

Abstract
Horns are a cranial appendage found exclusively in Bovidae, and play important roles in accessing resources and mates. In sheep (Ovies aries), horns vary from polled to six-horned, and human have been selecting polled animals in farming and breeding. Here, we conducted a genome-wide association study on 24 two-horned versus 22 four-horned phenotypes in a native Chinese breed of Sishui Fur sheep. Together with linkage disequilibrium (LD) analyses and haplotype-based association tests, we identified a genomic region comprising 132.0–133.1 Mb on chromosome 2 that contained the top 10 SNPs (including 4 significant SNPs) and 5 most significant haplotypes associated with the polycerate phenotype. In humans and mice, this genomic region contains the HOXD gene cluster and adjacent functional genes EVX2 and KIAA1715, which have a close association with the formation of limbs and genital buds. Our results provide new insights into the genetic basis underlying variable numbers of horns and represent a new resource for use in sheep genetics and breeding.
In mammals, most members of the infraorder Pecora, such as deer, antelope, cattle, goat, and sheep (Ovies aries), have cranial appendages. These appendages have four extant forms: antlers, horns, pronghorns and ossicones1. Horns, being exclusive to bovids, consist of a bony horn core covered by a scabbard-like keratinous sheath that is never shed1,2. Horns serve as a form of sexual weaponry for most domestic and wild animals; thus, larger horns have an advantage in strong intra-sexual competition3,4. However, horns may be disadvantages in livestock farming and breeding. For example, horned males may attack other animals or people and can get their heads stuck in fences and feeders. Therefore, hornless (or polled) individuals are preferred for animal farming and breeding.
A number of earlier studies have identified candidate genes for various traits in sheep, including coat colour, horn, meat production, growth and fecundity etc., using a genome-wide association approach5,6,7,8. In sheep, a majority of rams have strong and normal horns, while ewes can have smaller horns, scurs (deformed vestigial horns) or no horns (polled). Previous studies have primarily focused on polled versus horned phenotypes or on horn type, size and length in sheep and their wild relatives (e.g., Ovis canadensis)3,4,8,9,10. Horns, a single autosomal locus mapped to a 7.4 cM region on chromosome 10 (OAR10) in Soay sheep, has been demonstrated to control horn-type polymorphism and exerts a comparatively large effect on horn size3,8. Fine mapping studies identified relaxin-like receptor 2 (RXFP2), which contributes to main sex characters in humans and mice, as the major candidate gene for Horns in Soay sheep3,4. Quite recently, a 1.8-kb insertion in the 3′-UTR of RXFP2 was identified to be associated with polledness in sheep9. Also, whole-genome resequencing of a wild sheep Ovis canadensis revealed that multiple populations shared a common selective sweep at RXFP210.
However, the genetic basis of a different aspect of horn phenotype, the polycerate type (multiple horns), has not been examined. Globally, there are only a small number of polycerate native sheep breeds, including Jacob, Manx Loaghtan, Hebridean, Navajo-Churro, Icelandic sheep, Because nowadays there are no four-horned Finnish Landrace sheep. Altay sheep and Chinese Sishui Fur sheep (see Fig. 1). Horn phenotypes for the breeds range from polled to as many as six horns. Historically, four-horned sheep were widely distributed in both Europe and Asia11,12; however, they have become rare due to long-term artificial selection. Most of the four-horned sheep are rams, and four horns are rarely observed in ewes. The four-hornedness trait is dominant over two-hornedness, but recessive to polledness12. Sishui Fur sheep is a native sheep breed in eastern China, originating from ancient Mongolian sheep. They are fat-tailed and mainly distributed in pastoral and agricultural areas in Sishui County of Shandong Province. They can produce high-quality meat and carpet wool. Also, they have sound body conformation, strong walking ability and excellent adaptation to local ecological environments13. Rams of the breed typically have 0–6 horns, and the occurrence of 2 and 4 horns is most common (see Fig. 1). Ewes are always polled. However, the underlying mechanisms for the polycerate phenotype in rams of this and other sheep breeds have remained elusive.
Figure 1.

