 μl 0.5 M EDTA and purified using MinElute gel extraction kit (Qiagen). Subsequently, DNA fragments were end-repaired and A-tailed by incubation with 5 U Klenow fragment (New England Biolabs Inc.) and 0.5 mM dATP and 0.05 mM dGTP and dCTP at 30 °C for 20 min followed by 20 min at 37 °C. After purification using the MinElute gel extraction kit, methylated Illumina standard adapters were ligated to the fragments overnight at 16 °C using 400 U T4 ligase (New England Biolabs Inc.). The fragments were then separated on a 3 % Nusieve 3:1 agarose 0.5× TBE gel and 160–340 bp fragments were excised and purified using the MinElute gel extraction kit. The purified DNA was subjected to two rounds of bisulfite conversion using the EZ DNA methylation kit (Zymo Research). Subsequently, the final library was prepared by 19 cycles of PCR amplification and purification using the MinElute gel extraction kit. Libraries were sequenced on the Illumina Genome Analyser IIx following the manufacturer’s protocol at the D-BSSE, ETH Basel. These data have been deposited in NCBI's Gene Expression Omnibus [64] and are accessible through GEO Series accession number GSE72230 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72230).
RRBS methylation analyses
Mapping of obtained sequences was performed using the Genomatix mining station (http://www.genomatix.de/solutions/genomatix-mining-station.html). Annotation and correlation analyses were carried out using the Genomatix Regionminer. Statistical analyses were carried out using R [62] or GraphPad Prism. Statistical significance was assessed using the paired t test or Fisher exact test. The level of significance was selected as p < 0.05.
Bisulfite treatment and pyrosequencing
Genomic DNA (200–500 ng) was bisulfite treated and purified using the EZ DNA Methylation kit (Zymo Research). One micro litre of the converted DNA was used for nested PCR amplification (for primer sequence see Additional file 8), and the PCR product was sequenced by pyrosequencing in a Pyromark Q24 (Qiagen).
Methylation-sensitive restriction enzyme digest
Five micro gram of genomic DNA was digested with 50 U HpaII or 100 U MspI overnight. Subsequently, enzymes were removed by digest with proteinase K for 30 min at 40 °C and digested fragments were analysed by real-time PCR (primers listed in Additional file 8) using Rotor-Gene SYBR Green PCR Kit on a Rotor-Gene RG-3000 (Qiagen).
RNA isolation, cDNA production, and real-time PCR
RNA was isolated using Tri (Sigma) according to the manufacturer’s recommendations. One microgram of total RNA was treated with DNAseI (New England Biolabs Inc.) and reverse transcribed using random hexamer primers (Fermentas). One microlitre of the resulting cDNA was used for real-time PCR using Rotor-Gene SYBR Green PCR kit on a Rotor-Gene RG-3000 (Qiagen). Gene transcripts were normalised to the Gapdh RNA content (primers listed in Additional file 8). All results are based on the ΔΔCT method and represent the mean of at least three independent experiments.
Gene expression microarray analysis
Gene expression microarray data were generated using the Affymetrix® Mouse Gene 1.1. ST platform. The data were processed using the Affy package with software R. Global background correction was performed through the robust multi-array average (RMA), and the statistical significance was calculated by the empirical Bayes model. The expression probes with adjusted p value less than 0.01 and with at least 0.5-fold change between two conditions are defined as differentially expressed probes.
Motif enrichment analysis
Enriched transcription factor motifs as well as classical ERE motif finding were identified using Haystack software (PMID: 24395799) according to the manuals. Briefly, genomic regions containing 200 bp around the DMPs were selected for motif analysis and genomic regions containing 200 bp around the CpG sites that can be covered by RRBS experiments were chosen as the reference genome (background). The classical ERE motif was defined as the consensus sequence of GGTCAnnTGACC (PMID: 12824376).
