

Abstract
Autoantibodies to nuclear antigens arise in human autoimmune diseases, but a unifying pathogenetic mechanism remains elusive. Recently we reported that exposure of neutrophils to inflammatory conditions induces the citrullination of core histones by peptidylarginine deiminase 4 (PAD4) and that patients with autoimmune disorders produce autoantibodies that recognize such citrullinated histones. Here we identify histone H1 as an additional substrate of PAD4, localize H1 within neutrophil extracellular traps, and detect autoantibodies to citrullinated H1 in 6% of sera from patients with systemic lupus erythematosus and Sjögren's syndrome. No preference for deiminated H1 was observed in healthy control sera and sera from patients with scleroderma or rheumatoid arthritis. We map binding to the winged helix of H1 and determine that citrulline 53 represents a key determinant of the autoantibody epitope. In addition, we quantitate RNA for H1 histone subtypes in mature human neutrophils and identify citrulline residues by liquid chromatography and tandem mass spectrometry. Our results indicate that deimination of linker histones generates new autoantibody epitopes with enhanced potential for stimulating autoreactive human B cells.—Dwivedi, N., Neeli, I., Schall, N., Wan, H., Desiderio, D. M., Csernok, E., Thompson, P. R., Dali, H., Briand, J.-P., Muller, S., Radic, M. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity.
Keywords: inflammation, autoimmune disorders, peptidylarginine deiminase, chromatin
Autoantibodies in systemic lupus erythematosus (SLE), Sjögren's syndrome (SS), and related rheumatic disorders bind nuclear antigens such as DNA and histones (1–3). How such antigens engage B cells to induce autoantibodies remains unclear. One important possibility is that B cells respond to nuclear chromatin that is released from neutrophils during NETosis, a cell death that is induced by inflammatory conditions (4, 5) and that depends on peptidylarginine deiminase 4 (PAD4)-mediated conversion of arginine to citrulline (6, 7). Histones are the most abundant substrates of PAD4, and their post-translational modifications (PTMs) affect chromatin structure and may play an essential role during NETosis. NETosis is characterized by the release of neutrophil extracellular traps (NETs), flexible lattices that consist of chromatin fibers and associated microbicidal substances (8). NETs contribute to innate immunity because they immobilize and kill microbes (9). Although core histones are integral components of NETs, it is uncertain whether extranucleosomal H1 linker histones are substrates of PAD4 and whether they remain associated with NET chromatin (8–11).
In viable cells, the H1 linker histones bind DNA at the entry and exit points of the nucleosome core particle, thereby determining the trajectory of linker DNA that connects neighboring nucleosomes (12). By controlling the stacking of adjacent nucleosomes, H1 modulates the overall features of the chromatin fiber. In addition, H1 histone binding to DNA sequences that connect adjacent nucleosomes contributes to the regulation of gene expression (13). The importance of H1 for higher-order chromatin structure and gene transcription is reflected in the expression of multiple, structurally diverse, and cell-type-specific isoforms of H1 (14). The tendency of H1 histones to condense chromatin is regulated by a repertoire of PTM that fine-tune H1 affinity for DNA and mediate H1 interactions with other proteins (15, 16). Although a number of PTMs in H1 are known (15), it is not certain whether H1 deimination by PAD4 occurs under conditions that induce NETosis.
Recently we found that certain human autoantibodies preferentially bind deiminated core histones from activated neutrophils (17). Our observations suggested that covalent modifications of histones that arise in the course of neutrophil activation enhance immunogenicity of chromatin autoantigens (18, 19). Here we asked whether activated neutrophils also provide antigenic stimuli for H1-reactive B cells. This question is relevant because autoantibodies to linker histones are specific markers for the diagnosis of SLE (20) and display a strong association with disease activity (21), yet they also arise in other autoimmune conditions such as SS (22). Here we identify autoantibodies that preferentially bind deiminated human H1 linker histones and map the reactive H1 epitopes to H1 domains that contain citrulline residues. In addition, we observe preferential binding of H1-reactive sera to activated neutrophils and detect IgG reactivity with NETs. The data argue that H1-reactive B cells may be stimulated under circumstances that lead to neutrophil activation and NET chromatin release. These observations suggest that NETs may stimulate autoreactive B cells by providing an important source of nuclear autoantigens.
MATERIALS AND METHODS
Patient serum samples
Serum samples were obtained from French and German patients. We further analyzed sera from patients with SLE examined in Dwivedi et al. (17). Additional sera were collected by Dr. Jean-François Kleinmann (Strasbourg University Hospitals, Strasbourg, France) from volunteers attending the Rheumatology Clinics of Strasbourg University Hospitals. Informed consent was obtained from each individual in agreement with the Helsinki declaration, French legislation, and institutional review boards of the University of Tennessee (Memphis, TN, USA) and the University of Lübeck (Lübeck, Germany). In all, sera from 24 SLE, 20 SS, 20 scleroderma, and 20 rheumatoid arthritis (RA) patients and 20 age- and gender-matched controls were tested. All patients fulfilled the American College of Rheumatology classification criteria for SLE (23), SS (24), scleroderma (25), and RA (26).
Neutrophil isolation and stimulation
Neutrophils were isolated from healthy donor blood purchased from Keybiologics (Memphis, TN, USA) as described previously (4). Briefly, neutrophils were enriched using dextran sedimentation and recovered from an isolymph density gradient (Gallard-Schlesinger, Plainview, NY, USA) under endotoxin-free conditions. Erythrocytes were lysed in ice-cold, hypotonic buffer (0.2% NaCl) for 30 s, and lysis was stopped by adding hypertonic saline (1.6% NaCl). Neutrophils were suspended in HBSS (without Ca2+ or Mg2+, pH 7.4) with 0.1% glucose and 0.5% heat-inactivated human serum at a final concentration of 2 × 106 neutrophils/ml. Neutrophils were stimulated with calcium ionophore A23187 at 1 μM with or without the addition of 5 μM chelerythrine in HBBS containing 2 mM Ca2+ at 37°C for 2 h. Following incubations, neutrophils were pelleted and lysed in SDS-lysis buffer (2% SDS in 62.5 mM Tris, pH 6.8, supplemented with 5% 2-ME and 10% glycerol).
Western blot and competition assays
Proteins were resolved on 15% SDS-PAGE and transferred to nitrocellulose, as described previously (17). Membranes were blocked in 5% BSA and 0.1% Tween 20 in Tris-buffered saline (TBS) overnight at 4°C. The following day, the membranes were incubated with rabbit antibodies to deiminated histone H3 (ab5103, Abcam, Cambridge, MA, USA) or deiminated H4 (07-596, Millipore, Billerica, MA, USA), or a mouse monoclonal antibody to human histone H1 (05-457, Millipore). Alternatively, we used patient sera at 5 μg IgG per milliliter of TBS containing 2.5% BSA, 1% Nonidet P-40, and 0.1% SDS. After 2 h of incubation, membranes were washed with 1% Nonidet P-40 in TBS. Antibody binding was detected with species-appropriate anti-IgG-HRP conjugates at 1:40,000 in TBS containing 0.05% Tween 20, and blots were developed using chemiluminescence (PerkinElmer, Waltham, MA, USA). Band intensities were quantitated using the ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). Protein concentrations were measured in a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and equal loading of the samples was assayed by Coomassie blue staining.