Sishui Fur sheep with the (a) two-horned and (b) four-horned phenotypes.
Here, we carried out a genome-wide association (GWA) study and haplotype-based association tests to map a strong candidate genomic region for the polycerate phenotype in Sishui Fur sheep. Our results will help to elucidate the molecular mechanism underlying the trait and can be applied in molecular marker-assisted breeding programs.
Methods
The methods were carried out in accordance with the approved guidelines of the Good Experimental Practices adopted by the Institute of Zoology, Chinese Academy of Sciences. All experimental procedures and animal collections were conducted under a permit (No. IOZ13015) approved by the Committee for Animal Experiments of the Institute of Zoology, Chinese Academy of Sciences, China.
Sample collection and DNA extraction
Ear tissue samples were collected from a total of 60 rams of Sishui Fur sheep. Among them, 26 individuals with four horns were assigned as the case group, and the other 34 two-horned sheep were considered as the control group. All the samples were collected from stocks maintained on different farms in Sishui County, Shandong Province, China. Using both pedigree records and the farmers’ knowledge, particular efforts were made to ensure that all animals were typical of the breed and as unrelated as possible.
Genomic DNA was extracted from the ear tissues using a standard phenol/chloroform method14, and the DNA concentration was measured on a spectrophotometer (Nanodrop 2000, Wilmington, DE, USA). The extracted DNA was diluted to 100 ng/μl for SNP BeadChip genotyping.
SNP genotyping and quality control
All sample DNA met the concentrations recommended for the Illumina Ovine Infinium HD SNP BeadChip genotyping according to the manufacturer’s protocol. Details on creation of the ovine BeadChip (685,734 SNPs) and genotyping procedures were described in Anderson et al.15. Genotypes for a total of 606,006 scorable SNPs, which passed the manufacturer’s quality control, were available for further analysis.
Stringent quality control parameters were applied for both the samples and SNPs to ensure reliability of results. We implemented the quality control measures using the software PLINK v1.0716. SNPs or individuals who met any of the following criteria were removed: (1) no chromosomal or physical location; (2) minor allele frequency (MAF) <0.05; (3) individuals call rate <0.9; (4) missing genotype frequency for SNP >0.05; (5) Fisher’s exact test17 P-value for Hardy-Weinberg equilibrium (HWE) <0.001. We further estimated pairwise relatedness using KING18,19, and closely related animals (e.g., full-sibs) were excluded from further analyses. After filtering, a total of 491,507 SNPs and 50 individuals (two-horned: 28 animals; four-horned: 22 animals) were left for the within-breed population stratification analysis.
Population stratification and genome-wide association (GWA) analysis
Genetic differentiation between the case and control groups was assessed by Weir & Cockerham’s FST method20 using the program GENEPOP v4.221, and multidimensional scaling (MDS) analysis using the program PLINK v1.07. The MDS analysis were implemented on a set of 15,911 independent SNPs pruned using the option of “indep-pairwise 50 5 0.05” in PLINK. This function calculates pairwise linkage disequilibrium (LD) in a 50-SNPs-window, shifts at a pace of 5 SNPs and excludes one of a pair of SNPs if the LD estimate r2 > 0.05. Results of the MDS analysis were plotted in the GenABEL package22 for R v3.2.2 (http://www.r-project.org). We further removed the 4 separated individuals from the control group in the following analyses (see Results). Our final working data set contained 46 individuals (24 two-horned; 22 four-horned).
Because the phenotype (2 or 4 horns) is binary, we performed a genome-wide association (GWA) study using the case-control model in the GenABEL package for R v3.2.2. Given the 15,911 pruned independent SNPs, the statistical threshold of genome-wide significance after Bonferroni correction was 3.14 × 10−6 (0.05/15,911) at the empirical level of 0.053,23. To account for the GWA analysis confounded by population stratification, the genome-wide association P-values were corrected using the principal components (PCs)22,23. We used the top five principle components from the MDS analyses as covariates to adjust the systematic biases (see Results), which was also indicated by the Q-Q plot (quantile-quantile plot). We further conducted a resampling test (1,000 times) without replacement to determine the false positive rate (FPR) by chance using the function of “qtscore” in GenABEL. The command is detailed as follows:
qtscore(sample(phdata(data1)$dm2, replace = F), data1, trait.type = “binomial”), where “data1” is the gwaa.data object and “dm2” represents phenotype.