Chromatin immunoprecipitation
ChIP assays were performed as described [43] with minor modifications. Cells were grown to confluency on 15-cm dishes. Chromatin was cross-linked for 10 min with 1 % formaldehyde (Pierce Biotechnologies Inc.) and the reaction was stopped by addition of 125 mM glycine for 10 min. After washing twice with cold PBS, cells were harvested in PBS by centrifugation at 4 °C at 600g. Nuclei were isolated by sequential 5-min incubation on ice with 500 μl cold nucleus/chromatin preparation (NCP) buffer I (10 mM HEPES pH 6.5, 10 mM EDTA, 0.5 mM EGTA, 0.25 % Triton X-100) and twice cold NCP buffer II (10 mM HEPES pH 6.5, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl). Pelleted nuclei were lysed in 200–400 μl lysis buffer [50 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.5 % Triton X-100, 1 % SDS, 1 mM PMSF, 1× Complete (Roche)] for 10 min followed by sonication for 15 cycles (30 s on, 30 s off, power high) using a Bioruptor sonicator (Diagenode). After centrifugation at 4 °C and 14,000g for 10 min, chromatin concentration was estimated by absorbance at 260 nm. Hundred microgram (for HA, TDG and H3K9m3 ChIPs) or 50 µg (for H3K4m2 and H3K27m3 ChIPs) of chromatin were diluted ten times in IP buffer I (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 150 mM NaCl, 0.1 % Triton X-100, 1 mM PMSF, 1× Complete) for HA and TDG ChIPs or IP buffer II (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1 % Triton X-100, 1 mM PMSF, 1× complete) for histone ChIPs. Diluted chromatin was pre-cleared at 4 °C for 1 h with 40 µl of a 50 % slurry of magnetic Protein G beads (Invitrogen) preblocked with 1 mg/ml BSA and 1 mg/ml tRNA. Pre-cleared chromatin was incubated with 2–5 µg of the respective antibody (listed in Additional file 9) overnight at 4 °C and immuno-complexes were precipitated with 40 µl of a 50 % slurry of blocked Protein G beads at 4 °C for 2 h. Subsequently, beads were serially washed with 500 µl wash buffer I (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.1 % SDS, 1 % Triton X-100), 500 µl wash buffer II (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 500 mM NaCl, 0.1 % SDS, 1 % Triton X-100), and 500 µl wash buffer III (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 1 % sodium deoxycholate, 1 % NP-40). For TDG ChIPs, beads were washed once with 500 µl wash buffer I and twice with 500 µl wash buffer II. After two additional washes with 500 µl TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA), complexes were eluted by incubating twice with 250 µl extraction buffer (1 % SDS, 0.1 M NaHCO3) at 65 °C for 15 min. Crosslink was reversed by incubation at 65 °C for 4 h in the presence of 200 mM NaCl. Subsequently, proteins were removed by incubation with proteinase K (50 µg/ml) in the presence of 10 mM EDTA at 45 °C for 1 h, and DNA was purified by phenol/chloroform extraction and ethanol precipitation. The isolated DNA fragments were analysed by qPCR (primers listed in additional file 8) using Rotor-Gene SYBR Green PCR kit on a Rotor-Gene RG-3000 (Qiagen).
GST-pulldown assays
GST-ERβ and GST were expressed in E.coli BL21 at 15 °C overnight upon induction with 0.2 mM IPTG. Cells were harvested and lysed in 2 ml GST buffer [10 mM Tris–HCl, pH 8; 50 mM NaCl; 5 % Glycerol; 1 mM DTT; 0.1 mM PMSF; 1× complete (Roche)] by sonication in a Bioruptor® UDC-200 (Diagenode). After sonication, 1 % Triton X-100 was added to the lysates, and samples were gently mixed for 30 min at 4 °C and centrifuged at 12,000×g for 10 min at 4 °C. Lysates were centrifuged at 12,000×g for 10 min at 4 °C and supernatants transferred to fresh tubes and protein concentration was assessed using Bradford assay. Glutathione Sepharose high-performance beads (GE Healthcare) were prepared to 50 % slurry as described in the manufacturer’s protocol. The lysates were diluted in 500 µl GST buffer to a final total protein concentration of 0.8 µg/µl, 10 µl glutathione Sepharose 50 % slurry was added, and the mixture was incubated at 4 °C for 2 h with agitation. Three hundred and fifty nanogram recombinant hTDG was added, followed by incubation at 4 °C for 2 h with agitation. The beads were washed three times with GST washing buffer (10 mM Tris–HCl, pH 8; 80 mM NaCl; 5 % Glycerol) and once with GST wash buffer containing 150–300 mM NaCl. Proteins were eluted by boiling in Laemmli buffer at 95 °C for 5 min and analysed using SDS-PAGE followed by western blot. Band intensity was quantified using ImageJ.
Western blot and far-western blot analyses
The insoluble fraction of luciferase assay-lysates containing chromatin-bound proteins was used for protein expression analysis by immunoblotting using anti-TDG, anti-ERβ, and anti-Hsp90 antibodies.
