

Abstract
Using lentiviral vector products in clinical applications requires an accurate method for measuring transduction titer. For vectors lacking a marker gene, quantitative polymerase chain reaction is used to evaluate the number of vector DNA copies in transduced target cells, from which a transduction titer is calculated. Immune Design previously described an integration-deficient lentiviral vector pseudotyped with a modified Sindbis virus envelope for use in cancer immunotherapy (VP02, of the ZVex platform). Standard protocols for titering integration-competent lentiviral vectors employ commercial spin columns to purify vector DNA from transduced cells, but such columns are not optimized for isolation of extrachromosomal (nonintegrated) DNA. Here, we describe a 96-well transduction titer assay in which DNA extraction is performed in situ in the transduction plate, yielding quantitative recovery of extrachromosomal DNA. Vector titers measured by this method were higher than when commercial spin columns were used for DNA isolation. Evaluation of the method’s specificity, linear range, and precision demonstrate that it is suitable for use as a lot release assay to support clinical trials with VP02. Finally, the method is compatible with titering both integrating and nonintegrating lentiviral vectors, suggesting that it may be used to evaluate the transduction titer for any lentiviral vector.
Introduction
One of the battery of lot release tests performed on lentiviral vectors to allow use in clinical trials is a measure of cell transduction capability, commonly referred to as a transduction titer. Defined as the transfer of genetic material from the vector to a target cell, transduction is a multistep process. In the case of lentiviral vectors, this process involves the conversion of one of the two copies of the single-stranded RNA genome into double-stranded vector DNA, which then resides in the target cell nucleus and directs expression of the delivered transgene(s). For the typical lentiviral vector designed to express a recombinant protein, one of two methods of readout is typically used to quantify these transduction events: measuring vector DNA copies by quantitative polymerase chain reaction (qPCR) or enumerating transgene-expressing cells in the target cell population. qPCR is the more straightforward of these two methods, as it can be transgene independent and allows absolute quantitation of transduction events. In comparison, assays for transgene expression require sufficient sensitivity and specificity for the recombinant protein to detect single transduction events per cell and thus can be limited by available reagents and quantitative methods.
Assays to support lot release of products for use in humans must be capable of being validated, consistent with current good manufacturing practices (cGMP) requirements. For assays that measure product characteristics (such as transduction titer), the parameters to be demonstrated include specificity, linearity within a defined range, precision, and accuracy.1 Specificity is the ability to unequivocally distinguish signal from a vector transduction event from noise present in the sample being analyzed. This parameter is a particular concern in the case of lentiviral vectors prepared by transient transfection of plasmid DNA, due to the inability of qPCR to distinguish between vector DNA (resulting from a legitimate transduction event) and residual plasmid DNA which may be present in the vector sample. Linearity is defined as the ability to obtain a titer measurement directly proportional to the concentration of transduction-competent vector in the input sample. Precision is the repeatability of that titer measurement over multiple replicates, both within a single assay (within-day variability) and from assays performed at different times (day-to-day variability). Accuracy is the agreement between a measured value and an accepted or true value. There is currently no widely accepted lentiviral vector reference material which can be used to evaluate accuracy. Moreover, transduction is a relative measure, influenced by receptor density on cells, cell culture conditions, and so on, which may vary from one protocol to another. Thus, for evaluating transduction assay methods, our focus was on specificity, determining the linear range, and precision or repeatability. In addition to these parameters, ease of use and robustness are significant considerations in method development. An ideal method would be compatible with titering multiple vector preparations simultaneously, with as little hands-on time as possible.
We have developed an integration-deficient, HIV-1-based lentiviral vector pseudotyped with a modified Sindbis virus envelope (VP02, a member of the ZVex vector platform) as an immunotherapeutic for use in the field of cancer.2,3 The first product of this platform (LV305), which directs expression of the cancer testes antigen NY-ESO-1, is currently in clinical trials. In designing a transduction titer assay to support VP02 vectors, a transgene-independent qPCR-based method was the focus of assay development. Two unique aspects of VP02 significantly impacted method development: pseudotyping with the modified Sindbis virus glycoprotein E1001 and integration deficiency. The E1001 envelope glycoprotein has been designed to target VP02 to the dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) receptor on human dendritic cells (DCs) in vivo2,3; in comparison, traditional lentiviral vectors encode the pan-tropic vesicular stomatitis virus (VSVG) envelope glycoprotein. To increase safety for in vivo administration, VP02 is rendered integration deficient due to a D64V Integrase mutation within the gag/pol gene and the deletion of the 3′-polypurine tract within the vector genome.3 This latter modification promotes circularization of vector DNA in target cells following reverse transcription, resulting in extrachromosomal episomes instead of integrated proviral DNA.4 Thus, key aspects of a suitable qPCR-based transduction assay would be a cell line expressing the DC-SIGN receptor and an approach to quantitatively recover episomal DNA from transduced cells. Here, we describe a method suitable for determining the transduction titer of nonintegrating lentiviral vectors, including VP02, and evaluate its performance characteristics, including specificity, linear range, and precision.
Results
Evaluation of two DNA extraction methods
The initial objective in developing our assay was to identify a method of DNA extraction that was compatible with recovering nonintegrated vector DNA. We investigated two methods: (i) the traditional approach for integrating lentiviruses of DNA isolation by commercial spin column5–9 and (ii) an in situ approach in which transduction, cell lysis, and DNA extraction are all performed in the same well of a microtiter plate by the addition of a cell lysis buffer, followed by heat denaturation. As a starting point for the latter method, we used a protocol developed for measuring tissue culture infectious dose (TCID50) of adeno-associated virus (AAV), an episomal DNA virus10 (see protocol posted at http://www.atcc.org/~/media/AAV8_Information/AAV2_Information/AAV2_RSS_Infectious_titer_assays_V2.ashx). In a TCID50 assay, each sample is scored negative or positive, so while the in situ method described for AAV is clearly compatible with extracting episomal DNA from cells, it was uncertain whether recovery of lentiviral vector target DNA would be quantitative.