Linkage disequilibrium (LD) and haplotype-based association analyses
The gametic LD measure of r2 among SNPs within the candidate genomic region (132–133.1Mb on OAR2, see Results) was calculated and visualised using the program Haploview v4.224. LD blocks were defined based on the four-gamete rule algorithm25. Pairwise tests of LD for the most significant SNP rs420183358 with its flanking SNPs within approximately 1Mb upstream and downstream were obtained using PLINK v1.07. A regional association plot was generated using the R v3.2.2.
Further, we applied a haplotype-based association test to identify haplotypes significantly associated with the polycerate phenotype across OAR2. We first estimated the haplotypes and their frequencies in the LD blocks using the maximum likelihood (ML) method and expectation-maximization (EM) algorithm. Then, we performed the chi-square test for haplotype-phenotype association26, and statistical significance was also determined. We considered raw P-values (Praw) <1 × 10−7 as significant and P-values <0.05 after 100,000 permutations as genome-wide significant (i.e. Pgenome <0.05). All the analyses were carried out using Haploview.
Gene annotation
Genes within the candidate genomic region were determined using the Ovis aries assembly Oar_v4.0 (http://www.ncbi.nlm.nih.gov/genome/?term=ovis+aries/). However, given the lagging research of the sheep genome, the gene annotation in sheep is incomplete. We obtained additional functional information of orthologous genes in other species (e.g. human, mouse, chimpanzee, chicken and bovine) from UniProt (http://www.uniprot.org/) and published research regarding the candidate genes identified in sheep.
Results
Population stratification and significant SNPs by GWA analysis
We obtained a FST value of 0.011 for the genetic differentiation between the case and control groups. Also, the MDS plot (see Supplementary Fig. S1 online) indicated within-population stratification, which may bias the association test. In particular, 4 two-horned animals of the control group showed apparent genetic differentiation from the rest animals (see Supplementary Fig. S1 online) and, thus, were removed from the GWA analysis. The raw genomic inflation factor lambda was 1.352 in the initial GWA analysis and turned to be 1.103 after applying the first five PCs in the population stratification correction (Fig. 2a).
Figure 2. Q-Q plot and manhattan plot of genome-wide association (GWA) test.
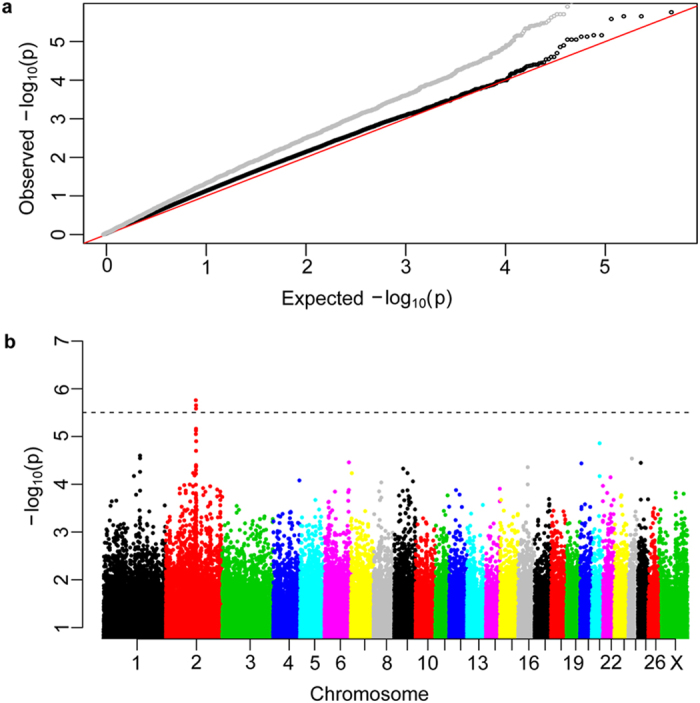
The “p” in the labels represents P-values of GWA analysis. (a) Q-Q plot: the grey and black dots denote association statistics before and after correction for population stratification, respectively; (b) Manhattan plot: the 5% genome-wide significant threshold value (P = 3.14 × 10−6) is presented by a black dashed line.
