

Abstract
The HIV-1 Nef accessory factor enhances viral replication and promotes immune system evasion of HIV-infected cells, making it an attractive target for drug discovery. Recently we described a novel class of diphenylpyrazolodiazene compounds that bind directly to Nef in vitro and inhibit Nef-dependent HIV-1 infectivity and replication in cell culture. However, these first-generation Nef antagonists have several structural liabilities, including an azo linkage that led to poor oral bioavailability. The azo group was therefore replaced with either a one- or two-carbon linker. The resulting set of non-azo analogs retained nanomolar binding affinity for Nef by surface plasmon resonance, while inhibiting HIV-1 replication with micromolar potency in cell-based assays without cytotoxicity. Computational docking studies show that these non-azo analogs occupy the same predicted binding site within the HIV-1 Nef dimer interface as the original azo compound. Computational methods also identified a hot spot for inhibitor binding within this site that is defined by conserved HIV-1 Nef residues Asp108, Leu112, and Pro122. Pharmacokinetic evaluation of the non-azo B9 analogs in mice showed that replacement of the azo linkage dramatically enhanced oral bioavailability without substantially affecting plasma half-life or clearance. The improved oral bioavailability of non-azo diphenylpyrazolo Nef antagonists provides a starting point for further drug lead optimization in support of future efficacy testing in animal models of HIV/AIDS.
Keywords: HIV-1, HIV Nef, Nef inhibitors, antiretroviral drug discovery
Graphical Abstract

Antiretroviral drug therapy has changed the prognosis of HIV infection from a life-threatening illness to a manageable chronic condition. While combinations of existing antiretroviral drugs suppress HIV replication to undetectable levels, they do not clear the virus from the patient and, therefore, require life-long administration to prevent relapse. Chronic exposure to antiretroviral drugs can result in metabolic changes, enhance susceptibility to cardiovascular disease, and drive drug resistance, highlighting the need for new approaches to combat HIV.1
Existing antiretroviral drugs inhibit critical HIV-1 enzymes including reverse transcriptase, integrase and protease, as well as viral entry by blocking fusion with host cell receptors.2 HIV-1 also expresses four accessory proteins (Vpr, Vpu, Vif, and Nef) that contribute to viral pathogenicity and represent underexplored targets for antiretroviral drugs.3-5 HIV-1 Nef is attractive in this regard because it enhances many aspects of the HIV-1 life cycle in vivo and also allows HIV-infected cells to escape adaptive immune responses.6-10 Nef is a relatively small (27-30 kDa), membrane-associated protein that is expressed at high levels soon after viral infection. Nef does not exhibit direct biochemical or enzymatic activities, and functions instead by interacting with a diverse group of host cell proteins involved in endocytic trafficking and signal transduction. For example, Nef induces down-regulation of cell-surface immune (MHC-I/II) and viral (CD4/CXCR4/CCR5) receptors, remodels the actin cytoskeleton, and stimulates host cell signaling pathways that favor viral replication.10 These Nef functions allow HIV-infected cells to avoid immune surveillance by the host, prevent viral superinfection, and enhance virion release. Pharmacological agents that inhibit these Nef functions have the potential to not only interfere with the HIV-1 life cycle, but also to enhance recognition of virally infected cells by the patient's immune system as part of a strategy to clear the virus.11
Recently we reported the discovery of diphenylpyrazolodiazene inhibitors of HIV-1 Nef function.12,13 The original hit compound, referred to as ‘B9’, consists of a three-ringed structure with a diazene linker connecting a pyrazole core to a chlorophenyl group (compound 1; Table 1)13. This compound binds directly to recombinant Nef with a KD value in the nanomolar range, and inhibits Nef-dependent enhancement of HIV-1 infectivity and replication in vitro. The parent compound, however, exhibits several medicinal chemistry liabilities, chief of which was the diazene linker which may cause carcinogenicity and impact bioavailability.14 Here we describe the chemical synthesis and characterization of non-azo analogs of this Nef inhibitor that maintain their antiretroviral activities and also display oral bioavailability.