A secondary objective was to identify a DNA extraction protocol compatible with maximizing sensitivity of the final transduction titer method. Assay sensitivity is directly correlated to the number of transduction events (and therefore the number of transduced cells) that can be analyzed in each qPCR reaction and therefore, given a fixed qPCR reaction volume, to the concentration of the isolated DNA from transduced cells. Thus, both DNA extraction methods were investigated for the ability to quantitatively recover episomal DNA in the context of maximizing cell number assayed per qPCR.
To model the recovery of episomal vector DNA from target cells, plasmid DNA of known concentration was “spiked” into a known quantity of assay target cells and total DNA was extracted using either the DNeasy Blood & Tissue kit (Qiagen) or a modified version of the AAV protocol for direct DNA extraction (in situ method). The assay target cell line consisted of adherent 293T cells engineered to express the VP02 receptor DC-SIGN (293T-MLV-DCSIGN). Previous work has demonstrated that this cell line is transduced by VP02 in a DC-SIGN-dependent manner.3,11 To maximize sensitivity of the spin column method, a wide range of cell inputs within the limits suggested by the manufacturer was evaluated (1.56 × 104 – 1.00 × 106 cells/column), each of which was eluted in a final fixed volume of 400 µl, as suggested by the manufacturer to maximize DNA yield. In comparison, in the in situ extraction method, the starting cell number is fixed. To increase sensitivity of this approach, we modified the original protocol described for TCID50 of AAV to maximize the proportion of a transduction well that could be analyzed without inhibiting the qPCR. In brief, we increased the volume of cell lysate analyzed per qPCR by greater than threefold and decreased the total DNA extraction volume by approximately half, thereby increasing the sample DNA concentration by approximately twofold. The result of these modifications was to increase the number of transduced cells that can be analyzed per qPCR (and therefore assay sensitivity) by greater than sevenfold (Figure 1).
Figure 1.
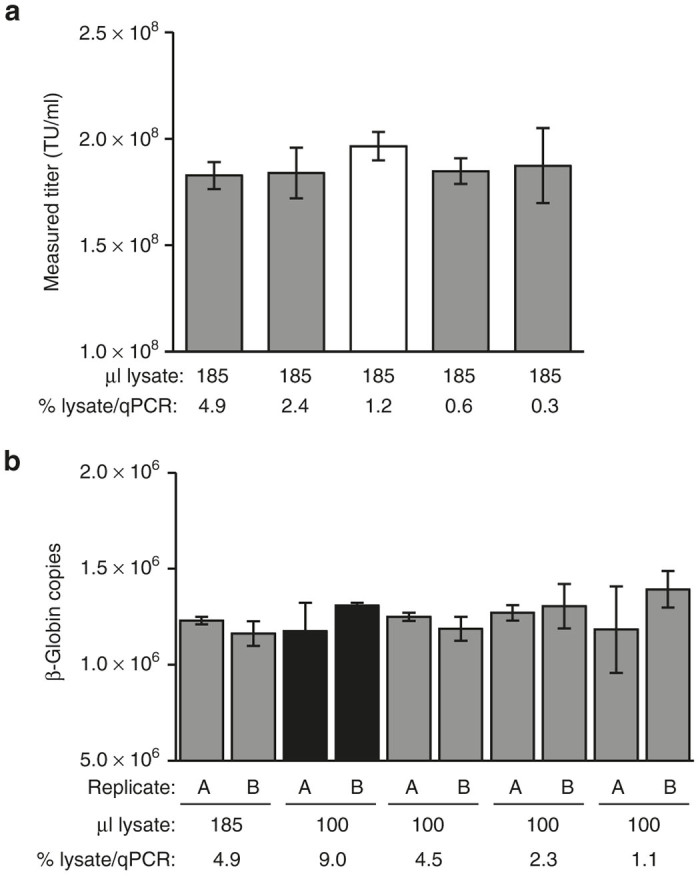
Optimization of cell lysis and qPCR conditions for direct DNA extraction in a microtiter plate. (a) 293T-MLV-DCSIGN assay cells were transduced with VP02 and lysed in a final volume of 185 µl. qPCR for vector sequence was performed on a range of input cell lysate volumes, expressed as a percentage of the total cell lysate analyzed per qPCR. White bar indicates conditions similar to those described in the TCID50 protocol for AAV.10 Error bars are mean ± SD for qPCR quadruplicates. (b) 293T-MLV-DCSIGN assay cells were lysed in a final volume of 100 or 185 µl and analyzed by qPCR for cellular β-globin as an indicator of DNA recovery. Cell lysis was performed in duplicate within a single experiment (replicates A, B); error bars are mean ± SD for qPCR triplicates. Black bars indicate DNA extraction conditions and subsequent qPCR sample volume chosen for in situ transduction titer assay. TU, transduction units.
Quantitative plasmid spike recovery was observed for both DNA extraction methods (Table 1). However, for the spin column, spike recovery varied considerably with input cell number and was optimal at an input of ≤6.25 × 104 cells/column. This limited sensitivity of the spin column method to ≤1.41 × 103 transduced cells/qPCR (assuming a 400 µl spin column elution volume and 9 µl sample/qPCR). In comparison, the modified in situ lysis protocol also yielded quantitative recovery and was predicted to be significantly more sensitive than the spin column method: 1.60 × 105 cells/extraction well corresponds to 1.44 × 104 transduced cells/qPCR.