All the top ten associated SNPs were identified as being located on OAR2 (Fig. 2b, Table 1) and extremely low FPR values (FPR <0.001) were observed for the SNPs after the bootstrapping test. We also detected statistically significant differences in allele frequencies of the ten SNPs between the case and control groups (paired t-test; P = 3.366 × 10−7, t = 13.128, d.f. = 9). Of the top ten SNPs, four (rs420183358, rs422753866, rs428812278 and rs428938943) showed significant associations with the polycerate phenotype at the genome-wide threshold of P < 0.05 (Table 1). SNP rs420183358 showed the most significant association (P = 1.74 × 10−6).
Table 1. The top ten SNPs associated with the polycerate phenotype.
| Chr. | SNP | Position | Allele | MAF | Frequency |
P-value | FPR | |
|---|---|---|---|---|---|---|---|---|
| Case | Control | |||||||
| 2 | rs420183358 | 132042535 | A:G | 0.222 | 0.405 | 0.062 | 1.74E-06 | <0.001 |
| 2 | rs422753866 | 133065296 | A:G | 0.402 | 0.727 | 0.104 | 2.221E-06 | <0.001 |
| 2 | rs428812278 | 133084414 | G:A | 0.402 | 0.727 | 0.104 | 2.221E-06 | <0.001 |
| 2 | rs428938943 | 133008470 | G:A | 0.413 | 0.727 | 0.125 | 2.601E-06 | <0.001 |
| 2 | rs429526398 | 132934057 | G:A | 0.413 | 0.705 | 0.146 | 6.876E-06 | <0.001 |
| 2 | rs419153948 | 132994229 | G:A | 0.413 | 0.705 | 0.146 | 6.876E-06 | <0.001 |
| 2 | rs416536940 | 132519432 | A:G | 0.217 | 0.386 | 0.062 | 7.482E-06 | <0.001 |
| 2 | rs403336550 | 133065371 | G:A | 0.446 | 0.75 | 0.167 | 7.518E-06 | <0.001 |
| 2 | rs418381175 | 132200980 | G:A | 0.391 | 0.818 | 0.417 | 8.912E-06 | <0.001 |
| 2 | rs408254270 | 132201113 | G:A | 0.391 | 0.818 | 0.417 | 8.912E-06 | <0.001 |
Chr. is the abbreviation format of chromosome. MAF represents minor allele frequency. P-value represents the corrected significance of GWA after principle component adjustment. FPR means false positive rate.
LD and haplotype-based association test
Pairwise tests of LD for the most significant SNP rs420183358 with neighbouring SNPs in its upstream and downstream regions indicated relatively strong LD (r2 > 0.6) with two SNPs (rs403275219 and rs416536940) (Fig. 3). A relatively high level of LD was observed across the pairwise SNPs in the candidate genomic region (Fig. 3). However, the two linked SNPs (rs403275219 and rs416536940) were distributed in a different haplotype block from that which contained the most significant SNP rs420183358.
Figure 3. Plot of regional association results for loci surrounding the most significant SNP rs420183358 (red rhombus).