For far-western blot analysis, GST-tagged proteins were expressed as described above. GST-ERβ was purified by affinity chromatography using glutathione Sepharose (GE Healthcare) according to the manufacturer’s protocol. Proteins were eluted by boiling in Laemmli buffer at 95 °C for 5 min. Proteins were fractionated by SDS-PAGE and transferred to a nitrocellulose membrane. The proteins on the membrane were denatured by incubation with 6 M guanidine-HCl (GuHCl) in AC buffer (20 mM Tris–HCl, pH 7.6; 100 mM NaCl; 10 % Glycerol; 0.1 % Tween-20; 2 % skim milk powder; 1 mM DTT; 0.5 mM EDTA) for 30 min at RT, and renatured by washing steps with 3 M GuHCl for 30 min at RT, 1 M GuHCl for 30 min at RT, 0.1 M GuHCl for 30 min at 4 °C, and AC buffer only for 1 h at 4 °C. Upon blocking with 5 % skim milk powder in TBST for 1 h at RT, the membrane was incubated at 4 °C overnight with gentle shaking in protein-binding buffer (20 mM Tris–HCl, pH 8; 50 mM NaCl; 10 % Glycerol; 0.1 % Tween-20; 2 % skim milk powder; 1 mM DTT) containing 7–9 µg recombinant TDG [63]. The membranes were washed thoroughly 3 times with TBS containing 0.2 % NP-40. Subsequently, bound proteins were detected using antibodies against TDG and GST.
Yeast two-hybrid analysis of ERβ–TDG interaction
The Matchmaker™ yeast two-hybrid system (Clontech) was used. Bait and trait proteins were cloned into plasmids encoding the binding and AD of the Gal4 protein, respectively. The S. cerevisiae strains AH109 (MATa, trp1–901, leu2–3, 112, ura3–52, his3–200, gal4Δ, gal80Δ, LYS2::GAL1UAS–GAL1TATA–HIS3, MEL1, GAL2UAS–GAL2TATA–ADE2, URA3::MEL1UAS–MEL1TATA–lacZ) and Y187 (MATα, ura3–52, his3–200, ade2–101, trp1–901, leu2–3, 112, gal4Δ, met−, gal80Δ, URA3::GAL1UAS–GAL1TATA–lacZ) were co-transformed with 50–500 ng of bait and trait plasmids according to the Clontech manual. For AH109, interactions were assessed by spotting serial dilutions of cells on selective medium (SC-LEU-TRP-ADE-HIS) and incubating them for 2–4 days at 30 °C. β-Galactosidase activity was assayed using the Y187 strain (Clontech manual). Briefly, 106 cells were dropped on SC medium selecting for the plasmids (SC-LEU-TRP) and grown for 24 h at 30 °C. Cells were transferred to filter paper (Filtrak, 80 g/m2) before snap-freezing in liquid nitrogen and subsequent thawing for cell lysis. The filter with the lysed cells was soaked with 2 ml of Z buffer (100 mM Na phosphate buffer pH 7.0, 10 mM KCl, 1 mM MgSO4, 33 µM β-mercaptoethanol, 817 µM X-Gal) and incubated at 30 °C for up to 17 h.
Luciferase assays
Four hours after transfection, 10 nM E2 was added to the medium. The next day, luciferase reporter assays were performed using the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s protocol: cells were lysed in passive lysis buffer (Promega) and firefly and renilla luciferase activity measured in 96-well plates using a luminometer Centro LB 960 (Berthold technologies).
Authors’ contributions
YL participated in designing experiments for the revision, and conducted gene expression analyses by Affymetrix microarrays and transcription factor motif finding. WD conducted far-western blot analyses, yeast 2-hybrid assays, and the luciferase assays. CK conducted yeast 2-hybrid assays. NB and CZ carried out bioinformatics on the RRBS data. GB and MV conducted the experiments in the βerko ESCs. PS participated in the design and coordination of the study. JR conceived the study, carried out RRBS, DNA methylation, gene expression, and ChIP assays, performed data analyses, and wrote the manuscript. All authors contributed to manuscript writing. All authors read and approved the final manuscript.
Acknowledgements
The authors thank Dr. Ivan Nalvarte for thorough review of the manuscript and valuable scientific inputs, Dr. Faiza Noreen for statistical advice, and Dr. Rabi Murr for providing methylated Illumina standard adapters. This work was supported by the Swiss National Science Foundation, the University of Basel Research Fund, the Swedish Research Council Formas (JR), and the National Natural Science Foundation Grant 31471212 (YL).