Table 1. Evaluation of plasmid DNA recovery using DNeasy DNA extraction columns or direct DNA extraction in situ.
|
Recovered plasmid copies |
||||||
|---|---|---|---|---|---|---|
| DNA extraction method | Cell count | Spiked plasmid copies | Replicate 1 | Replicate 2 | Average recovered plasmid copies | % Recovery |
| DNeasy Blood & Tissue | 1.00 × 106 | 1.33 × 106 | 3.43 × 105 | 6.35 × 105 | 4.89 × 105 | 36.8 |
| 2.50 × 105 | 1.33 × 106 | 1.02 × 106 | 3.95 × 105 | 7.08 × 105 | 53.2 | |
| 6.25 × 104 | 1.33 × 106 | 1.44 × 106 | 1.49 × 106 | 1.46 × 106 | 110 | |
| 1.56 × 104 | 1.33 × 106 | 1.61 × 106 | 1.73 × 106 | 1.67 × 106 | 126 | |
| in situ | ~1.60 × 105 | 1.33 × 106 | 1.23 × 106 | 1.25 × 106 | 1.24 × 106 | 93.5 |
Plasmid DNA of known concentration was added to the indicated number of 293T-MLV-DCSIGN assay cells in duplicate (replicates 1 and 2). DNA was extracted using the DNeasy Blood & Tissue kit or 96-well in situ DNA extraction method, and qPCR performed to detect the plasmid spike. % yield is expressed relative to the number of spiked plasmid copies and reflects the average yield of the two replicates. Cell count for the in situ DNA extraction method was estimated based on one cell doubling between cell seeding and DNA extraction.
We next performed a side-by-side comparison of the two DNA extraction methods in a transduction titer assay. Integration-deficient VP02 and a matched E1001-pseudotyped, integration-competent vector were assayed in parallel (Figure 2). 293T-MLV-DCSIGN assay cells were transduced in 12-well plates and DNA isolated from 5 × 104 transduced cells using a DNeasy spin column; alternatively, assay cells were transduced in a 96-well plate and DNA isolated using the modified in situ DNA extraction method. Following direct extraction, no difference in titer was observed between the integration-competent and -deficient vectors, suggesting that both integrated provirus and extrachromosomal vector DNA were recovered with equal efficiency (Figure 2a). In contrast, DNA extraction using a spin column appeared to favor recovery of integrated vector DNA, as evidenced by the approximately twofold higher transduction titer measured for integration-competent vector using this method (Figure 2b). In addition, overall higher transduction titers (greater than fivefold) were measured using the modified in situ DNA extraction method. These results demonstrate that the modified in situ DNA extraction method is compatible with titering both integration-competent and -deficient lentiviral vectors and yields higher titers than when DNA is extracted using a traditional spin column. Based on these experiments, direct extraction was favored over the use of the spin column for increased sensitivity, reduced sample manipulations, labor and cost, and easier scalability in the cell culture portion of the final transduction titer assay.
Figure 2.
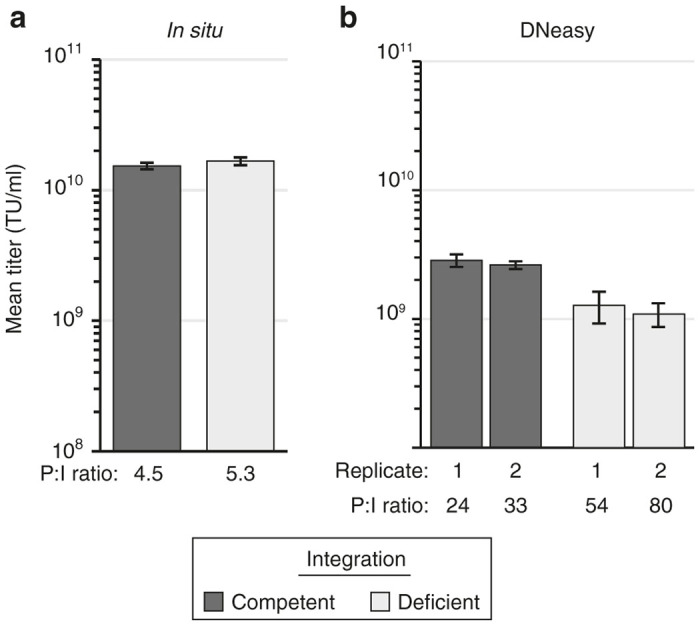
Comparison of vector transduction titers measured following in situ versus spin column (DNeasy) DNA extraction. 293T-MLV-DCSIGN assay cells were transduced with equivalent amounts of integration-competent or -deficient vector particles (as measured by vector genomes). Following an overnight incubation, DNA was isolated, and samples were analyzed by qPCR to detect vector sequence. (a) Extraction performed using in situ method. Each bar represents the mean ± SD of nine transductions performed in parallel (n = 3 qPCR replicates/transduction). (b) Extraction performed using DNeasy column (5 × 104 cells loaded per column). Each bar represents the mean ± SD of qPCR triplicates for a single transduction; two transductions were performed in parallel for each vector tested (replicates 1, 2). Particle-to-infectivity (P:I) ratios were calculated by dividing the physical particle titer (measured by RNA genome content per vector) by the measured transduction titer. Results in both panels are representative of two independent experiments. TU, transduction units.