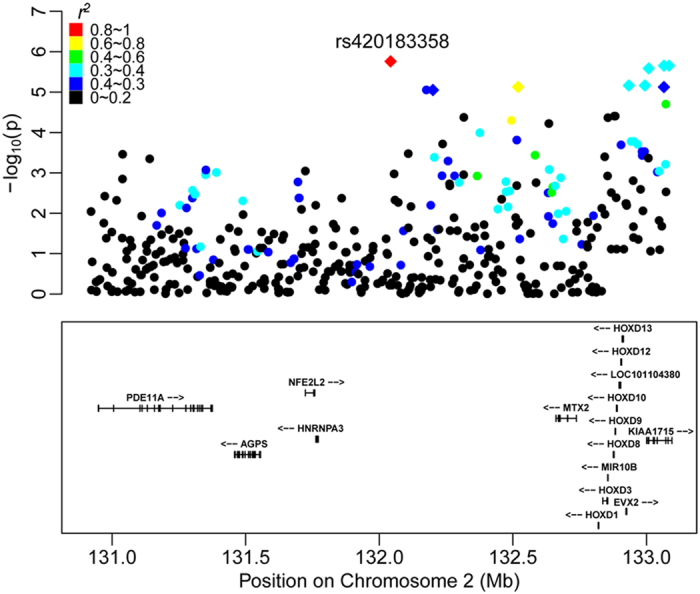
Different colours represent the r2 values of pair-wise LD estimates. Functional genes in this region are plotted in the box. The rhombus represents the top ten SNPs of GWA analysis. The yellow dot represents rs403275219.
Four-gamete rule algorithm identified a total of 1,552 LD blocks containing 44,373 haplotypes on OAR2. The chi-square test revealed that after 100,000 permutations 20 haplotypes were significant (P < 0.05) at the genome-wide threshold on the chromosome (Table 2). The most significant five haplotypes were distributed in 5 adjacent and strongly linked blocks (Table 2, Supplementary Fig. S2 online). Of the 5 most significant haplotypes, the top three haplotype of blocks 6085 (AGAG, P < 1 × 10−5), 6086 (AGGGA, P < 1 × 10−5) and 6084 (GAGCA, P < 1 × 10−5) had the same frequencies of 0.727 in the case groups versus 0.104, 0.104 and 0.125 in the control groups, respectively (Table 2). The results indicated that the region 132.9–133.1 Mb most probably harbours the causative mutations accounting for the polycerate phenotype.
Table 2. The top ten significant haplotypes with the polycerate phenotype.
| LD Block | Haplotype | Position |
Frequency |
P-value | P100,000 | ||
|---|---|---|---|---|---|---|---|
| first | last | Case | Control | ||||
| 6085 | AGAG | 133040292 | 133065679 | 0.727 | 0.104 | 1.14E-09 | <0.00001 |
| 6086 | AGGGA | 133072339 | 133084414 | 0.727 | 0.104 | 1.14E-09 | <0.00001 |
| 6084 | GAGCA | 133008423 | 133017771 | 0.727 | 0.125 | 4.61E-09 | <0.00001 |
| 6081 | ACCGA | 132934057 | 132951006 | 0.705 | 0.146 | 5.43E-08 | 0.0005 |
| 6083 | AAAG | 132978078 | 132994332 | 0.705 | 0.146 | 5.43E-08 | 0.0005 |
| 11095 | GA | 240198021 | 240198140 | 0.136 | 0.646 | 6.42E-07 | 0.0096 |
| 6125 | GGAG | 133917957 | 133928107 | 0.455 | 0.021 | 7.38E-07 | 0.0105 |
| 6107 | GGGG | 133560053 | 133564962 | 0.841 | 0.333 | 8.60E-07 | 0.0120 |
| 11110 | AGACC | 240440866 | 240460254 | 0.592 | 0.106 | 8.82E-07 | 0.0120 |
| 6097 | GCGGGG | 133339051 | 133365628 | 0.523 | 0.062 | 9.72E-07 | 0.0128 |
P100,000 represents P-value with 100,000 permutations in the association tests; LD denotes linkage disequilibrium.