For competition assays, synthetic peptides matching human H1.2 were used. Competitor A had arginine 32 of H1.2 replaced with citrulline (KKAGGTP-Cit-KASGPPVS), whereas competitor B had arginine 53 of H1.2 replaced with citrulline (KAVAASKE-Cit-SGVSLAA). The peptides were incubated for 1 h with 5 μg IgG/ml before use in Western blots.
Confocal microscopy
Neutrophils were isolated as described above and allowed to settle for 30 min onto poly-l-lysine-coated glass coverslips as described previously (4). Cells were stimulated with A23187 calcium ionophore with 5 μM chelerythrine for 1 h. Alternatively, we also used lipopolysaccharide (LPS; 100 ng/ml) for 1 h at 37°C. Coverslips were washed with ice-cold HBSS. Cells were fixed in HBSS containing 4% paraformaldehyde and blocked overnight in HBSS with 10% FBS, 1% BSA, 0.05% Tween 20, and 2 mM EDTA. The coverslips were washed in HBSS with 3% FBS and incubated with patient sera (20 μg IgG/ml) or primary anti-histone antibodies indicated above at a 1:100 dilution. Coverslips were washed and incubated with AF647-conjugated goat anti-human IgG, AF488-conjugated goat anti-rabbit IgG, and Sytox Orange (all from Invitrogen, Carlsbad, CA, USA) for 30 min at 4°C. Coverslips were washed and mounted on glass slides using mounting medium (wash buffer with 50% glycerol) and analyzed with an LSM510 microscope (Zeiss, Jena, Germany).
Linker histone H1 extraction and purification
Perchloric acid extraction of H1 was performed as described earlier (15). Briefly, 5 × 107 neutrophils were resuspended in 0.5 ml of 5% HClO4 and subjected to 3 cycles of freezing, thawing, and vortexing (10 s). The final suspension was centrifuged at 15,000 g for 10 min. The clear supernatants were mixed with an equal volume of ice-cold 66% (v/v) trichloroacetic acid. The linker histones were precipitated for 30 min on ice and collected by centrifugation at 15,000 g for 10 min. The pellets were washed with 0.2% HCl in acetone, then twice with pure acetone, and vacuum dried. The dried pellets were reconstituted in high salt buffer (25 mM HEPES, pH 7.6, with 550 mM NaCl, 12.5 mM MgCl2, and 0.1 mM EDTA).
In vitro deimination of recombinant H1.2
Recombinant human histone H1.2 (Axxora, San Diego, CA, USA), was dissolved in PAD4 reaction buffer (100 mM HEPES, 100 mM NaCl, 10 mM CaCl2, 0.1 mM EDTA, and 2 mM dithiothreitol) to a final concentration of 0.1 mM. The reaction mixture was incubated with recombinant PAD4 at 0.2 μM (27), and deimination was assessed by Western blotting using antibody to chemically modified citrulline residues (Millipore), according to the manufacturer's instructions.
Preparation of protease-digested peptides
Approximately 1.5 μg of acid-extracted or recombinant histone H1.2 was desalted using a 3-kDa-cutoff centrifugal filter device (Millipore) according to the manufacturer's instructions. Protease digestion of H1.2 was performed overnight at 37°C with 50 ng of Lys-C, and the digestion was stopped with the addition of trifluoroacetic acid (TFA; 1% v/v). The solution was dried in a Vacufuge Concentrator (Eppendorf, Westbury, NY, USA), and the dried pellet was reconstituted in 20 μl of 0.1% TFA solution. The Lys-C digested peptides were loaded on a C18 Ziptip column (Millipore), washed with 0.1% TFA, and eluted with 10 μl of a 50% acetonitrile solution. The eluent was near-dried in a vacufuge concentrator, and the residue was reconstituted in 4 μl of a 0.1% formic acid solution. This solution (2 ml) was used for the MS analysis.
Liquid chromatography-mass spectrometry (LC-MS) and database search
The H1.2 peptides were resolved by HPLC with a linear gradient from solvent A (0.1% formic acid solution) to solvent B (0.1% formic acid in 90% methanol solution) within 60 min. Peptide masses were determined with a Q-TOF2 mass spectrometer (Micromass, Manchester, UK) in the positive-ion mode. The nano-electrospray ionization source temperature was set at 90°C and capillary and cone voltages to 3.0 kV and 30 V, respectively. Mass calibrations were done with polyalanine (Michrom Bioresources, Auburn, CA, USA). Double- and triple-charged precursor ions with a count ≥20 were selected for tandem mass spectrometry (MS/MS). MS/MS scans (5 s) were followed by an MS scan (1 s). Masslynx 3.5 software (Waters Corp., Milford, MA, USA) was used to process the chromatographic and mass spectrometric data. To identify proteins, SwissProt (European Molecular Biology Laboratory–European Bioinformatics Institute, Hinxton, UK; http://www.ebi.ac.uk/uniprot/) and U.S. National Center for Biotechnology Information (NCBI; Bethesda, MD, USA; http://www.ncbi.nlm.nih.gov) nonredundant databases were searched with Bioworks 3.2 (Thermo Fisher) and Mascot (web version) software. The precursor ion (MS) and product-ion (MS/MS) mass-error tolerances were both set to 0.1 Da. The modification of arginine was set to 0.98 Da because the monoisotopic mass difference between citrulline (C6H13N3O3) and arginine (C6H14N4O2) is 0.98 Da.
RNA extraction and microarray analysis
Total mRNA was isolated from neutrophils using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) as suggested by the manufacturer. Genomic DNA was eliminated by RNase-free DNase I (Qiagen) digestion during the isolation procedure. Isolated total RNA was analyzed on an Agilent 2100 Bioanalyzer using RNA 6000 Pico Labchip Kit (Agilent Technologies, Santa Clara, CA, USA), and the relative integrity of the sample was routinely >8.0. Reverse transcription into cDNA was performed at 50°C for 1 h followed by 85°C for 5 min with the Transcriptor cDNA Synthesis Kit (Roche, Indianapolis, IN, USA) using random hexamer and anchored-oligo (dT) primers (Roche). Gene-specific RNA was quantified on a Whole-Transcript Human Gene 1.0 ST array (Affymetrix, Santa Clara, CA, USA).
RESULTS
Deimination of core and linker histones and association with NETs
NETosis can be induced in vitro by various stimuli, most of which also induce core histone deimination by PAD4 (28). Notably, compounds that increase intracellular calcium are potent stimulators of PAD4 in NETosis, and we recently reported that a further enhancement of histone deimination is achieved by inhibition of PKCα with chelerythrine (29). To examine the fate of linker histone H1 during NETosis, we stimulated neutrophils with ionophore and chelerythrine and observed NET release by immunofluorescence with antibodies to deiminated histone H3 (Fig. 1A) and deiminated histone H4 (Fig. 1B). These antibodies intensely reacted with released NET chromatin and showed precise colocalization with extracellular DNA. Because there is no available antibody to deiminated H1, we used a mouse anti-H1 monoclonal IgG to test whether linker histone is associated with NET chromatin (Fig. 1C). Indeed, the released NET chromatin reacted with excellent specificity with the anti-H1 IgG. Untreated control neutrophils were relatively inert to binding by any of the antibodies used in this experiment (Fig. 1D), in accord with the observation that NET chromatin is more accessible to antibodies than the compact chromatin of intact nuclei (30).