Specificity
Our final transduction titer assay format, which employs the modified in situ DNA extraction protocol, is diagrammed in Figure 3a. The assay cell line comprised 293T-MLV-DCSIGN cells. Vector is incubated with target cells for 1 day prior to harvest and analysis, for ease of use. No significant difference in vector DNA signal was observed when assay cells were incubated with vector for longer periods of time prior to harvest (data not shown). The qPCR primer and probe set was designed to specifically amplify an 89-bp sequence within the lentiviral vector genome located between the packaging signal and the Rev-response element. This sequence is contained within all Immune Design lentiviral vectors and is absent from the genome of the assay cell line. Quantitation of transduction events in extracted DNA is performed relative to a seven-point qPCR standard curve comprised of vector plasmid DNA (Figure 3b).
Figure 3.
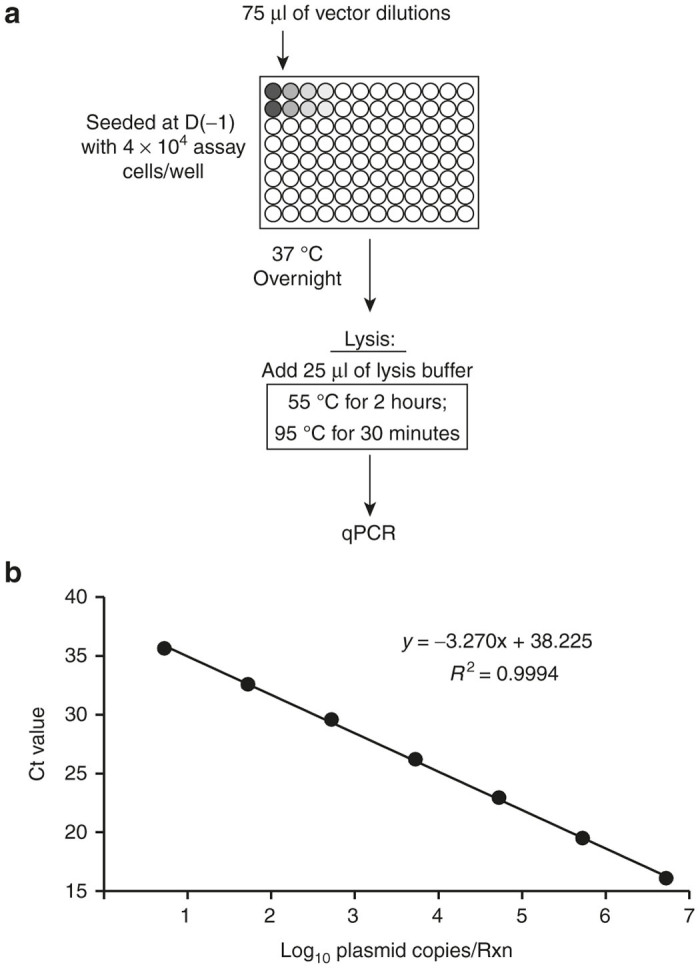
Design of a 96-well lentiviral vector transduction assay using an in situ DNA extraction method. (a) Schematic of the method. Assay cells seeded in 96-well microtiter plates are transduced with serial dilutions of vector (75 µl/well final volume). Following an overnight incubation, cells are lysed by the addition of 25 µl of lysis buffer containing detergents and proteinase K (100 µl total cell lysate). Plates are then sealed and incubated at the indicated temperature hold steps to degrade proteins and denature the DNA, after which samples are analyzed by qPCR (9 µl sample/rxn) to detect vector sequence. (b) Ct values (mean and SD) of the seven-point qPCR standard curve (n = 3 qPCR replicates; symbol size was larger than error bars).
Specificity is a significant challenge for any qPCR-based transduction titer assay when vector test article is produced by transient transfection, since vector DNA (resulting from a transduction event) cannot be distinguished from residual plasmid in the test vector, as the qPCR amplicon is present in both. The significance of this problem depends on the purity level of the vector being tested. It is currently common practice to treat preparations of harvested lentiviral vector with Benzonase nuclease to digest residual plasmid DNA. However, we have found that Benzonase treatment alone is not sufficient to degrade the totality of plasmid DNA into pieces smaller than the 89-bp amplicon that is targeted in our qPCR assay. Therefore, additional purification measures are required to isolate the vector to be assayed from the contaminating plasmid DNA fragments. This can be done by ultracentrifugation through a sucrose cushion (research-grade vector) or via column chromatography (GMP-grade vector).
Given this issue of residual plasmid DNA, the transduction assay by nature cannot be specific for vector transduction events. To compensate for this lack of specificity, we performed the method in the presence or absence of nevirapine, a reverse-transcriptase inhibitor. In the presence of nevirapine, lentiviral vector transduction is blocked at the point of RNA vector genome conversion into vector DNA, which is the target for qPCR. Therefore, nevirapine-treated samples are useful for identifying when the measured vector DNA titer is transduction-specific or due to pseudotransduction (transfer of residual plasmid DNA present in lentiviral vector preparations from transient transfection systems). As demonstrated in Figure 4a, a representative preparation of research-grade vector contains a measurable amount of residual plasmid DNA (pseudotransduction), as evidenced by the signal observed in the presence of nevirapine. By comparison, signal from nevirapine-treated, GMP-manufactured VP02 is below the assay lower limit of detection (data not shown).
Figure 4.

Evaluation of specificity and linear response range of the 96-well transduction titer assay. (a) Research-grade lentiviral vector treated with benzonase and purified by centrifugation through a sucrose cushion was analyzed using the 96-well transduction titer assay performed in the presence or absence of nevirapine, an inhibitor of reverse transcriptase. Error bars are mean of n = 4 transduction replicates performed in a single experiment ± SD (n = 3 qPCR replicates/transduction). (b) Serial dilutions of GMP-manufactured VP02 vector were titered using the method described in Figure 3a. Error bars are mean of n = 3 transduction replicates ± SD in a single experiment (n = 3 qPCR replicates/transduction). Graph is representative of four independent experiments. (c) Dilutional linearity analysis for data points indicated by horizontal bar in b. The slope of the line is not significantly different from zero (P = 0.1), as analyzed by GraphPad Prism 6. Vector DNA copies/cell assumes two cell doublings between cell seeding and time of harvest. The approximate MOI was calculated from the measured transduction titer at the corresponding vector input amount, assuming one cell doubling at time of transduction. MOI, multiplicity of infection; TU, transduction units.