Table 1.
Structures, in vitro Nef-binding activity, calculated physiochemical properties, antiretroviral activity and cytotoxicity of non-azo diphenylpyrazolo Nef inhibitors.
| # | Structure | Nef bindinga KD ± SE, nM | MW | Log D (pH 7.4) | Log P | TPSA (A2) | Acidic pKa | Basic pKa | Infectivityb IC50, μM | Cytotoxicityc CC50, μM |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 (B9) |
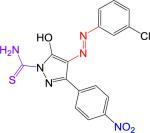
|
81.9 ± 6.2 | 402.81 | 3.41 | 4.92 | 132 | 2.4 | −0.7 | 2.53 | 10 |
| 2 |
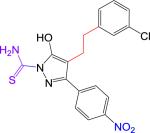
|
159 ± 0.03 | 402.85 | 3.43 | 5.05 | 107 | 4.9 | −0.3 | 0.69 | > 10 |
| 3 |
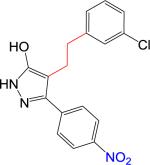
|
NB | 343.77 | 3.80 | 5.18 | 92 | 5.9 | 2.6 | > 10 | > 10 |
| 4 |
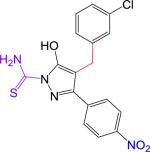
|
150 ± 59 | 388.83 | 2.97 | 4.61 | 107 | 4.6 | −0.4 | 3.54 | > 10 |
| 5 |
|
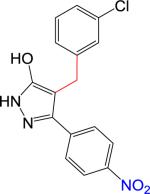
NB | 329.74 | 2.85 | 4.55 | 92 | 3.8 | 2.6 | >10 | > 10 |
SPR analysis was performed in triplicate and the resulting binding curves were best-fit using a heterogeneous ligand model; the higher affinity components are shown. NB, no binding observed at a compound concentration of > 50 μM. Representative SPR traces are presented in Figure S1 (Supplementary Material).
Infectivity was determined in HIV-1-infected TZM-bl reporter cells as described in the text and Supplementary Information.
Cytotoxicity was evaluated in TZM-bl cells using the Cell Titer Blue assay (Promega); CC50 represents the concentration required for 50% loss of viability in this assay.
We synthesized B9 analogs in which the diazene linker was replaced with either a one or two-carbon bond. In this context, we also prepared derivatives without the thioamide. As shown in Scheme 1, the ethyl 4-nitrobenzoylacetate 6 was alkylated with m-chlorobenzyl bromide to provide 7 or with m-chlorophenethyl bromide to provide 8 in moderate yields. Compounds 7 and 8 were further reacted with thiosemicarbazide to provide target compounds 2 and 4 in 30-35% yields. Reaction of 7 and 8 with hydrazine provided good yields of the corresponding 3-hydroxypyrazoles 3 and 5. Synthetic details for compound 2 along with the analytical data for compounds 2-5 are given in the Supplementary Material.
Scheme 1.

Synthesis of B9 analogs in which the diazene linker is replaced with either a one- or two carbon bond.
The four non-azo analogs, together with the B9 parent compound, were tested for direct binding to Nef using a surface plasmon resonance (SPR) assay13. For these studies, recombinant purified Nef was immobilized on the surface of a CM5 biosensor chip, and each of the analogs was injected over a range of concentrations. The binding isotherms were best-fit using a heterogeneous ligand model, and the resulting kinetic constants were used to determine the KD values. As shown in Table 1, replacement of the diazene linker with a two-carbon bond (compound 2) reduced the binding affinity for Nef by only 2-fold relative to the B9 parent compound, demonstrating that the diazene linker is dispensable for interaction with Nef. The analog with a one-carbon linker replacing the diazene also bound to Nef with similar nanomolar affinity (compound 4). Removal of the thioamide from either of these analogs (compounds 3 and 5) resulted in a dramatic loss of Nef binding, consistent with predicted contacts made by this moiety in the docking models (see below). Representative sensorgrams for the interaction of each compound with Nef are presented in Figure S1.