Figure 1.

NETosis and histone deimination. A–D) NETosis was induced by treating purified neutrophils with calcium ionophore and chelerythrine for 1 h. Cells were fixed and bound by antibodies to deiminated histone H3 (dH3; A), to deiminated histone H4 (dH4; B), and to the linker histone H1 (C). Antibody binding was detected with fluorescence-conjugated secondary antibodies, (displayed in red), and DNA is shown in blue. The overlap between antibody binding and DNA staining thus yields violet. Control cells reacted poorly with antibody to dH3 (D). E, F) The extent of histone deimination in cells incubated in calcium buffer (Ca), ionophore (Io), or ionophore with chelerythrine (I/C) was detected with antibody to dH3 (E) or antibody to dH4 (F). Bands whose migration corresponds to dH3 and dH4 are marked by arrowheads. G) The total cell lysates (Tot. Prot.) from cells treated as in E and F were analyzed by SDS-PAGE, as were acid-extracted (Ac. Ex.) linker histones H1. Bands corresponding to H1 are indicated by a bracket. H) The same fractions as are shown in G were blotted to membrane, chemically modified, and probed with an antibody to modified citrulline. I) Only H1 histones from cells treated with ionophore and chelerythrine reacted with antibody to modified citrullines.
To confirm histone deimination under these experimental conditions, we prepared cell lysates and compared the binding of antibodies to deiminated histone H3 (Fig. 1E) and H4 (Fig. 1F). Western blots revealed the near absence of binding of either antibody to cell lysates from neutrophils incubated in buffer alone. In contrast, H3 and H4 histones from cells treated with ionophore were recognized efficiently by the citrulline-specific antibodies, and more intense binding was observed with histones from cells treated with ionophore and chelerythrine. Next, we assessed the protein composition of total lysates from cells treated to induce NETosis (or controls) and compared them to cell extracts that were enriched for histone H1 (Fig. 1G). The protein profiles were similarly complex in the total cell lysates, but perchloric acid extraction resulted in substantial enrichment of H1. The same protein preparations were also blotted onto membranes, the membranes were incubated with diacetyl monoxime and antipyrine (31), and citrullines that were modified by these reagents were detected with a modified citrulline-specific antibody (Fig. 1H, I). We observed immunoreactive proteins with a mobility of linker histones in total cell lysates, but core histones H3 and H4 were more avid PAD4 substrates (Fig. 1H). Additional proteins, ranging in size from 40 to 60 kDa, were also deiminated in cells treated with ionophore and chelerythrine. Because the acid-enriched H1 preparation from activated neutrophils was more immunoreactive than the unfractionated lysates, we conclude that ionophore and chelerythrine treatment induces H1 deimination in parallel with PAD4 activation and NETosis.
MS/MS analysis of H1 histones from activated neutrophils
Having developed a procedure to purify deiminated H1 histones, we next analyzed H1 arginine residues that are converted to citrulline under conditions leading to NETosis. We used MS to analyze sequences of peptides from H1 histones purified from activated neutrophils. Our approach also allowed us to sample the complement of H1 subtypes expressed in mature neutrophils. We observed peptides that identified unique subtypes as well as peptides that are shared by more than one H1 subtype (Table 1). One peptide that may derive from H1.2, H1.3, or H1.4, ER*SGVSLAALK, was a potential candidate to contain citrulline (R*). To test this peptide for arginine deimination, we performed MS/MS of the (M+2H)2+ precursor ion and observed 3 b-series ions (b2–b10) whose mass was consistent with citrulline. The b2-NH3, b4-H2O, and b10 ions were found at m/z 270.11, 413.21, and 985.60, respectively. The mass errors were 0.02, 0.03, and 0.07 Da, respectively, for an average error of 0.04 Da (Fig. 2A). Evidence of deimination was also obtained for the unique peptide (ER*NGLSLAALK; Fig. 2C) derived from histone H1.5. Masses that corresponded to its b3, b7, b8, b9, and b10 ions closely matched those that were predicted from the replacement of arginine by citrulline.
Table 1.
Arginine-containing peptides detected in H1 extracted from activated neutrophils
| Deiminated peptide | Nonmodified peptide | Corresponding H1 subtype |
|---|---|---|
| ER*SGVSLAALK | ERSGVSLAALK | H1.2–H1.4 |
| ER*NGLSLAALK | ERNGLSLAALK | H1.5 |
| GVGASGFR*LAK | – | H1.0 |
| R*LVTTGVLK | – | H1.0 |
| – | NNSRIK | H1.1–H1.5 |
Asterisks indicate the position of citrulline residues.
Figure 2.
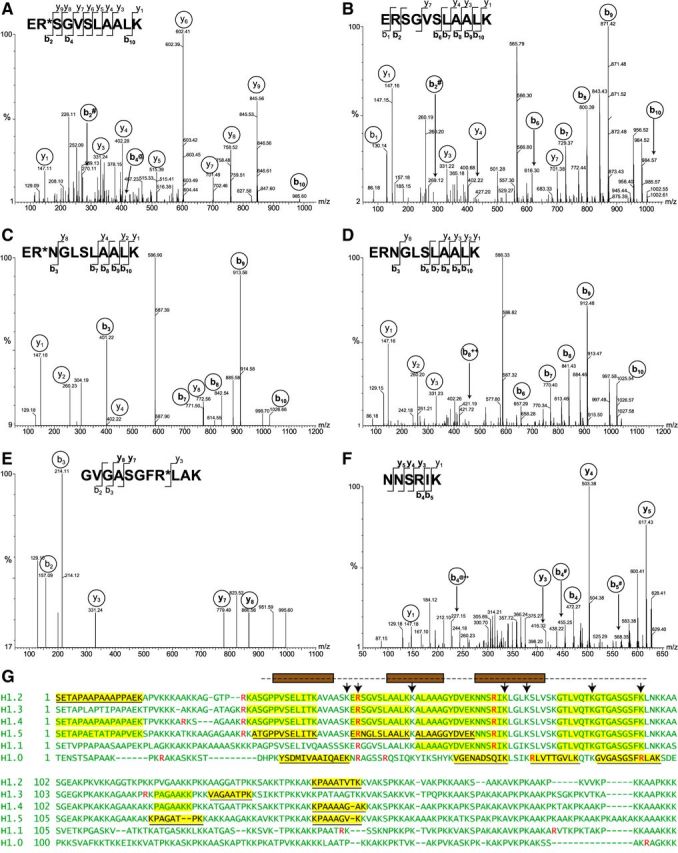
MS/MS analysis of H1 peptides from activated neutrophils A–F) MS/MS spectra of peptides yielding doubly charged precursor ions of H1 histone from Ca2+ ionophore-treated neutrophils. Fragmented ions (b series and y series) are marked on the peptide sequences or spectral peaks. Both peptide ER*SGVSLAALK (A) and ERSGVSLAALK (B) were observed, with a +1 Da shift resulting from deimination of arginine in evidence in b2 and b10 ions. The peptide ERSGVSLAALK is conserved between H1.2, H1.3, and H1.4 subtypes. Both ER*NGLSLAALK (C) and its nondeiminated form ERNGLSLAALK (D) derived from H1.5 were identified. Again, the mass shift of corresponding b ions is consistent with arginine deimination. Arginine-containing peptide GVGASGFR*LAK (E) from H1.0 was also identified, whereas peptide NNSRIK (F), conserved in subtypes H1.1–H1.5, was found only in its nondeiminated form. # indicates -NH3; @ indicates -H2O; ++ indicates a doubly charged ion. G) Summary of results obtained by MS is shown using sequences of 6 human H1 histones. Sequences of somatic linker histone H1 subtypes were aligned to maximize homology. Arginine residues are indicated in red. Peptides highlighted in yellow were identified by mass spectrometry, and H1 subtype-specific peptide sequences are underlined. The globular domain of H1 is indicated by a broken line above the sequences. Brown boxes represent helices that arrange into the H1 winged helix. Residues contributing to DNA binding (50) are indicated with arrows.