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- 5caC
5-carboxylcytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- AD
activation domain
- AP2
activator protein 2
- ARNT
aryl hydrocarbon receptor nuclear translocator
- BD
binding domain
- BSA
bovine serum albumin
- CGI
CpG island
- ChIP
chromatin immunoprecipitation
- DMEM
Dulbecco’s modified eagle medium
- DMPs
differentially methylated positions
- DNMT
DNA methyl-transferase
- DPN
diarylpropionitrile
- E2
17-β-estradiol
- EDTA
ethylene diamine tetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ERα
oestrogen receptor α
- ERβ
oestrogen receptor β
- ERE
oestrogen response element
- ESCs
embryonic stem cells
- FCS
foetal calf serum
- Glut4
glucose transporter 4
- GO
gene ontology
- GST
glutathion-S-transferase
- H3K4m3
lysine 4 tri-methylation at histone H3
- H3K27m3
lysine 27 tri-methylation at histone H3
- H3K9m3
lysine 9 tri-methylation at histone H3
- HA
hemagglutinin
- IPTG
isopropyl-β-d-thiogalactopyranoside
- LIF
leukaemia inhibitory factor
- LMR
low methylated regions
- MEFs
mouse embryonic fibroblasts
- Myog
myogenin
- NPC
neuronal precursor cell
- NR
nuclear receptor
- NRF1
nuclear respiratory factor-1
- PMSF
phenylmethylsulfonyl fluoride
- Rfx
regulatory factor for X-box
- RRBS
reduced representation bisulfite sequencing
- SDS
sodium dodecyl sulphate
- shRNA
small hairpin RNA
- RXR
retinoid X receptor
- Stat
signal transducers and activators of transcription
- SUMO
small ubiquitin-related modifier
- TDG
thymine DNA glycosylase
- TET
ten-eleven translocation
- wt
wildtype
Additional files
10.1186/s13072-016-0055-7 Additional DNA methylation and gene expression analyses. A: DNA methylation of regions surrounding DMPs. DNA methylation of four additional differentially methylated genes in wt, βerko, and βerkohERβ MEFs, assessed by pyrosequencing of bisulfite-treated DNA. Black arrows mark DMPs identified by RRBS. B: Effect of the ERβ-agonist DPN on DNA methylation of identified target genes. DNA methylation of differentially methylated genes in wt, βerko, and βerkohERβ MEFs upon 0, 6, 24, and 96 h treatment with 10 nM DPN, assessed by pyrosequencing of bisulfite-treated DNA. C: Effect of the ERβ-agonist DPN on expression of identified target genes. Gene expression analysis in wt, βerko, and βerkohERβ MEFs upon a 0 and 6 h treatment with 10 nM DPN, analysed by RT-qPCR.
10.1186/s13072-016-0055-7 Genes differently expressed in wt and βerko MEFs. List of genes that show changed expression in βerko compared to wt MEFs assessed by microarray gene expression analyses using the Affymetrix® Mouse Gene 1.1. ST platform. The list includes fold change, p value, chromosomal location, gene name and annotation.
10.1186/s13072-016-0055-7 Genes differently expressed in βerko and βerkohERβ MEFs. List of genes that show changed expression in βerkohERβ compared to βerko MEFs assessed by microarray gene expression analyses using the Affymetrix® Mouse Gene 1.1. ST platform. The list includes fold change, p value, chromosomal location, gene name and annotation.
10.1186/s13072-016-0055-7 Genomic location of the identified EREs.
10.1186/s13072-016-0055-7 Links to enriched transcription factor motifs identified using Haystack software.
10.1186/s13072-016-0055-7 Links to enriched transcription factor motifs identified using Haystack software.
10.1186/s13072-016-0055-7 Model of ERβ-TDG interaction and its effects. ERβ binds to regulatory regions of target genes and recruits TDG to these places. This interaction enhances gene expression on one hand and affects DNA methylation on the other hand. The effects on the DNA methylation pattern depends on the activity of the respective gene in a given cell type.
10.1186/s13072-016-0055-7 Primers used for real-time PCR and Pyrosequencing. MSD: methylation sensitive restriction enzyme digest, pyro: pyrosequencing.
10.1186/s13072-016-0055-7 Antibodies used for western blotting and ChIP.