Docking studies presented in the original report describing the discovery of B9 showed that the most energetically favorable binding pose for this inhibitor maps to the Nef dimer interface13. To determine whether non-azo B9 analogs dock to Nef in a similar manner, we repeated the docking study using the same crystal structure of the Nef dimer (PDB: 1EFN15) and the Schrödinger Small Molecule Drug Discovery Suite. This analysis predicted a very similar binding interaction of B9 with the Nef dimer interface as originally reported, involving a reciprocal network of polar contacts between the ligand and the side chains of conserved residues Gln104, Gln107, and Asn126 from both Nef monomers (Figure 1A). Non-azo analog 2, with a 2-carbon linker, docked in an almost identical orientation as the B9 parent compound (Figure 1B), with the nitro and thioamide groups positioned to make a similar set of polar interactions with these three Nef residues. Analog 4 also adopted the same overall orientation (Figure 1C), although the central hydroxypyrazolo ring and thioamide group are more centrally positioned most likely as a result of the shorter one-carbon linker replacing the original azo group. For both non-azo analogs, the thioamide group is positioned to make polar interactions with the side chains of both Gln107 and Asn126. SPR analysis shows that analogs lacking the thioamide moiety (3 and 5) fail to bind to Nef (Table 1), demonstrating the importance of a polar constituent in this area for potential interaction with this region of Nef.
Figure 1.

Computational docking predicts binding of non-azo diphenylpyrazolo inhibitors to the Nef dimer interface. Docking models were generated using the Schrödinger Small Molecule Drug Discovery Suite and the crystal structure of the HIV-1 Nef protein dimer (PDB ID: 1EFN) as described in the Supplementary Information. All four panels show a close-up view of the Nef dimer interface, which is formed by nearly orthogonal interactions of α-helices from each Nef subunit (rendered in blue and green, respectively). A) The parent compound B9 (1, cyan) docks to the Nef dimer interface, forming predicted contacts with conserved Nef residues Gln104, Gln107, and Asn126. This result is virtually identical to a previous docking model produced with Nef and this compound using AutoDock Vina. B and C) Non-azo B9 analogs 2 and 4 dock in a similar pose as B9 at the Nef dimer interface. D) Identification of a hot spot for Nef antagonist binding within the dimer interface (grey mesh). This pocket is defined by the side chains of Nef residues Asp108, Leu112, and Pro122 which accommodate the chlorophenyl group found in all three ligands.
We also performed grand canonical Monte Carlo simulations with annealing of chemical potential that focused on identification of potential hot spots for Nef inhibitor binding. Hot spots are sites on proteins that have high interaction energies with small molecules and identification of these sites helps to evaluate druggability. Using a hot-spot algorithm16, we identified a portion of the previously described docking site as a potential drug binding pocket within the dimer interface. This hot spot is defined by conserved residues Asp108, Leu112, and Pro122 from each Nef monomer (Figure 1D). All of the docked poses of each active compound found this site even though the grid was large enough to cover the complete dimer interface and almost the entire dimer complex. Specifically, the azo linker of B9 (compound 1) directs the 3-chlorophenyl group into this pocket. In compound 2, the two-carbon linker that replaces the azo group also directs the 3-chlorophenyl group into the core of the hot spot. Despite a shorter methylene linker, compound 4 also positions its 3-chlorophenyl group in the predicted hot spot, although the chlorine atom is flipped relative to its orientation in the compounds with 2-atom linkers.
Docking studies described above predict similar binding modes for compounds 1, 2, and 4 and the rigid docking scores showed that both non-azo compounds had lower docking scores than compound 1 (Table S1), consistent with the SPR binding data (Table 1). To confirm and extend these results, we repeated the in silico analysis using induced-fit docking (IFD) to better sample the binding of each compound. The IFD poses were similar to those obtained with rigid docking, but the scores were more consistent with the experimental binding data which showed only modest differences in binding affinities among the active compounds. The two-carbon linker found in compound 2 is likely to add more flexibility to the molecule, resulting in an increase in entropic penalty upon binding. A rigid rotor-harmonic oscillator calculation predicted that compound 2 would have the largest penalty going from the unbound to bound state, consistent with this idea (Table S1). This finding may account for the observation that compound 2 exhibits a 2-fold reduction in binding affinity by SPR despite its structural and docking similarities to the B9 parent compound.