Gene annotation
Based on the lists of significant SNPs identified by GWA study and the haplotype-based association test, the 132.0–133.1Mb region on OAR2 was identified as a strong candidate genomic region for the polycerate phenotype. Thirteen genes have been annotated within this genomic region (see Supplementary Table S1 online). Metaxin 2 (MXT2) is a protein that is located on the cytosolic face of the mitochondrial outer membrane and works with metaxin 1 to import mitochondrial preproteins into mammalian mitochondria27. The HOXD gene cluster belongs to the homeobox family of genes, which encode a highly conserved family of transcription factors that specify differences in morphogenesis in all multicellular organisms28,29. Earlier studies in humans, mice and chicks have concluded that 5′HOXD genes are closely associated with the development of limbs and genitalia30,31,32,33,34. EVX2 is adjacent to HOXD13, i.e., 8 kb upstream, and forms the EVX2-HOXD13 intergenic region, which behaves as a spatio-temporal boundary element35. KIAA1715 is an orthologous gene to LUNPARK (LNP) in the human and mouse genomes, and it plays an important role in the development of digits and the central nervous system, along with EVX2 and the HOXD cluster30. The most significant SNP rs420183358 identified here lies downstream of the HOXD genes, and the other three significant SNPs (rs422753866, rs428812278 and rs428938943) lie within the gene KIAA1715.
Discussion
Understanding the genetic architecture of horn phenotypic variation in wild and domestic vertebrates is one of the fundamental goals in evolutionary genetics. In this study, we identified a genomic region for the polycerate phenotype using a GWA study approach.
The most significant SNP (rs420183358) is not located in any annotated gene, but downstream of the HOXD gene cluster. Thus, rs420183358 may be the causal mutation, which has a regulatory function. Alternatively, it is also possible that the causal mutation is located inside the HOXD gene cluster and is closely linked to the target SNP rs420183358. The candidate genomic region (132–133.1 Mb) revealed in this investigation is in accordance with an ongoing investigation on the genetic cause for the polycerate trait in Navajo-Churro and Jacob sheep, where a genomic region of 132Mb on OAR2 was identified as being associated with this trait36. This region is well-known for the HOXD gene cluster, which comprises nine genes (HOXD1, HOXD3, HOXD4 and HOXD8-HOXD13) in order from 3′ to 5′. HOXD genes are the primary determinants of the anterior-posterior body axis in all bilaterians, including the trunk or appendicular axis37,38. HOXD11 is expressed along the primary body axis, and HOXD11 mutant mice (or mice with a deletion of HOXD13 to HOXD11) exhibit a supernumerary lumbar vertebra39,40. HOXD13 and HOXA13 are most strongly expressed at the distal ends of limbs and genital buds, and the absence of the two genes could affect the development of hands and feet31,32,34. Synpolydactyly, a limb malformation, is caused by HOXD13 mutations41, and later research suggested that removing HOXD9-HOXD13 and EVX2 also led to synpolydactyly35. EVX2 is located 8kb upstream of HOXD13 and responds to the digit enhancer in a similar way as the HOXD genes35,42.
The significantly associated haplotypes were distributed in several LD blocks (see Supplementary Fig. S2 online). The three most significant haplotypes (in blocks 6084–6086) containing three significant SNPs (rs422753866, rs428812278 and rs428938943) were located within the gene KIAA1715, which has the functions associated with the formation of limb digits and genital buds30,41,43. The HOXD gene cluster and genes EVX2 and KIAA1715 are located in the candidate genomic region in a successive order (Fig. 3). All these genes have analogous functions in limb and digit development, and the number of digits and lumbar vertebras are controlled by this region30. Also, previous work localized a “digit enhancer” upstream from the HOXD cluster39. Subsequent studies demonstrated that a global control region (GCR) defined a chromosomal regulatory landscape containing the HOXD gene cluster, with LNP, EVX2 and HOXD-specific patterns30. Therefore, the GCR could also regulate the polycerate phenotype.
Our approaches for detecting the candidate genomic region underlying the polycerate phenotype were efficient, although the relatively small sample size was one possible potential limitation of this study. Future investigations with a larger sample size in other sheep breeds and deep-sequencing of the candidate genomic region are necessary to confirm and extend our findings. In conclusion, our results provided strong evidence that the genomic region 132.0–133.1 Mb on chromosome 2 is associated with the polycerate phenotype.
Additional Information
How to cite this article: Ren, X. et al. A genome-wide association study identifies a genomic region for the polycerate phenotype in sheep (Ovis aries). Sci. Rep. 6, 21111; doi: 10.1038/srep21111 (2016).