Interestingly, the H1 histones that were purified from activated neutrophils also yielded peptides that were identical to the peptides described above, except that they contained arginine residues (Fig. 2B, D). There the b- and y-series ion masses were consistent with the presence of unmodified arginine. Those data indicated that a 2 h treatment of neutrophils with ionophore does not completely convert histone H1 arginine residues to citrullines. Furthermore, we observed the deimination of arginine residues in unique peptides derived from histone H1.0: GVGASGFR*LAK (Fig. 2E) and R*LVTGVLK (Table 1). In contrast, we found no evidence for deimination of the arginine in NNSRIK that is part of the winged helix of histone H1 (Fig. 2F). Our results suggest that multiple arginine residues in the globular domain and the flexible tails of H1 are modified to citrulline residues following neutrophil activation (Fig. 2G). In addition, MS indicated that at least 5 different H1 proteins are present in mature neutrophils. We identified peptides whose sequences unambiguously matched unique sequences of the H1.0, H1.2, H1.3, H1.4, and H1.5 subtypes (Fig. 2G). Overall, protease cleavage and MS provided 30–44% sequence coverage for each of the H1 proteins, although we could not unambiguously assign each peptide to a specific H1 subtype (Fig. 2G). Collectively, the data provide strong evidence that NETosis proceeds in parallel with the deimination of multiple arginine residues in a variety of linker H1 subtypes expressed in neutrophils (Table 1).
Deimination of recombinant H1.2 in vitro
To screen patient sera for binding to deiminated H1, we sought to define a renewable and defined supply of precisely characterized antigen. We selected to use recombinant human H1.2, and we deiminated H1.2 with recombinant PAD4. We made this choice because bacterially expressed H1.2 allows us to sidestep confounding contributions of other eukaryotic PTM. Moreover, H1.2 is the most prototypical H1 subtype in humans, and it contains 3 conserved arginine residues, including the most conserved H1 arginine, arginine 53. To assay the extent of H1.2 deimination in vitro, we subjected the deiminated H1 to Lys C protease cleavage and analyzed the proteolytic fragments by quadrupole time of flight (Q-TOF) MS and MS/MS. One of the peptides was measured at 1130.70 Da, a mass that is close to the predicted mass of the Lys C fragment containing citrulline at position 53 (ER*SGVSLAALK, predicted mass 1130.63 Da). In contrast, a mass of 1129.65 Da was predicted for the arginine-containing peptide. Similarly, a second peptide was measured at 686.40 Da, a mass that closely matched the expected mass of AGGTPR*K, a peptide with a citrulline at position 32.
More definitive support for deimination of arginines 32 and 53 came from MS/MS analysis. We observed multiple peptide fragments (including the b2 and y9 ions; Fig. 3A) that were consistent with deimination in the amino-terminal dipeptide of ER*SGVSLAALK. In addition, we identified citrulline at position 32 of H1.2 with MS/MS of the AGGTPR*K peptide (Fig. 3B). In contrast, the mass of peptide NNSRIK was measured at 730.40, a close approximation of the predicted mass of an arginine-containing peptide (730.41), a result that was consistent with MS/MS (Fig. 3C). Thus, the MS and MS/MS experiments identified citrulline residues at positions 32 and 53 of H1.2, whereas arginine was detected at position 78. These modifications closely matched those of the H1 deimination products that we identified in H1 from stimulated neutrophils. Therefore, our recombinant antigen closely resembled the H1.2 from neutrophils undergoing NETosis.
Figure 3.

MS/MS analysis of in vitro deiminated recombinant H1.2. MS/MS spectra of doubly charged precursor ions of peptides obtained by LysC protease digestion of deiminated histone H1.2. Fragmented ions (b series and y series) are marked on respective peptide sequences and on the MS/MS spectra. A, B) Two of 3 arginines in H1.2, those contained in peptide ER*SGVSLAALK (A) and AGGTPR*K (B), showed mass consistent with deimination to citrulline. C) The peptide NNSRIK showed no evidence of deimination during the in vitro PAD4 reaction. # indicates -NH3; @ indicates -H2O; ++ indicates a doubly charged ion.
Identification of autoantibodies to deiminated H1.2
To characterize autoantibody binding to deiminated H1.2 linker histone, we screened the sera of patients with SLE (n=24), SS (n=20), scleroderma (n=20), RA (n=20), or control individuals (n=20) for IgG reactivity with deiminated and unmodified H1.2 histone. A detailed account of patient and control reactivities will be presented elsewhere. Western blotting, followed by quantitative densitometry (Fig. 4), revealed that 4 of 72 anti-H1.2-positive sera (∼6%) bound deiminated H1.2 with at least 2-fold preference over nondeiminated H1.2, whereas the remaining sera bound comparably or did not bind to either antigen. Our approach likely underestimates the occurrence of autoantibodies to deiminated linker histones because we analyzed only binding to H1.2, yet other H1 subtypes also contain unique citrulline epitopes (Fig. 2G) that may function as autoantibody targets (14). Nevertheless, the fact that autoantibodies to deiminated H1.2 are present in SLE and SS but not in RA and scleroderma indicates that these autoantibodies are relatively disease-specific markers of low clinical incidence. The antibodies detected by our assay are not broadly cross-reactive ACPA, as several sera with ACPA did not preferentially bind to deiminated H1.2.
Figure 4.

Preferential binding of autoimmune sera to deiminated H1.2. Equal amounts of nondeiminated (H) and deiminated (C) H1.2 were blotted on nitrocellulose and probed with sera from SLE and SS patients. Five representative blots from each set are shown. The ratio of binding to deiminated vs. nondeiminated H1.2 (C/H) was calculated, and values are listed below each blot. Two sera each of SLE and SS syndrome showed a >2.0 binding preference for deiminated H1.2.