Linearity
To evaluate the linearity of the method, VP02 was assayed over a three-log range of input vector and the titer back-calculated, accounting for dilution factor (Figure 4b). Dilutional linearity was consistently observed at <1 vector DNA copy detected per target cell as evidenced by the slope of the line from these points being not significantly different from zero (P = 0.1, Figure 4c). This corresponds to multiplicity of infection (MOI) of vector input ≤ 1. At higher concentrations of input vector, the measured titer increased with increasing MOI. Therefore, we defined the linear response range of the method as restricted to vector input of MOI ≤ 1.
Precision
Intra- and inter-assay variability of the 96-well vector transduction assay were evaluated. A six-point dilution series of VP02 was prepared, and three transduction replicates were performed for each dilution on a single transduction plate (18 transductions total). For each transduction well, the titer was calculated from the average of qPCR triplicates. This analysis was repeated for a total of three assays (Table 2). At vector input levels shown in the previous section to have a linear response, the coefficient of variation of back-calculated transduction titer for replicate transduction wells ranged from 11 to 47% (intra-assay variability). For each assay, the vector titer was expressed as the average of all 15 transductions that fell within the linear response range. The coefficient of variation of the vector transduction titers measured across three assays was 3.9% (inter-assay variability; Table 3). This low inter-assay variability demonstrates that the assay exhibits good precision.
Table 2. Evaluation of intra-assay variation.
|
Transduction titer (TU/ml) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Vector genomes | Approximate MOI | Series 1 | Series 2 | Series 3 | Average | SD | % CV | |
| Assay 1 | 2.2 × 107 | 4.3 | 3.4 × 108 | 2.1 × 108 | 2.6 × 108 | 2.7 × 108 | 6.7 × 107 | 25 |
| 7.3 × 106 | 1.0 | 1.9 × 108 | 1.4 × 108 | 1.2 × 108 | 1.5 × 108 | 3.2 × 107 | 21 | |
| 2.4 × 106 | 0.3 | 1.7 × 108 | 6.3 × 107 | 1.0 × 108 | 1.1 × 108 | 5.2 × 107 | 47 | |
| 1.2 × 106 | 0.1 | 1.6 × 108 | 6.4 × 107 | 9.6 × 107 | 1.1 × 108 | 4.8 × 107 | 45 | |
| 6.1 × 105 | 0.06 | 1.5 × 108 | 7.6 × 107 | 8.9 × 107 | 1.1 × 108 | 3.9 × 107 | 38 | |
| 3.1 × 105 | 0.04 | 1.5 × 108 | ND | 9.2 × 107 | 1.2 × 108 | 4.3 × 107 | 35 | |
| Assay 2 | 2.2 × 107 | 4.3 | 1.9 × 108 | 1.5 × 108 | 1.6 × 108 | 1.6 × 108 | 3.4 × 107 | 22 |
| 7.3 × 106 | 1.0 | 1.4 × 108 | 1.4 × 108 | 1.3 × 108 | 1.3 × 108 | 1.7 × 107 | 13 | |
| 2.4 × 106 | 0.3 | 1.5 × 108 | 1.3 × 108 | 1.2 × 108 | 1.2 × 108 | 3.6 × 107 | 31 | |
| 1.2 × 106 | 0.1 | 1.1 × 108 | 1.2 × 108 | 1.0 × 108 | 1.0 × 108 | 2.0 × 107 | 20 | |
| 6.1 × 105 | 0.06 | 1.2 × 108 | 9.4 × 107 | 9.9 × 107 | 9.9 × 107 | 2.0 × 107 | 20 | |
| 3.1 × 105 | 0.04 | 1.2 × 108 | 9.0 × 107 | 1.1 × 108 | 1.1 × 108 | 1.7 × 107 | 15 | |
| Assay 3 | 2.2 × 107 | 4.3 | 1.6 × 108 | 1.6 × 108 | 1.6 × 108 | 1.6 × 108 | 1.2 × 106 | 0.7 |
| 7.3 × 106 | 1.0 | 1.3 × 108 | 9.9 × 107 | 1.4 × 108 | 1.2 × 108 | 2.1 × 107 | 17 | |
| 2.4 × 106 | 0.3 | 1.0 × 108 | 1.1 × 108 | 1.3 × 108 | 1.1 × 108 | 1.3 × 107 | 11 | |
| 1.2 × 106 | 0.1 | 1.3 × 108 | 1.2 × 108 | 1.5 × 108 | 1.3 × 108 | 1.7 × 107 | 13 | |
| 6.1 × 105 | 0.06 | 1.2 × 108 | 1.1 × 108 | 9.0 × 107 | 1.1 × 108 | 1.6 × 107 | 15 | |
| 3.1 × 105 | 0.04 | 9.7 × 107 | 1.2 × 108 | 1.6 × 108 | 1.3 × 108 | 3.2 × 107 | 25 | |
Multiple independent dilution series of a single vector sample were titered on one assay plate (series 1, series 2, and series 3). This assay was performed a total of three times on different days (assay 1, assay 2, and assay 3). Italicized numbers are outside the linear response range established in Figure 4c.
CV, coefficient of variation; MOI, multiplicity of infection; TU, transduction units.