To provide experimental validation of the docking model, we performed SPR experiments with mutant Nef proteins in which conserved Nef residue Asn126 is substituted with either alanine or glutamine. The Asn126 side chains from each ‘half’ of the Nef dimer are predicted to make contacts with the nitro and thioamide groups present in the B9 parent compound (Figure 1A). Previous work showed that mutagenesis of this position reduced the Nef-binding activity of B9 by SPR.13 To test the role of Nef Asn126 in non-azo analog binding, recombinant wild-type, N126A, and N126Q Nef proteins were immobilized on a biosensor chip, and B9 as well as compounds 2 and 4 were injected at a final concentration of 10 μM. Both mutants showed a significant reduction in binding to all three compounds, with glutamine substitution producing a greater impact (Figure 2). This finding supports an important role for Nef Asn126 in the interaction with the non-azo B9 analogs.
Figure 2.

Interaction of Nef inhibitors with wild-type and mutant forms of Nef by surface plasmon resonance (SPR). Wild-type and mutant Nef proteins (N126A and N126Q) were expressed in bacteria and purified to homogeneity. SPR assays were performed using a Biacore T100 instrument (GE Healthcare). Nef proteins were covalently attached to a CM5 biosensor chip, and compounds 1 (B9), 2 and 4 were injected in triplicate at a final concentration of 10 μM. Compound association with Nef was followed until equilibrium was reached and the resulting binding curves were fit using a 1:1 Langmuir model to estimate the binding maximum (RUmax). Data are presented as the percent of maximum binding observed with each compound relative to the wild-type Nef protein ± S.E.
We next evaluated the Nef-dependent antiretroviral activity of these four analogs in cell culture models of HIV-1 infectivity and replication. For infectivity assays, we used TZM-bl reporter cells in which the HIV-1 LTR drives expression of a luciferase reporter gene17. Short-term infection of these cells with HIV-1 models early steps in the viral life cycle from entry through viral gene expression. Previous studies have shown that B9 (compound 1) inhibits Nef-dependent HIV-1 infectivity in this system and was included as a positive control.12,13 Analogs with one- and two-carbon linkers (compounds 2 and 4) suppressed Nef-mediated enhancement of HIV-1 infectivity with IC50 values of 0.69 μM and 3.54 μM, respectively (Table 1). These values agree favorably with results from the B9 parent compound, which yielded an IC50 value of 2.53 μM. In contrast, non-azo analogs lacking the thioamide moiety were inactive in the HIV-1 infectivity assay, consistent with the inability of these analogs to bind to Nef by SPR. These results show that the diazene linker present in the parent compound is dispensable for Nef-dependent antiretroviral activity. We also evaluated the cytotoxicity of each compound in the TZM-bl reporter cell line. None of the non-azo compounds showed evidence of toxicity in these cells (CC50 > 10 μM; Table 1). This represents an improvement over the B9 parent compound, which showed evidence of cytotoxicity at higher concentrations (CC50 = 10 μM).
To assess the impact of the non-azo B9 analogs on Nef-dependent HIV replication, we used an astroglioma cell line (U87MG) which has been engineered to express cell-surface HIV receptors CD4 and CXCR4. Previous studies have shown that Nef strongly enhances HIV-1 replication in this cell line12,13,18-20, making it a valuable model to study the antiretroviral impact of Nef inhibitors in cell culture. As shown in Figure 3, non-azo analogs 2 and 4 both suppressed Nef-dependent enhancement of HIV-1 replication to the same extent as B9 at 3 μM, while the non-binding analogs lacking the thioamide moiety (3 and 5) had no activity at this concentration. No cytotoxicity was observed with these compounds in U87MG/CD4/R4 cells up to 10 μM (data not shown).
Figure 3.