Identification of histone H1.2 autoepitopes
To learn how antibody preference for deiminated histone H1.2 arises, we asked which H1.2 citrulline forms part of the antibody binding determinant. We addressed the question by using SLE6, SS63, and SS64 sera, the sera with the most pronounced bias toward deiminated H1.2 histone (Fig. 4), and synthetic peptides as inhibitors of antibody binding (Fig. 5A). To compete binding to the deiminated histone H1.2, we incubated the sera with either of two 16-mer peptides that matched H1.2 and centered around citrulline 32 (competitor A) or 53 (competitor B). The preference of SLE6 and SS63 for deiminated H1.2 was maintained after incubation with competitor A, whereas preferential binding was eliminated after incubation with competitor B (Fig. 5B). Thus, the SLE6 and SS63 epitope likely includes citrulline 53, whereas the SS64 epitope could not be determined by this experimental approach.
Figure 5.

Peptide inhibition of SLE6 binding to deiminated H1.2. A) The preferential binding of autoimmune sera to deiminated H1.2 was assessed after inhibition by either of 2 synthetic peptides. Equal amounts of deiminated (Cit H1.2) and nondeiminated (H1.2) linker histone were analyzed by SDS-PAGE (Coomassie) or blotted and probed with SLE6, SS63, and SS64 sera. Panels show results of Western blots performed following a preincubation in the presence of competitor A or B. Competitor B (R53Cit) was more effective at blocking preferential binding of SLE6 and SS63 to deiminated H1.2. B) The peptides were designed to match H1.2 with citrulline residues substituting for arginine 32 or 53.
IgG autoantibodies bind NETs and histone H1-sized protein in neutrophil cell lysates
Having identified sera that preferentially reacted with deiminated recombinant H1.2, we assayed binding to activated neutrophils and neutrophil lysates. To examine the distribution of SLE6-reactive antigens in cells, we activated neutrophils with LPS, a stimulus that induces deimination and chromatin release from neutrophils (4), and analyzed patient IgG binding to LPS-treated and untreated neutrophils by confocal microscopy. Binding of diluted patient sera was analyzed relative to a rabbit antibody to deiminated H3 and the DNA stain sytox orange (Fig. 6A–C). Antibody binding was stronger to neutrophils with decondensed chromatin (Fig. 6B) than to cells that maintained a lobed nucleus (Fig. 6A), as SLE6 IgG antibodies extensively colocalized with deiminated H3 in cells with decondensed intracellular chromatin (Fig. 6B). Colocalization of SLE6 IgG antibodies with deiminated H3 was particularly notable along NETs (Fig. 6C). The intense granular SLE6 IgG antibody staining in the cytoplasm of activated neutrophils suggested that immunoreactive epitopes accumulate outside of the nucleus prior to NET chromatin release (Fig. 6C).
Figure 6.

Neutrophil activation increases SLE6 reactivity. A–C) SLE6 IgG binding (red) to unstimulated or LPS-treated neutrophils was compared to the binding of antideiminated H3 antibody (green) and the DNA binding dye, sytox orange (blue). A) Faint SLE6 binding is observed to the cytoplasm of untreated neutrophils. B, C) SLE6 binding is increased in LPS-treated neutrophils that have lost the integrity of nuclear membrane (B) and show increased reactivity with the antideiminated H3 antibody (C). D) SLE6 IgG binds to NETs that react with the antideiminated H3, but prominent cytoplasmic clusters of SLE6 binding were also observed. The micrographs are representative of SLE6 IgG binding to activated neutrophils. Lysates of unstimulated (U) and Ca2+ ionophore-treated (S) neutrophils were probed with SLE6 serum. Enhanced binding of IgG to a doublet of proteins migrating to a position near the 37kD marker is indicated (arrow). E) Equal amounts of nondeiminated (H) and deiminated (C) H1.2 and BSA were analyzed by Coomassie staining or probed with SLE6 serum. Preferential binding of SLE6 to deiminated H1.2 over nondeiminated H1.2 and no detectable binding to deiminated BSA indicate the contribution of flanking protein sequences to the specificity determinants of SLE6 IgG (B).
Next, we used the SLE6 serum to assess whether other proteins in cell lysates from unstimulated or Ca-ionophore stimulated human neutrophils bind this serum on Western blots. The SLE6 IgG antibodies bound more strongly to a protein whose relative mobility approximated histone H1 in lysates from activated neutrophils, whereas the reactivity of other proteins did not appreciably change (Fig. 6D). SLE6 IgG did not react with proteins with the mobility of core histones and thus appeared quite selective for deiminated H1. We also wished to determine the ability of the SLE6 serum to discriminate between citrulline containing proteins in general, so we deiminated BSA and added it to deiminated H1. On Western blots, SLE6 bound preferentially to deiminated H1.2 with little to no binding to deiminated BSA (Fig. 6E). Collectively, the data support the interpretation that the SLE6 patient IgG antibodies preferentially recognize deiminated histone H1 and bind to decondensed nuclear and NET chromatin.
Neutrophils express multiple H1 mRNAs and proteins
Chromatin structure in neutrophils undergoes dramatic changes and affects the multilobed structure of the nucleus, neutrophil extravasation, and NETosis, yet an analysis of H1 subtypes expressed in neutrophils has not been performed. To survey H1 subtype expression in blood neutrophils, we quantified different H1 mRNA by hybridization to expression microarrays (Fig. 7). H1.4 was the most actively transcribed linker histone, followed by H1.0 and H1.2. In contrast, H1.1, H1.3, and H1.5 mRNA expression was near background levels. Histone H1.2 was expressed at levels similar to the housekeeping gene, HGPRT (Fig. 5A). These data indicate that mature neutrophils express a limited set of linker histones and that the bulk of transcripts derives from H1.4 and, to a lesser extent, from H1.0.
Figure 7.

Analysis of linker histone transcripts from purified neutrophils. Relative mRNA levels of different H1 subtypes were determined from total mRNA extracted from neutrophils. cDNA was prepared by reverse transcription, and expression levels were measured on an Affymetrix Whole-Transcript Human Gene 1.0 ST Array. Relative fluorescence intensities were plotted for each H1 subtype.
DISCUSSION
Here we examine conditions leading to H1 deimination and identify human IgG autoantibodies that react with deiminated H1.2. These data indicate that deiminated histone H1, a presumptive NET component, elicits the production of autoantibodies. Previously we presented evidence that NETs stimulate autoantibody responses in susceptible individuals. In sera from patients with SLE, RA, and Felty's syndrome, we detected autoantibodies that preferentially bound deiminated histones (17). Consistent results were obtained by 2 other groups of researchers. Pratesi et al. (32) identified autoantibodies to deiminated histone H4 in a majority of patients with RA, and Sokolove et al. (33) detected autoreactivity to a deiminated peptide matching the H2A amino terminus in sera of individuals during the preclinical phase of RA. Thus, mounting evidence suggests that NETs participate in the induction of autoimmune responses (34).