Table 3. Evaluation of inter-assay variation.
| Assay | Titer (TU/ml)a | Average titer | SD | %CV |
|---|---|---|---|---|
| 1 | 1.2 × 108 | 1.2 × 108 | 4.5 × 106 | 3.9 |
| 2 | 1.1 × 108 | — | — | — |
| 3 | 1.2 × 108 | — | — | — |
CV, coefficient of variation; TU, transduction units.
Mean titer from all dilutions within the linear response range of the assays described in Table 2.
Discussion
A transduction titer assay was developed that is compatible with measuring the titer of nonintegrating lentiviral vectors such as VP02, an E1001-pseudotyped enhanced third-generation lentiviral vector designed to target the DC-SIGN receptor on human DCs in vivo. In this method, DNA extraction is performed directly in the transduction plate by the addition of a lysis buffer containing proteinase K, followed by incubation at 55 °C to degrade proteins, and finally heat denaturation. The resulting cell lysates are directly analyzed by qPCR to detect vector DNA sequence.
The method of DNA isolation was a primary concern during initial assay development. Commercial spin columns are commonly used for DNA extraction but pose several challenges for vector titering. First, the efficiency of sample recovery may vary from column to column and is not quantitative. In the case of Qiagen DNeasy columns, recovery of ~60–80% of sample DNA is anticipated, according to the manufacturer. Since titer assignment is directly linked to sample recovery (back-calculated to account for input vector), it stands to reason that titers measured using a spin column DNA extraction method are likely to be an underestimate. Indeed, this is what we observed when DNeasy columns were compared to direct lysis of vector-transduced cells in situ (Figure 2). Second, manipulating multiple spin columns becomes cumbersome when dealing with >18–24 samples (the number that can fit in a standard microfuge rotor). While 96-well high-throughput versions are available for many DNA isolation columns (including DNeasy), scaling up the cell culture portion of the assay in parallel becomes burdensome if 12-well scale transductions are required (as was performed here). Finally, for vectors that do not integrate, including VP02 lentiviral vectors, we were concerned that spin columns designed to isolate genomic DNA would be suboptimal for recovery of episomal DNA. Indeed, we observed that the efficiency of plasmid spike recovery from DNeasy columns was inversely proportional to the number of input cells on the column (Table 1). Presumably, this was due to competition between genomic DNA and plasmid DNA for column binding. The result of this competition was an apparent difference in particle-to-infectivity (P:I) ratios between integration-competent versus integration-deficient vector preparations when titers were measured following spin column DNA extraction of transduced cells (Figure 2b).
The choice of assay target cell type was another significant consideration. VP02 has been designed to transduce human DCs, which are nondividing cells. Arguably, using human DCs in our transduction titer assay may yield more a physiologically relevant titer assignment for VP02 vectors than the engineered 293T-MLV-DCSIGN cell line chosen. However, donor-to-donor variability poses a significant challenge for the use of human primary cells in quality control lot release assays for products intended for clinical use, such as VP02. To minimize this variability, we chose to develop our transduction titer assay using a defined cell line, from which we will derive a master and working cell bank and which can ultimately be certified. One disadvantage of using a dividing cell line is that replication of target cells between transduction and harvest may result in loss of episomal vector DNA copies. To mitigate this, in our assay, cell harvest is performed at ~18 hours posttransduction, during the course of which only a single cell division may be expected to occur. Notably, we have titered VP02 on primary human DCs utilizing the method described herein and found no difference in the number of transduction events measured, in comparison to assaying the same vector on 293T-MLV-DCSIGN cells (data not shown). These data suggest that cell division is not contributing significantly to loss of vector DNA signal in the chosen assay cell line.
Products for use in humans must meet lot release criteria that are set based on the performance characteristics of the assays employed for lot release. Our final transduction titer assay was evaluated for the following standard assay performance characteristics: specificity, linear response range, and precision (repeatability). While several groups have described transduction titer assays for HIV-1-based lentiviral vectors,6,7,9 to date none has examined the robustness of these assays using standard metrics applied to quality control lot release assays. In our study, we were surprised to note that the linear response range of our 96-well transduction assay was limited to vector inputs of MOI ≤ 1 (corresponding to detection of <1 copy of vector DNA/cell). Our findings suggest that at higher MOIs, vector transduction events are cooperative, resulting in an artificially inflated titer. One possible explanation for this finding is that vector particles whose transduction would otherwise be aborted are rescued by co-transduction with transduction-competent particles. Regardless of mechanism, the lack of dilutional linearity at MOI > 1 likely applies to any protocol employed to titer lentiviral vectors and should be an avenue of investigation for researchers using alternative transduction titering protocols.
While variability is inherent with any biological assay, in the case of our new transduction titering method we found that precision increases with the number of transduction wells used to calculate the final vector titer (Tables 2 and 3). This includes both the number of dilutions of input vector as well as the number of transduction replicates per dilution. One advantage of the 96-well format of our transduction titer assay is that it is well suited for assaying many sample replicates simultaneously, with little added labor. Assuming 15 transduction wells per test article (a five-point dilution series within the linear response range, with three transduction replicates per dilution, as in Table 2), it is possible to assay five or six vector test articles per transduction plate, depending on whether the user employs a 384-well qPCR machine or is limited by fitting both samples and standard curve replicates within a 96-well qPCR format. For test articles that do not require as precise a final titer measurement (i.e., vectors for research that do not require quality control lot release), fewer transductions can be performed per test article, thereby greatly increasing the number of vector samples that may be analyzed simultaneously.
In conclusion, we have developed a sensitive, reproducible, and robust transduction titer assay for lentiviral vectors, which employs direct DNA extraction of vector-transduced cells in situ. Initially designed for titering integration-deficient VP02, this new transduction titering method is also compatible with integrating lentiviral vectors and has the potential to be adapted more broadly for the analysis of replication-competent retroviruses.