Non-azo B9 analogs inhibit Nef-dependent HIV-1 replication. Compounds 1-5 were added to cultures of U87MG/CD4/CXCR4 cells at a final concentration of 3 μM followed by infection with wild-type or ΔNef HIV-1NL4-3 one hour later. Viral replication was assessed after 4 days by p24 Gag ELISA (AlphaLISA method, Perkin Elmer), and data are shown as mean percent of viral replication observed in DMSO control cells ± S.E. (n=4). Additional details of the HIV assay methods are presented in the supplementary information.
Finally, we evaluated the pharmacokinetics and oral bioavailability of B9 (compound 1) and the corresponding active non-azo analogs (2 and 4) in mice. Groups of C3H mice were dosed IV and PO with each compound, and plasma levels were determined by LC-MS over time. As shown in Table 2, the parent compound showed a plasma half-life of just over 2 hours following IV administration but very low bioavailability (2%), which may reflect poor absorption or ease of first-pass metabolism of the azo linker, which may lead to the generation of free radicals and other toxic intermediates.14 Substitution of the azo linker with carbon in compounds 2 and 4 dramatically enhanced oral bioavailability, with %F values ranging from 53 to 85% for compound 2 and 57 to 100% for compound 4, depending upon the PO formulation. Each of the non-azo compounds showed a similar plasma half-life as the B9 parent compound (ranging from 1.53 to 2.70 h), with low plasma clearance and a volume of distribution similar to the total body water. Subsequent experiments with higher PO doses of 2 and 4 (100 mg/kg in suspension) resulted in proportionally higher plasma drug concentrations (data not shown). No weight loss or other signs of acute toxicity were observed in C3H mice treated daily by oral administration of compounds 1, 2 or 4 at various doses for up to twelve days (Table S2).
Table 2.
Impact of diazene linker replacement on the pharmacokinetics and oral bioavailability of selected diphenylpyrazolo Nef antagonists.a
| Compound | Route | Vehicleb | Dose mg/kg | Tmax h | Cmax ng/mL | AUClast h•ng/mL | AUCinf h•ng/mL | T1/2 h | Vss L/kg | Cl mL/min/kg | %Fc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (B9) | IV | solution | 2 | 3959.35 | 9783.74 | 9787.22 | 2.13 | 0.37 | 3.41 | ||
| PO | solution | 10 | 0.50 | 418.11 | 962.95 | 1016.51 | 2 | ||||
| 2 | IV | solution | 2 | 11214.62 | 2397.55 | 2471.34 | 2.70 | 0.83 | 13.49 | ||
| PO | solution | 10 | 0.25 | 2368.83 | 6302.36 | 6425.52 | 53 | ||||
| IV | solution | 2 | 4416.32 | 2133.83 | 2153.46 | 1.53 | 0.91 | 15.48 | |||
| PO | suspension | 10 | 0.50 | 5610.83 | 9034.36 | 9483.82 | 85 | ||||
| 4 | IV | solution | 2 | 2804.97 | 2961.35 | 3003.31 | 1.66 | 0.79 | 11.19 | ||
| PO | solution | 10 | 1.00 | 5806.68 | 18606.95 | 18830.31 | ~100 | ||||
| IV | solution | 2 | 3877.31 | 3765.96 | 3894.75 | 1.58 | 0.95 | 8.56 | |||
| PO | suspension | 10 | 0.5 | 3397.40 | 10650.23 | 11538.33 | 57 | ||||
Two independent studies of compounds 2 and 4 were performed and results of each trial are shown.
Compounds were administered PO in solution (5% N-methyl-2-pyrrolidone and 5% solutol in saline) or in suspension (0.5% w/v sodium-carboxymethyl cellulose with 0.1% v/v Tween 80) as indicated.
AUClast used for calculation of oral bioavailability.