Additional observations strengthen the notion that NETs drive autoimmune B-cell responses. Autoantibodies frequently target NET components, including DNA, the NET-associated neutrophil proteases elastase, cathepsin G, and proteinase 3 (35–37), as well as various bactericidal peptides that are stored in granules and copurify with NETs (38, 39). These findings also suggest a scenario whereby infections could contribute to the induction of autoimmunity. NETs are released under inflammatory conditions, as may arise during infections (40–43), and NET chromatin intimately associates with bacteria (9, 44). The microbes may act as adjuvants to promote autoreactivity (45).
Progression of autoimmune disease may benefit from the inappropriate induction of NETosis. Neutrophils from juvenile SLE patients show an enhanced response to anti-RNP immune complexes (46), and RA neutrophils are more likely than control neutrophils to spontaneously release NETs (11). Increased release of NETs may aggravate inflammation by stimulating plasmacytoid dendritic cells to secrete type I interferon (39). This may further enhance the tendency of patient neutrophils to undergo NETosis. Such a vicious cycle may sustain the disease process and be a key factor in rendering autoimmune diseases resistant to treatment.
Development of new therapies for autoimmune disease will depend on a deeper understanding of mechanisms leading to NETosis. PAD4 converts the positively charged arginine to the neutral citrulline in a variety of substrates, including PAD4 itself (47). Thus, it is plausible that histone deimination loosens charge-mediated interactions between histones and DNA and unfolds nuclear chromatin into the extended structure of NETs (48), as shown previously for other H1 PTM (16). Consistent with this role, PAD4 treatment of a nucleosome array decreased its capacity for compaction in the presence of linker histone H5 (5). Our previous studies focused on the role of deiminated core histones in NETosis (4), and others subsequently used deiminated histones as markers of NET chromatin (49, 50). However, it is equally likely that NET chromatin unfolds in response to deimination of linker histones.
To understand how H1 deimination contributes to NET release, it is necessary to know which H1 subtypes are expressed in human neutrophils. We identified no less than 5 H1 proteins in mature, circulating neutrophils (Fig. 2G), even though we found that H1.4 gene transcripts dominate H1 subtype expression in neutrophils (Fig. 7). Peptides matching unique sequences of H1.3 and H1.5 were identified by MS, despite the fact that very little to no mRNA transcripts for these proteins were detected. This suggests that H1.3 and H1.5 are synthesized during neutrophil development but replaced by H1.4 and H1.0 during neutrophil maturation. Changes in H1 subtype expression may affect chromatin conformation, packing, and nuclear morphology.
Any change in the global properties of neutrophil chromatin may expose H1's arginine 53, the residue that, when converted to citrulline, yields the preferred target of SLE6 and SS63 IgG. Arginine 53 is located within the winged helix, the most highly conserved domain of H1, and it is invariant in all of the H1 subtypes present in humans (14). This residue is also conserved in organisms from yeast to higher vertebrates. One reason for the conservation of arginine 53 may be its key role in regulating overall chromatin structure. Chrostophorou et al. (51) discovered that deimination of H1.2 parallels the acquisition of cellular pluripotency. PAD4-mediated deimination of arginine 53 in H1.2 led to the displacement of this linker histone from chromatin with concomitant chromatin decondensation and an increase in nuclear volume. Because H1.2 is depleted from active promoters (52), H1.2 deimination may open access to inactive gene promoters, as a requisite for pluripotency. It will be interesting to determine whether PAD4-mediated histone H1 deimination also regulates gene expression in neutrophils.
However, DNA binding may not be the only function of arginine 53. In histone H1.0, arginine 53 contributes to DNA binding (53), but, in H1.2, replacement of arginine 53 with alanine did not appreciably affect chromatin binding in vivo (54). This difference indicates that H1.0 binds DNA more tightly than H1.2. The role of arginine 53 in DNA binding may be controlled by the overall structure of the winged helix. If arginine 53 does not contribute to DNA binding, it must carry out an alternative function that accounts for its evolutionary conservation. Arginine 53 may contribute to protein interactions with core histones in chromatin (55), interactions with RNA polymerase (56), with HMGB1 (57), or transcription or splicing factors (58, 59). As such, H1 is intimately associated with other well-known nuclear autoantigens.
Our study also suggests that, under certain circumstances, deimination of H1 histones helps to break immune tolerance. Support for this idea is provided by the remarkable success of PAD4 inhibitors in the treatment of experimental autoimmunity. Chemical inhibitors of PAD4 have been used in mice with collagen-induced arthritis (60), in mice with induced colitis (34), and in a spontaneous model of murine lupus (61). In each of these, PAD4 inhibition led to clear and quantifiable improvement of disease symptoms. New inhibitors of PAD4 were used to prevent autoimmune neurodegeneration (62), and thrombosis severity was diminished in the absence of PAD4 (52). There are numerous ways in which PAD4 could contribute to autoimmune disease because PAD4 affects transcription (63), cytokine activity (64), and PTM of the extracellular matrix (65). However, one direct way in which PAD4 may contribute to autoimmunity is by covalent modification of nuclear autoantigens, which could pose a challenge to immune tolerance.
Acknowledgments
The authors gratefully acknowledge Dr. Jean-François Kleinmann (Strasbourg University Hospitals, Strasbourg, France), for providing patient sera used in these studies; the insightful and critical comments of Dr. Marc Monestier (Temple University, Philadelphia, PA, USA); and the technical expertise of Tim Higgins (Senior Illustrator, University of Tennessee Health Science Center, Memphis, TN, USA).
This work was supported by research funding provided by the Lupus Research Institute of New York and the Dana Foundation Program in Human Immunology. Research was supported in part by the French Centre National de la Recherche Scientifique, the Laboratory of Excellence Medalis (ANR-10-LABX-0034), Initiative of Excellence (IdEx), Strasbourg University, and ImmuPharma France.