Materials and Methods
Cell lines
293T-MLV-DCSIGN assay cells constitutively express human DC-SIGN, the cellular receptor for VP02 (ref. 3). This cell line was maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum.
Plasmid spike recovery
To test spike recovery in the DNeasy Blood & Tissue kit (Qiagen, Valencia, CA), the indicated number of 293T-MLV-DCSIGN cells were pelleted in microfuge tubes and resuspended in 200 µl of PBS in duplicate. 1.33 × 106 copies of plasmid DNA were added to each tube and total DNA extracted following the manufacturer’s instructions for tissue culture cells (eluted in a final volume of 400 µl). To test the in situ method for DNA extraction, 8 × 104 293T-MLV-DCSIGN cells were seeded in 96-well microtiter plates and left to grow overnight (to yield ~1.6 × 105 cells at time of harvest) to model the final target cell number at time of lysis in the final transduction assay. The indicated copies of plasmid DNA were added to each well, and total DNA extracted following the transduction assay procedure described below. For both methods, extracted DNA was analyzed by qPCR to detect spiked plasmid; total recovered plasmid copies was back calculated based on the percentage of isolated DNA analyzed per qPCR.
Vectors
The design and production of VP02 has previously been described.2,3,11 In brief, VP02 is produced via transient transfection of 293T cells with five component plasmids consisting of the transfer vector (lentiviral vector genome encoding the transgene of interest), a modified gag/pol transcript, the accessory protein Rev from HIV-1, the accessory protein Vpx from SIVmac, and the E1001 envelope glycoprotein. The VP02 genome encodes cis-elements that are derived from HIV-1 important for packaging and splicing, as well as the Rev-response element. A modified ubiquitin promoter transcribes the genome, which is flanked by a 5′LTR (R and U5) and 3′LTR (self-inactivating U3, R, and U5). VP02 is rendered integration deficient because of a D64V Integrase mutation within the gag/pol gene and the deletion of the 3′-polypurine tract within the vector genome. Unless otherwise noted, VP02 vector was used throughout these studies. In Figure 2 the integration-competent vector consisted of VP02 modified to encode wild-type Integrase and an intact 3′-polypurine tract. The vectors used encoded the human NY-ESO-1 gene, with the exception of Figure 2, in which both the integration-competent and -deficient vectors encoded firefly luciferase.
Transductions were performed in EasyFill Cell Factories (research-grade vector) (Thermo Scientific, Waltham, MA) or HYPERStack-36 layer vessels (GMP-grade vector) (Corning, Corning, NY). Kifunensine (Glycosyn, Gracefield, New Zealand) was added to the culture media at 5 hours posttransfection to a final concentration of 1 µg/ml. At 2 or 3 days posttransfection, harvested vector was treated with Benzonase nuclease (EMD Millipore, Billerica, MA) to digest residual input plasmid DNA. Research-grade vector was then purified by ultra-centrifugation through a sucrose cushion. Alternatively, GMP-grade VP02 was purified through a series of steps, including chromatography and tangential flow filtration, to remove nucleic acid and process-related impurities. Research-grade vector was used in Figures 2 and 4a. All other figures used GMP-grade VP02.
Vector quantification: 96-well transduction titer assay
293T-MLV-DCSIGN assay cells were seeded at 4 × 104 cells/well in 50 µl in 96-well tissue culture microtiter plates and left to adhere overnight. Vector diluted in complete medium was added to each well (25 µl/well); where indicated in the text, nevirapine was added to a final concentration of 10 µmol/l (Sigma-Aldrich, St Louis, MO). At 18 hours posttransduction, 25 µl of lysis buffer (0.14% sodium deoxycholate, 1% Tween-20, 25 mmol/l Hepes (pH 8.0), 2 mmol/l Tris–HCl (pH 8.0), 2 mmol/l EDTA, 0.002% SDS, 0.43 mg/ml Proteinase K) was added to each well and pipetted to mix. An adhesive PCR plate seal was applied (Bio-Rad, Hercules, CA) to prevent evaporation, and the plate was then incubated at 55 °C for 2 hours, followed by a 30-minute hold at 95 °C. After cooling to room temperature, the resulting samples were analyzed directly by qPCR using the following primer/probe set (purchased from Integrated DNA Technologies, Coralville, IA, and BioSearch Technologies, Petaluma, CA): forward primer, 5′-GGCAAGCAGGGAGCTAGAAC-3′; reverse primer, 5′-GTTGTAGCTGTCCCAGTATTTGTC-3′; probe, 5′-(FAM)-TCGCAGTTAATCCTGGCCTGTTAGA-(BHQ)-3′. This primer/probe set amplifies an 89-bp sequence between the vector packaging signal and the Rev-response element, homologous to a fragment of HIV-1 gag. Quantitation was performed relative to a standard curve comprised of plasmid DNA containing the target sequence and diluted over a 7-log range (5.3 – 5.3 × 106 copies/reaction). Reactions were performed using EXPRESS qPCR Supermix Universal (Life Technologies, Carlsbad, CA), 500 nmol/l forward primer, 500 nmol/l reverse primer, 300 nmol/l probe, and 9 µl of sample in a 25 µl reaction volume. After initial incubations at 50 °C for 2 minutes and 95 °C for 3 minutes, 40 cycles of amplification were carried out at 95 °C for 15 seconds followed by 60 °C for 30 seconds. Reactions were analyzed using the Bio-Rad CFX96 or CFX384 system (Bio-Rad, Hercules, CA).