Footnotes
Yun Liu and William Duong have contributed equally to this work
Contributor Information
Yun Liu, Email: yliu39@fudan.edu.cn.
William Duong, Email: william.duong@gmx.ch.
Claudia Krawczyk, Email: claudiakrawczyk@web.de.
Nancy Bretschneider, Email: bretschneider@genomatix.de.
Gábor Borbély, Email: gabor.borbely@swetox.se.
Mukesh Varshney, Email: mukesh.varshney@ki.se.
Christian Zinser, Email: zinser@genomatix.de.
Primo Schär, Email: primo.schaer@unibas.ch.
Joëlle Rüegg, Email: joelle.ruegg@swetox.se.
References
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15(2):93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen M, Zhang L. Epigenetic mechanisms in developmental programming of adult disease. Drug Discovery Today. 2011;16(23–24):1007–1018. doi: 10.1016/j.drudis.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patra SK, Patra A, Rizzi F, Ghosh TC, Bettuzzi S. Demethylation of (Cytosine-5-C-methyl) DNA and regulation of transcription in the epigenetic pathways of cancer development. Cancer Metastasis Rev. 2008;27(2):315–334. doi: 10.1007/s10555-008-9118-y. [DOI] [PubMed] [Google Scholar]
- 5.Saitou M, Kagiwada S, Kurimoto K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development. 2012;139(1):15–31. doi: 10.1242/dev.050849. [DOI] [PubMed] [Google Scholar]
- 6.Coskun V, Tsoa R, Sun YE. Epigenetic regulation of stem cells differentiating along the neural lineage. Curr Opin Neurobiol. 2012;22(5):762–767. doi: 10.1016/j.conb.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10(16):2662–2668. doi: 10.4161/cc.10.16.17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013;25(3):289–296. doi: 10.1016/j.ceb.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao F, Xia Y, Wang J, Luo H, Gao Z, Han X, et al. Integrated detection of both 5-mC and 5-hmC by high-throughput tag sequencing technology highlights methylation reprogramming of bivalent genes during cellular differentiation. Epigenetics Off J DNA Methylation Soc. 2013;8(4):421–430. doi: 10.4161/epi.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 11.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286(41):35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs AL, Schar P. DNA glycosylases: in DNA repair and beyond. Chromosoma. 2012;121(1):1–20. doi: 10.1007/s00412-011-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470(7334):419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 14.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146(1):67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung HL, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153(3):692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153(3):678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 2011;43(11):1091–1097. doi: 10.1038/ng.946. [DOI] [PubMed] [Google Scholar]
- 18.Feldmann A, Ivanek R, Murr R, Gaidatzis D, Burger L, Schubeler D. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013;9(12):e1003994. doi: 10.1371/journal.pgen.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24(20):2264–2269. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Tang H, Wang Z, Zhang B, Liu W, Lu H, et al. MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Mol Cancer. 2011;10:124. doi: 10.1186/1476-4598-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guseva N, Mondal T, Kanduri C. Antisense noncoding RNA promoter regulates the timing of de novo methylation of an imprinting control region. Dev Biol. 2012;361(2):403–411. doi: 10.1016/j.ydbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Arab K, Park YJ, Lindroth AM, Schafer A, Oakes C, Weichenhan D, et al. Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol Cell. 2014;55(4):604–614. doi: 10.1016/j.molcel.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 24.Martens JH, Rao NA, Stunnenberg HG. Genome-wide interplay of nuclear receptors with the epigenome. Biochim Biophys Acta. 2011;1812(8):818–823. doi: 10.1016/j.bbadis.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Ruegg J, Cai W, Karimi M, Kiss NB, Swedenborg E, Larsson C, et al. Epigenetic regulation of glucose transporter 4 by estrogen receptor beta. Mol Endocrinol. 2011;25(12):2017–2028. doi: 10.1210/me.2011-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomassin H, Flavin M, Espinas ML, Grange T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 2001;20(8):1974–1983. doi: 10.1093/emboj/20.8.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452(7183):45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 28.Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452(7183):112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 29.Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, et al. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461(7266):1007–1012. doi: 10.1038/nature08456. [DOI] [PubMed] [Google Scholar]