Footnotes
- LPS
- lipopolysaccharide
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- NET
- neutrophil extracellular trap
- PAD
- peptidylarginine deiminase
- PTM
- post-translational modification
- RA
- rheumatoid arthritis
- SLE
- systemic lupus erythematosus
- SS
- Sjögren's syndrome
- TBS
- Tris-buffered saline
REFERENCES
- 1. Muller S., Dieker J., Tincani A., Meroni P. L. (2008) Pathogenic anti-nucleosome antibodies. Lupus 17, 431–436 [DOI] [PubMed] [Google Scholar]
- 2. Fauchais A. L., Martel C., Gondran G., Lambert M., Launay D., Jauberteau M. O., Hachulla E., Vidal E., Hatron P. Y. (2010) Immunological profile in primary Sjögren syndrome: clinical significance, prognosis and long-term evolution to other auto-immune disease. Autoimm. Rev. 9, 595–599 [DOI] [PubMed] [Google Scholar]
- 3. El-Azhary R. A., Aponte C. C., Nelson A. M., Weaver A. L., Homburger H. A. (2006) Antihistone antibodies in linear scleroderma variants. Int. J. Dermatol. 45, 1296–1299 [DOI] [PubMed] [Google Scholar]
- 4. Neeli I., Khan S. N., Radic M. (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180, 1895–1902 [DOI] [PubMed] [Google Scholar]
- 5. Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S. A., Allis C. D., Coonrod S. A. (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li P., Li M., Lindberg M. R., Kennett M. J., Xiong N., Wang Y. (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hemmers S., Teijaro J. R., Arandjelovic S., Mowen K. A. (2011) PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 6, e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Urban C. F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P. R., Zychlinsky A. (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5, e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., Weinrauch Y., Zychlinsky A. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 [DOI] [PubMed] [Google Scholar]
- 10. Daigo K., Hamakubo T. (2012) Host-protective effect of circulating pentraxin 3 (PTX3) and complex formation with neutrophil extracellular traps. Front. Immunol. 3, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khandpur R., Carmona-Rivera C., Vivekanandan-Giri A., Gizinski A., Yalavarthi S., Knight J. S., Friday S., Li S., Patel R. M., Subramanian V., Thompson P., Chen P., Fox D. A., Pennathur S., Kaplan M. J. (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 5, 178ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Syed S. H., Goutte-Gattat D., Becker N., Meyer S., Shukla M. S., Hayes J. J., Everaers R., Angelov D., Bednar J., Dimitrov S. (2010) Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl. Acad. Sci. U.S.A. 107, 9620–9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phair R. D., Scaffidi P., Elbi C., Vecerova J., Dey A., Ozato K., Brown D. T., Hager G., Bustin M., Misteli T. (2004) Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24, 6393–6402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Happel N., Doenecke D. (2009) Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 431, 1–12 [DOI] [PubMed] [Google Scholar]
- 15. Wisniewski J. R., Zougman A., Kruger S., Mann M. (2007) Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteomics 6, 72–87 [DOI] [PubMed] [Google Scholar]
- 16. Muller S., Mazen A., Martinage A., Van Regenmortel M. H. (1984) Use of histone antibodies for studying chromatin topography and the phosphorylation of chromatin subunits. EMBO J. 3, 2431–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dwivedi N., Upadhyay J., Neeli I., Khan S., Pattanaik D., Myers L., Kirou K. A., Hellmich B., Knuckley B., Thompson P. R., Crow M. K., Mikuls T. R., Csernok E., Radic M. (2012) Felty's syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum. 64, 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radic M., Muller S. (2013) Epigenetics of autoantigens: new opportunities for therapy of autoimmune diseases. Genetics Epigenetics 5, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dwivedi N., Radic M. (2014) Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann. Rheum. Dis. 73, 483–491 [DOI] [PubMed] [Google Scholar]
- 20. Schett G., Smolen J., Zimmermann C., Hiesberger H., Hoefler E., Fournel S., Muller S., Rubin R. L., Steiner G. (2002) The autoimmune response to chromatin antigens in systemic lupus erythematosus: autoantibodies against histone H1 are a highly specific marker for SLE associated with increased disease activity. Lupus 11, 704–715 [DOI] [PubMed] [Google Scholar]
- 21. Sun X. Y., Shi J., Han L., Su Y., Li Z. G. (2008) Anti-histones antibodies in systemic lupus erythematosus: prevalence and frequency in neuropsychiatric lupus. J. Clin. Lab. Anal. 22, 271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stemmer C., Briand J. P., Muller S. (1994) Mapping of linear epitopes of human histone H1 recognized by rabbit anti-H1/H5 antisera and antibodies from autoimmune patients. Mol. Immunol. 31, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 23. Tan E. M., Cohen A. S., Fries J. F., Masi A. T., McShane D. J., Rothfield N. F., Schaller J. G., Talal N., Winchester R. J. (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 25, 1271–1277 [DOI] [PubMed] [Google Scholar]
- 24. Vitali C., Bombardieri S., Jonsson R., Moutsopoulos H. M., Alexander E. L., Carsons S. E., Daniels T. E., Fox P. C., Fox R. I., Kassan S. S., Pillemer S. R., Talal N., Weisman M. H. (2002) Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 61, 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subcommittee (1980) Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 23, 581–590 [DOI] [PubMed] [Google Scholar]
- 26. Aletaha D., Neogi T., Silman A. J., Funovits J., Felson D. T., Bingham C. O., 3rd, Birnbaum N. S., Burmester G. R., Bykerk V. P., Cohen M. D., Combe B., Costenbader K. H., Dougados M., Emery P., Ferraccioli G., Hazes J. M., Hobbs K., Huizinga T. W., Kavanaugh A., Kay J., Kvien T. K., Laing T., Mease P., Menard H. A., Moreland L. W., Naden R. L., Pincus T., Smolen J. S., Stanislawska-Biernat E., Symmons D., Tak P. P., Upchurch K. S., Vencovsky J., Wolfe F., Hawker G. (2010) 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62, 2569–2581 [DOI] [PubMed] [Google Scholar]
- 27. Kearney P. L., Bhatia M., Jones N. G., Yuan L., Glascock M. C., Catchings K. L., Yamada M., Thompson P. R. (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44, 10570–10582 [DOI] [PubMed] [Google Scholar]
- 28. Rohrbach A. S., Slade D. J., Thompson P. R., Mowen K. A. (2012) Activation of PAD4 in NET formation. Front. Immunol. 3, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neeli I., Radic M. (2013) Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brinkmann V., Goosmann C., Kuhn L. I., Zychlinsky A. (2012) Automatic quantification of in vitro NET formation. Front. Immunol. 3, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Senshu T., Akiyama K., Kan S., Asaga H., Ishigami A., Manabe M. (1995) Detection of deiminated proteins in rat skin: probing with a monospecific antibody after modification of citrulline residues. J. Invest. Dermatol. 105, 163–169 [DOI] [PubMed] [Google Scholar]
- 32. Pratesi F., Dioni I., Tommasi C., Alcaro M. C., Paolini I., Barbetti F., Boscaro F., Panza F., Puxeddu I., Rovero P., Migliorini P. (2013) Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. [E-pub ahead of print] Ann. Rheum. Dis. 10.1136/annrheumdis-2012-202765 [DOI] [PubMed] [Google Scholar]
- 33. Sokolove J., Bromberg R., Deane K. D., Lahey L. J., Derber L. A., Chandra P. E., Edison J. D., Gilliland W. R., Tibshirani R. J., Norris J. M., Holers V. M., Robinson W. H. (2012) Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE 7, e35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Radic M., Marion T. N. (2013) Neutrophil extracellular chromatin traps connect innate immune response to autoimmunity. Semin. Immunopathol. 35, 465–480 [DOI] [PubMed] [Google Scholar]