In summary, these studies show that the undesirable azo linker present in the original diphenylpyrazolodiazene Nef inhibitor scaffold can be replaced without loss of Nef binding or antiretroviral activity. Replacement of the azo linker with a one- or two-carbon bond also led to a dramatic enhancement in oral bioavailability. These non-azo analogs represent an important step forward in the development of Nef antagonists, and will guide the synthesis of next-generation compounds with appropriate pharmacological profiles for assessment of antiretroviral activity in vivo.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grants AI57083, AI102704 and GM112516.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary information
Supplementary experimental procedures and data associated with this article can be found in the online version.
References
- 1.Deeks SG, Lewin SR, Havlir DV. Lancet. 2013;382:1525. doi: 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Temesgen Z, Warnke D, Kasten MJ. Expert.Opin.Pharmacother. 2006;7:1541. doi: 10.1517/14656566.7.12.1541. [DOI] [PubMed] [Google Scholar]
- 3.Malim MH, Emerman M. Cell Host Microbe. 2008;3:388. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Ali A, Wang J, Nathans RS, Cao H, Sharova N, Stevenson M, Rana TM. ChemMedChem. 2012;7:1217. doi: 10.1002/cmdc.201200079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang W, Zuo T, Luo X, Jin H, Liu Z, Yang Z, Yu X, Zhang L, Zhang L. Chem.Biol Drug Des. 2013;81:730. doi: 10.1111/cbdd.12119. [DOI] [PubMed] [Google Scholar]
- 6.Arora VK, Fredericksen BL, Garcia JV. Microbes.Infect. 2002;4:189. doi: 10.1016/s1286-4579(01)01527-1. [DOI] [PubMed] [Google Scholar]
- 7.Saksela K. Curr.HIV.Res. 2011;9:531. doi: 10.2174/157016211798842107. [DOI] [PubMed] [Google Scholar]
- 8.Joseph AM, Kumar M, Mitra D. Curr.HIV.Res. 2005;3:87. doi: 10.2174/1570162052773013. [DOI] [PubMed] [Google Scholar]
- 9.O'Neill E, Kuo LS, Krisko JF, Tomchick DR, Garcia JV, Foster JL. J.Virol. 2006;80:1311. doi: 10.1128/JVI.80.3.1311-1320.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster JL, Garcia JV. Retrovirology. 2008;5:84. doi: 10.1186/1742-4690-5-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smithgall TE, Thomas G. Drug Discov Today: Technol. 2013;10:523. doi: 10.1016/j.ddtec.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer PC, Zhao J, Emert-Sedlak LA, Moore KK, Smithgall TE, Day BW. Bioorg.Med.Chem.Lett. 2014;24:1702. doi: 10.1016/j.bmcl.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emert-Sedlak LA, Narute P, Shu ST, Poe JA, Shi H, Yanamala N, Alvarado JJ, Lazo JS, Yeh JI, Johnston PA, Smithgall TE. Chem.Biol. 2013;20:82. doi: 10.1016/j.chembiol.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith GF. Prog.Med.Chem. 2011;50:1. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 15.Lee C-H, Saksela K, Mirza UA, Chait BT, Kuriyan J. Cell. 1996;85:931. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- 16.Kulp JL, III, Kulp JL, Jr., Pompliano DL, Guarnieri F. J Am.Chem.Soc. 2011;133:10740. doi: 10.1021/ja203929x. [DOI] [PubMed] [Google Scholar]
- 17.Gervaix A, West D, Leoni LM, Richman DD, Wong-Staal F, Corbeil J. Proc.Natl.Acad.Sci.U.S.A. 1997;94:4653. doi: 10.1073/pnas.94.9.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emert-Sedlak L, Kodama T, Lerner EC, Dai W, Foster C, Day BW, Lazo JS, Smithgall TE. ACS Chem.Biol. 2009;4:939. doi: 10.1021/cb900195c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trible RP, Narute P, Emert-Sedlak LA, Alvarado JJ, Atkins K, Thomas L, Kodama T, Yanamala N, Korotchenko V, Day BW, Thomas G, Smithgall TE. Retrovirology. 2013;10:135. doi: 10.1186/1742-4690-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narute PS, Smithgall TE. PLoS.One. 2012;7:e32561. doi: 10.1371/journal.pone.0032561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.