Supplementary Material
Acknowledgments
This work was supported by the 100-talent Program of the Chinese Academy of Sciences (CAS), the National High Technology Research and Development Program of China (i.e., 863 Program, grant No. 2013AA102506), grants from the National Natural Science Foundation of China (grants Nos. 31272413 and U1303284), the National Transgenic Breeding Project of China (2014ZX0800952B) and the Academy of Finland (grants No. 250633 and No. 256077).
Footnotes
Author Contributions M.-H.L. conceived and designed the project. G.-L.Y. and F.-A.W. collected the samples. W.-F.P., Y.-X.Z., M.Z. and Z.-H.C extracted the DNA. J.K. helped to produce the SNP chip data. X.R., G.-L.Y. and M.S. analysed the data. X.R. and G.-L.Y. wrote the paper, with contributions from M.-H.L. All authors reviewed and approved the final manuscript.
References
- Davis E. B., Brakora K. A. & Lee A. H. Evolution of ruminant headgear: a review. Proc. R. Soc. Lond. B Biol. Sci. 278, 2857–2865 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove W. F. The physiology of horn growth: a study of the morphogenesis, the interaction of tissues, and the evolutionary processes of a Mendelian recessive character by means of transplantation of tissues. J. Exp. Zool. 69, 347–405 (1935). [Google Scholar]
- Johnston S. E. et al. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol. Ecol. 20, 2555–2566 (2011). [DOI] [PubMed] [Google Scholar]
- Johnston S. E. et al. Life history trade-offs at a single locus maintain sexually selected genetic variation. Nature 502, 93–96 (2013). [DOI] [PubMed] [Google Scholar]
- Li M. H., Tiirikka T. & Kantanen J. A genome-wide scan study identifies a single nucleotide substitution in ASIP associated with white versus non-white coat-colour variation in sheep (Ovis aries). Heredity 112, 122–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demars J. et al. Genome-wide association studies identify two novel BMP15 mutations responsible for an atypical hyperprolificacy phenotype in sheep. PLoS Genet. 9, e1003482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS One 8, e66569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S. E., Beraldi D., McRae A. F., Pemberton J. M. & Slate J. Horn type and horn length genes map to the same chromosomal region in Soay sheep. Heredity 104, 196–205 (2010). [DOI] [PubMed] [Google Scholar]
- Wiedemar N. & Drögemüller C. A 1.8-kb insertion in the 3′-UTR of RXFP2 is associated with polledness in sheep. Anim. Genet. 46, 457–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos M. et al. Whole-genome resequencing uncovers molecular signatures of natural and sexual selection in wild bighorn sheep. Mol. Ecol. 24, 5616–5632 (2015). [DOI] [PubMed] [Google Scholar]
- Ryder M. L. in Sheep and man, (Duckworth, London, 1983). [Google Scholar]
- Dýrmundsson Ó. R. Four-hornedness; a rare peculiarity still found in Icelandic sheep. The Icelandic Sheep Breeders of North America Newsletter 9, 6–8 (2005). [Google Scholar]
- China National Commission of Animal Genetic Resources. in Animal genetic resources in China sheep and goats, 56–58 (China Agriculture Press, Beijing, 2011). [Google Scholar]
- Köchl S., Niederstätter H. & Parson W. DNA extraction and quantitation of forensic samples using the phenol-chloroform method and real-time PCR. Methods Mol. Biol. 297, 13–29 (2005). [DOI] [PubMed] [Google Scholar]
- Anderson R. Development of a high density (600K) Illumina Ovine SNP chip and its use to fine map the yellow fat locus. in Plant and Animal Genome XXII Conference (Plant and Animal Genome, 2014).
- Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M. & Rousset F. An exact test for population differentiation. Evolution 49, 1280–1283 (1995). [DOI] [PubMed] [Google Scholar]
- Manichaikul A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. H., Stranden I., Tiirikka T., Sevon-Aimonen M. L. & Kantanen J. A comparison of approaches to estimate the inbreeding coefficient and pairwise relatedness using genomic and pedigree data in a sheep population. PLoS One 6, e26256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir B. S. & Cockerham C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984). [DOI] [PubMed] [Google Scholar]
- Raymond M. & Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995). [Google Scholar]
- Aulchenko Y. S., Ripke S., Isaacs A. & van Duijn C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007). [DOI] [PubMed] [Google Scholar]
- Johnston S. E. et al. Genome-wide SNP analysis reveals a genetic basis for sea-age variation in a wild population of Atlantic salmon (Salmo salar). Mol. Ecol. 23, 3452–3468 (2014). [DOI] [PubMed] [Google Scholar]
- Barrett J. C., Fry B., Maller J. & Daly M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
- Wang N., Akey J. M., Zhang K., Chakraborty R. & Jin L. Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am. J. Hum. Genet. 71, 1227–1234 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. et al. Genome-wide haplotype association study identifies BLM as a risk gene for prostate cancer in Chinese population. Tumor Biol. 36, 2703–2707 (2015). [DOI] [PubMed] [Google Scholar]
- Armstrong L. C., Saenz A. J. & Bornstein P. Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J. Cell Biochem. 74, 11–22 (1999). [PubMed] [Google Scholar]
- Krumlauf R. Hox genes in vertebrate development. Cell 78, 191–201 (1994). [DOI] [PubMed] [Google Scholar]
- Acampora D. et al. The human HOX gene family. Nucleic acids Res. 17, 10385–10402 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F., Gonzalez F. & Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell 113, 405–417 (2003). [DOI] [PubMed] [Google Scholar]
- Delpretti S., Zakany J. & Duboule D. A function for all posterior Hoxd genes during digit development. Dev. Dynam. 241, 792–802 (2012). [DOI] [PubMed] [Google Scholar]
- Andrey G. et al. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340, 1234167 (2013). [DOI] [PubMed] [Google Scholar]
- Kmita M., Fraudeau N., Herault Y. & Duboule D. Serial deletions and duplications suggest a mechanism for the collinearity of Hoxd genes in limbs. Nature 420, 145–150 (2002). [DOI] [PubMed] [Google Scholar]
- Cobb J. & Duboule D. Comparative analysis of genes downstream of the Hoxd cluster in developing digits and external genitalia. Development 132, 3055–3067 (2005). [DOI] [PubMed] [Google Scholar]
- Goodman F. R., Majewski F., Collins A. L. & Scambler P. J. A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am. J. Hum. Genet. 70, 547–555 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield T. The polycerate trait in Jacob and Navajo-Churro sheep may be located on Ovine Chromosome 2. in Plant and Animal Genome XXIII Conference (Plant and Animal Genome, 2015).
- Zha Y. et al. Functional dissection of HOXD cluster genes in regulation of neuroblastoma cell proliferation and differentiation. PLoS One 7, e40728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordino P. & Duboule D. A molecular approach to the evolution of vertebrate paired appendages. Trends Ecol. Evol. 11, 114–119 (1996). [DOI] [PubMed] [Google Scholar]
- Spitz F. et al. Large scale transgenic and cluster deletion analysis of the HoxD complex separate an ancestral regulatory module from evolutionary innovations. Genes Dev. 15, 2209–2214 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P. & Capecchi M. R. Axial homeosis and appendicular skeleton defects in mice with a targeted disruption of hoxd-11. Development 120, 2187–2198 (1994). [DOI] [PubMed] [Google Scholar]
- Akarsu A. N., Stoilov I., Yilmaz E., Sayli B. S. & Sarfarazi M. Genomic structure of HOXD13 gene: a nine polyalanine duplication causes synpolydactyly in two unrelated families. Hum. Mol. Genet. 5, 945–952 (1996). [DOI] [PubMed] [Google Scholar]
- Yamagishi T., Ozawa M., Ohtsuka C., Ohyama-Goto R. & Kondo T. Evx2-Hoxd13 intergenic region restricts enhancer association to Hoxd13 promoter. PLoS One 2, e175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugaszewska B. et al. Breakpoints around the HOXD cluster result in various limb malformations. J. Med. Genet. 43, 111–118 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.