In Figure 1b, qPCR for β-globin was performed using the following primer/probe set (purchased from Integrated DNA Technologies, Coralville, IA, and BioSearch Technologies, Petaluma, CA): forward primer, 5′-ACACAACTGTGTTCACTAGC-3′; reverse primer, 5′-CAACTTCATCCACGTTCACC-3′; probe, 5′(CAL Fluor Red 610)-CATGGTGCATCTGACTCCTGAGGA-(BHQ2)-3′. Quantitation was performed relative to a standard curve comprised of a synthetic oligonucleotide containing the target sequence (gBlock, Integrated DNA Technologies) and diluted over a 7-log range (101 – 107 copies/reaction). Reactions were performed using EXPRESS qPCR Supermix Universal (Life Technologies), 500 nmol/l forward primer, 500 nmol/l reverse primer, 300 nmol/l probe, and 9 µl of sample in a 25 µl reaction volume. After initial incubations at 50 °C for 2 minutes and 95 °C for 3 minutes, 40 cycles of amplification were carried out at 95 °C for 15 seconds followed by 50 °C for 30 seconds. Reactions were analyzed using the Bio-Rad CFX96 system.
In Figure 2b, assay cells seeded in 12-well plates were transduced with equivalent amounts of integration-competent or -deficient vector (as measured by vector genomes). At 18 hours posttransduction, DNA was purified from 5 × 104 harvested cells using the DNeasy Blood & Tissue kit (Qiagen), following the manufacturer’s protocol. Purified DNA was analyzed by qPCR following the method described above.
Vector quantification: genomes assay
Genomic RNA was isolated from vector particles using the QIAamp Viral RNA Mini kit (Qiagen). To eliminate contaminating DNA, the extracted nucleic acid was then digested with DNAseI (Life Technologies) following the manufacturer’s directions. Samples were analyzed by quantitative reverse transcription polymerase chain reaction using the RNA Ultrasense One-Step Quantitative RT-PCR System (Life Technologies) and vector-specific primers and probe. The RNA genome copy number was calculated in reference to a standard curve comprised of plasmid DNA containing the target sequences, diluted over a 7-log range (5.3 – 5.3 × 106 copies/reaction). The genome titer as expressed here reflects the number of physical vector particles, calculated based on genomes, with each vector particle predicted to contain two single-stranded copies of genomic RNA.
Statistical analysis
Dilutional linearity analysis was performed using Prism 6 (GraphPad Software, La Jolla, CA). To evaluate assay variability, the coefficient of variation was calculated as the percentage of the SD to the mean. For intra-assay variability, the mean encompasses n = 3 wells transduced with the same vector input. For inter-assay variability, the mean was evaluated for n = 3 assay runs, in which each assay run was expressed as the mean of all transduction wells within the linear response range of the method.
Acknowledgments
The authors thank Barbara A. Thorne for insightful scientific discussions and review of the manuscript.
Footnotes
M.E.M., C.D.V., M.M.S., W.R.G., and B.K.-C. are at least one of the following: a current employee of Immune Design and a holder of stock and/or options in Immune Design.
References
- International conference on the harmonization of technical requirements for the registration of pharmaceuticals for human use. (2005). ICH Harmonized Tripartite Guideline Validation of Analytical Procedures: Text and Methodology Q2 (R1). ICH, pp. 1–13. [Google Scholar]
- Odegard, JM, Kelley-Clarke, B, Tareen, SU, Campbell, DJ, Flynn, PA, Nicolai, CJ et al. (2015). Virological and preclinical characterization of a dendritic cell targeting, integration-deficient lentiviral vector for cancer immunotherapy. J Immunother 38: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen, SU, Kelley-Clarke, B, Nicolai, CJ, Cassiano, LA, Nelson, LT, Slough, MM et al. (2014). Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther 22: 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor, B, Bayer, M, Ma, H, Samulski, J, Li, C, McCown, T et al. (2011). Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther 19: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, SL, Hansen, MS and Bushman, FD (2001). A quantitative assay for HIV DNA integration in vivo. Nat Med 7: 631–634. [DOI] [PubMed] [Google Scholar]
- Charrier, S, Stockholm, D, Seye, K, Opolon, P, Taveau, M, Gross, DA et al. (2005). A lentiviral vector encoding the human Wiskott-Aldrich syndrome protein corrects immune and cytoskeletal defects in WASP knockout mice. Gene Ther 12: 597–606. [DOI] [PubMed] [Google Scholar]
- Lizée, G, Aerts, JL, Gonzales, MI, Chinnasamy, N, Morgan, RA and Topalian, SL (2003). Real-time quantitative reverse transcriptase-polymerase chain reaction as a method for determining lentiviral vector titers and measuring transgene expression. Hum Gene Ther 14: 497–507. [DOI] [PubMed] [Google Scholar]
- Martin-Rendon, E, White, LJ, Olsen, A, Mitrophanous, KA and Mazarakis, ND (2002). New methods to titrate EIAV-based lentiviral vectors. Mol Ther 5: 566–570. [DOI] [PubMed] [Google Scholar]
- Sastry, L, Johnson, T, Hobson, MJ, Smucker, B and Cornetta, K (2002). Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther 9: 1155–1162. [DOI] [PubMed] [Google Scholar]
- Lock, M, McGorray, S, Auricchio, A, Ayuso, E, Beecham, EJ, Blouin-Tavel, V et al. (2010). Characterization of a recombinant adeno-associated virus type 2 reference standard material. Hum Gene Ther 21: 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley, DC, McCloskey, L, Thorne, BA, Tareen, SU, Nicolai, CJ, Campbell, DJ et al. (2015). Development of a replication-competent lentivirus assay for dendritic cell-targeting lentiviral vectors. Mol Ther Methods Clin Dev 2: 15017. [DOI] [PMC free article] [PubMed] [Google Scholar]