- 35. Manolova I., Dancheva M., Halacheva K. (2001) Antineutrophil cytoplasmic antibodies in patients with systemic lupus erythematosus: prevalence, antigen specificity, and clinical associations. Rheumatol. Int. 20, 197–204 [DOI] [PubMed] [Google Scholar]
- 36. Pradhan V. D., Badakere S. S., Bichile L. S., Almeida A. F. (2004) Anti-neutrophil cytoplasmic antibodies (ANCA) in systemic lupus erythematosus: prevalence, clinical associations and correlation with other autoantibodies. J. Assoc. Physicians India 52, 533–537 [PubMed] [Google Scholar]
- 37. Tamiya H., Tani K., Miyata J., Sato K., Urata T., Lkhagvaa B., Otsuka S., Shigekiyo S., Sone S. (2006) Defensins- and cathepsin G-ANCA in systemic lupus erythematosus. Rheumatol. Int. 27, 147–152 [DOI] [PubMed] [Google Scholar]
- 38. Froy O., Sthoeger Z. M. (2009) Defensins in systemic lupus erythematosus. Ann. N.Y. Acad. Sci. 1173, 365–369 [DOI] [PubMed] [Google Scholar]
- 39. Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., Bassett R., Amuro H., Fukuhara S., Ito T., Liu Y. J., Gilliet M. (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kessenbrock K., Krumbholz M., Schonermarck U., Back W., Gross W. L., Werb Z., Grone H. J., Brinkmann V., Jenne D. E. (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuchs T. A., Brill A., Duerschmied D., Schatzberg D., Monestier M., Myers D. D., Jr., Wrobleski S. K., Wakefield T. W., Hartwig J. H., Wagner D. D. (2010) Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clark S. R., Ma A. C., Tavener S. A., McDonald B., Goodarzi Z., Kelly M. M., Patel K. D., Chakrabarti S., McAvoy E., Sinclair G. D., Keys E. M., Allen-Vercoe E., Devinney R., Doig C. J., Green F. H., Kubes P. (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 [DOI] [PubMed] [Google Scholar]
- 43. Berends E. T., Horswill A. R., Haste N. M., Monestier M., Nizet V., von Kockritz-Blickwede M. (2010) Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2, 576–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Von Kockritz-Blickwede M., Goldmann O., Thulin P., Heinemann K., Norrby-Teglund A., Rohde M., Medina E. (2008) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111, 3070–3080 [DOI] [PubMed] [Google Scholar]
- 45. Brinkmann V., Zychlinsky A. (2007) Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 5, 577–582 [DOI] [PubMed] [Google Scholar]
- 46. Garcia-Romo G. S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R. L., Barrat F. J., Banchereau J., Pascual V. (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andrade F., Darrah E., Gucek M., Cole R. N., Rosen A., Zhu X. (2010) Autocitrullination of human peptidyl arginine deiminase type 4 regulates protein citrullination during cell activation. Arthritis Rheum. 62, 1630–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leshner M., Wang S., Lewis C., Zheng H., Chen X. A., Santy L., Wang Y. (2012) PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 3, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Munks M. W., McKee A. S., Macleod M. K., Powell R. L., Degen J. L., Reisdorph N. A., Kappler J. W., Marrack P. (2010) Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood 116, 5191–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Y., Liu B., Fukudome E. Y., Lu J., Chong W., Jin G., Liu Z., Velmahos G. C., Demoya M., King D. R., Alam H. B. (2011) Identification of citrullinated histone H3 as a potential serum protein biomarker in a lethal model of lipopolysaccharide-induced shock. Surgery 150, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Christophorou M. A., Castelo-Branco G., Halley-Stott R. P., Oliveira C. S., Loos R., Radzisheuskaya A., Mowen K. A., Bertone P., Silva J. C., Zernicka-Goetz M., Nielsen M. L., Gurdon J. B., Kouzarides T. (2014) Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Millan-Arino L., Islam A. B., Izquierdo-Bouldstridge A., Mayor R., Terme J. M., Luque N., Sancho M., Lopez-Bigas N., Jordan A. (2014) Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2.[E-pub ahead of print] Nucleic Acids Res. 10.1093/nar/gku079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown D. T., Izard T., Misteli T. (2006) Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol. 13, 250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. George E. M., Izard T., Anderson S. D., Brown D. T. (2010) Nucleosome interaction surface of linker histone H1c is distinct from that of H1(0). J. Biol. Chem. 285, 20891–20896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vogler C., Huber C., Waldmann T., Ettig R., Braun L., Izzo A., Daujat S., Chassignet I., Lopez-Contreras A. J., Fernandez-Capetillo O., Dundr M., Rippe K., Langst G., Schneider R. (2010) Histone H2A C-terminus regulates chromatin dynamics, remodeling, and histone H1 binding. PLoS Genet. 6, e1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng Y., John S., Pesavento J. J., Schultz-Norton J. R., Schiltz R. L., Baek S., Nardulli A. M., Hager G. L., Kelleher N. L., Mizzen C. A. (2010) Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol. 189, 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Polyanichko A., Wieser H. (2005) Fourier transform infrared/vibrational circular dichroism spectroscopy as an informative tool for the investigation of large supramolecular complexes of biological macromolecules. Biopolymers 78, 329–339 [DOI] [PubMed] [Google Scholar]
- 58. Kim K., Choi J., Heo K., Kim H., Levens D., Kohno K., Johnson E. M., Brock H. W., An W. (2008) Isolation and characterization of a novel H1.2 complex that acts as a repressor of p53-mediated transcription. J. Biol. Chem. 283, 9113–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mackey-Cushman S. L., Gao J., Holmes D. A., Nunoya J. I., Wang R., Unutmaz D., Su L. (2011) FoxP3 interacts with linker histone H1.5 to modulate gene expression and program Treg cell activity. Genes Immun. 12, 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Willis V. C., Gizinski A. M., Banda N. K., Causey C. P., Knuckley B., Cordova K. N., Luo Y., Levitt B., Glogowska M., Chandra P., Kulik L., Robinson W. H., Arend W. P., Thompson P. R., Holers V. M. (2011) N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 186, 4396–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Knight J. S., Zhao W., Luo W., Subramanian V., O'Dell A. A., Yalavarthi S., Hodgin J. B., Eitzman D. T., Thompson P. R., Kaplan M. J. (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Invest. 123, 2981–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wei L., Wasilewski E., Chakka S. K., Bello A. M., Moscarello M. A., Kotra L. P. (2013) Novel inhibitors of protein arginine deiminase with potential activity in multiple sclerosis animal model. J. Med. Chem. 56, 1715–1722 [DOI] [PubMed] [Google Scholar]
- 63. Zhang X., Gamble M. J., Stadler S., Cherrington B. D., Causey C. P., Thompson P. R., Roberson M. S., Kraus W. L., Coonrod S. A. (2011) Genome-wide analysis reveals PADI4 cooperates with Elk-1 to activate c-Fos expression in breast cancer cells. PLoS Genet. 7, e1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Proost P., Loos T., Mortier A., Schutyser E., Gouwy M., Noppen S., Dillen C., Ronsse I., Conings R., Struyf S., Opdenakker G., Maudgal P. C., Van Damme J. (2008) Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J. Exp. Med. 205, 2085–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vossenaar E. R., Nijenhuis S., Helsen M. M., van der Heijden A., Senshu T., van den Berg W. B., van Venrooij W. J., Joosten L. A. (2003) Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 48, 2489–2500 [DOI] [PubMed] [Google Scholar]