- 30.Marques M, Laflamme L, Gaudreau L. Estrogen receptor alpha can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barros RP, Gustafsson JA. Estrogen receptors and the metabolic network. Cell Metab. 2011;14(3):289–299. doi: 10.1016/j.cmet.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115(6):751–763. doi: 10.1016/S0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 34.Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012;53(3–4):289–305. doi: 10.1093/ilar.53.3-4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernal AJ, Jirtle RL. Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res A Clin Mol Teratol. 2010;88(10):938–944. doi: 10.1002/bdra.20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grober OM, Mutarelli M, Giurato G, Ravo M, Cicatiello L, De Filippo MR, et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011;12:36. doi: 10.1186/1471-2164-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sengupta PK, Fargo J, Smith BD. The RFX family interacts at the collagen (COL1A2) start site and represses transcription. J Biol Chem. 2002;277(28):24926–24937. doi: 10.1074/jbc.M111712200. [DOI] [PubMed] [Google Scholar]
- 39.Zhao M, Sun Y, Gao F, Wu X, Tang J, Yin H, et al. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4 + T cells. J Autoimmun. 2010;35(1):58–69. doi: 10.1016/j.jaut.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Faulds MH, Pettersson K, Gustafsson JA, Haldosen LA. Cross-talk between ERs and signal transducer and activator of transcription 5 is E2 dependent and involves two functionally separate mechanisms. Mol Endocrinol. 2001;15(11):1929–1940. doi: 10.1210/mend.15.11.0726. [DOI] [PubMed] [Google Scholar]
- 42.Lee SK, Choi HS, Song MR, Lee MO, Lee JW. Estrogen receptor, a common interaction partner for a subset of nuclear receptors. Mol Endocrinol. 1998;12(8):1184–1192. doi: 10.1210/mend.12.8.0146. [DOI] [PubMed] [Google Scholar]
- 43.Ruegg J, Swedenborg E, Wahlstrom D, Escande A, Balaguer P, Pettersson K, et al. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor beta-selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Mol Endocrinol. 2008;22(2):304–316. doi: 10.1210/me.2007-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. 2003;278(40):38586–38592. doi: 10.1074/jbc.M304286200. [DOI] [PubMed] [Google Scholar]
- 45.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21(6):1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Legler J, van den Brink CE, Brouwer A, Murk AJ, van der Saag PT, Vethaak AD, et al. Development of a stably transfected estrogen receptor-mediated luciferase reporter gene assay in the human T47D breast cancer cell line. Toxicol Sci Off J Soc Toxicol. 1999;48(1):55–66. doi: 10.1093/toxsci/48.1.55. [DOI] [PubMed] [Google Scholar]
- 48.Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M, et al. TGF-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012;46(5):636–649. doi: 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 49.Leger H, Smet-Nocca C, Attmane-Elakeb A, Morley-Fletcher S, Benecke AG, Eilebrecht S. A TDG/CBP/RARalpha ternary complex mediates the retinoic acid-dependent expression of DNA methylation-sensitive genes. Genomics Proteomics Bioinformatics. 2014;12(1):8–18. doi: 10.1016/j.gpb.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruegg J, Cai W, Karimi M, Kiss NB, Swedenborg E, Larsson C, et al. Epigenetic regulation of glucose transporter 4 by estrogen receptor beta. Mol Endocrinol. 2011;25(12):2017–2028. doi: 10.1210/me.2011-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucey MJ, Chen D, Lopez-Garcia J, Hart SM, Phoenix F, Al-Jehani R, et al. T:G mismatch-specific thymine-DNA glycosylase (TDG) as a coregulator of transcription interacts with SRC1 family members through a novel tyrosine repeat motif. Nucleic Acids Res. 2005;33(19):6393–6404. doi: 10.1093/nar/gki940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147(10):4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 53.Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014;159(2):358–373. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franco MM, Prickett AR, Oakey RJ. The role of CCCTC-binding factor (CTCF) in genomic imprinting, development, and reproduction. Biol Reprod. 2014;91(5):125. doi: 10.1095/biolreprod.114.122945. [DOI] [PubMed] [Google Scholar]
- 55.Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell. 2012;45(6):814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, et al. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7(4):e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol. 2000;58(3):584–590. doi: 10.1124/mol.58.3.584. [DOI] [PubMed] [Google Scholar]
- 58.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, Macdougall E et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. doi:10.1038/nature09672. [DOI] [PubMed]
- 59.Ying QL, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–341. doi: 10.1016/S0076-6879(03)65023-8. [DOI] [PubMed] [Google Scholar]
- 60.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453(7194):519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 62.Team RC. R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2014.
- 63.Steinacher R, Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol. 2005;15(7):616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 64.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]