


Abstract
Analysis of RNA-seq data often detects numerous ‘non-co-linear’ (NCL) transcripts, which comprised sequence segments that are topologically inconsistent with their corresponding DNA sequences in the reference genome. However, detection of NCL transcripts involves two major challenges: removal of false positives arising from alignment artifacts and discrimination between different types of NCL transcripts (trans-spliced, circular or fusion transcripts). Here, we developed a new NCL-transcript-detecting method (‘NCLscan’), which utilized a stepwise alignment strategy to almost completely eliminate false calls (>98% precision) without sacrificing true positives, enabling NCLscan outperform 18 other publicly-available tools (including fusion- and circular-RNA-detecting tools) in terms of sensitivity and precision, regardless of the generation strategy of simulated dataset, type of intragenic or intergenic NCL event, read depth of coverage, read length or expression level of NCL transcript. With the high accuracy, NCLscan was applied to distinguishing between trans-spliced, circular and fusion transcripts on the basis of poly(A)- and nonpoly(A)-selected RNA-seq data. We showed that circular RNAs were expressed more ubiquitously, more abundantly and less cell type-specifically than trans-spliced and fusion transcripts. Our study thus describes a robust pipeline for the discovery of NCL transcripts, and sheds light on the fundamental biology of these non-canonical RNA events in human transcriptome.
INTRODUCTION
Technological advances in transcriptome sequencing have enabled biologists to better understand transcriptomes in a global manner. Comparative transcriptomics through the lens of high-throughput RNA sequencing (RNA-seq) has discovered a considerable number of ‘non-co-linear’ (NCL) transcripts, which are composed of sequence segments that are topologically inconsistent with the corresponding DNA sequences in the reference genome. An NCL transcript may consist of sequence segments either from a single gene but ordered inconsistently with the reference genome (i.e., intragenic NCL transcripts) or from two or more non-adjacent genes with distant regions of DNA (i.e., intergenic NCL transcripts), by means of genetic rearrangements or post-transcriptional events. Genetic rearrangement events may generate fusion transcripts/genes at the DNA level, while post-transcriptionally NCL transcripts (PtNCL transcripts) are produced during post-transcriptional RNA processing (e.g., trans-splicing or cis-backsplicing). Fusion and trans-spliced transcripts are usually called ‘chimeric transcripts’ or ‘chimeras’ because both of them comprise of sequence segments from different genes or precursor mRNAs (pre-mRNAs) (1). Figure 1 summarizes the categorization of NCL transcripts in transcriptome.
Figure 1.

Categorization of NCL transcripts.
Fusion genes have been reported to be associated with malignant hematological disorders and sarcomas (2–5). The most prominent example is the BCR-ABL1 fusion gene, which is a key factor of adult acute lymphoblastic leukemia cases and an effective biomarker for chronic myeloid leukemia (6–9). Other examples, such as ETV6-NTRK3 in breast carcinoma (10), BCAS4-BCAS3 in breast cancer (11), TMPRSS2-ERG/ETS in prostate cancer (12,13), EML4-ALK in lung cancer (14), MYB-NFIB in head and neck tumors (15) and VTI1A-TCF7L2 in colorectal cancer (16), emphasize the critical importance of fusion genes in cancer detection and diagnosis.
PtNCL transcripts, as mentioned above, can be generated through trans-splicing or cis-backsplicing. Trans-splicing can take place between separate pre-mRNAs either of a single gene (i.e., intragenic trans-splicing) or among different genes (i.e., intergenic trans-splicing) (17,18). Two prominent examples of intergenic trans-spliced RNAs are JAZF1-SUZ12 and SLC45A3-ELK4, which have been reported to be associated with anti-apoptotic function (1,19,20) and prostate cancer (1,21), respectively. In addition, tsRMST, which is produced via intragenic trans-splicing, plays a role in pluripotency maintenance of human embryonic stem cells, as evidenced by our recent discovery (22). On the other hand, cis-backsplicing occurs within a single pre-mRNA, leading to the formation of circular RNA (circRNA) (23,24). CircRNAs have been observed in diverse species (25–28), some of them are evolutionarily conserved between species (26,28–32). CDR1as/ciRS-7 and circRNA of Sry, two representative examples of circRNA in mammals, were demonstrated to function as microRNA sponges (29,33). In addition, circRNAs may play regulatory roles during aging of the central nervous system (31) or cell proliferation (34), and may store, sort or localize RNA-binding proteins (29,35). These findings suggest that circular RNA is an ancient, essential and fine-tuned member in the roster of functional transcripts.
Computational strategies for the identification of NCL transcripts have been proposed to facilitate RNA-seq-based studies (36–42) and have enabled the discovery of thousands of NCL transcript candidates in diverse species (43–53). However, false positives arising from sequencing or alignment errors appear to be unavoidable (48,54,55). Discrepancies among the NCL transcript candidates identified by different strategies imply that a large proportion of candidates may be spurious (40,56–58). Similarities among paralogous genes or repetitive sequences often lead to ambiguities during short-read mapping, which are often misinterpreted as NCL events (59). It remains a major challenge to effectively eliminate such false calls and detect genuine ones without losing sensitivity. Also, most previous studies focus on identifying either intergenic or intragenic NCL events. There thus remains a need for a robust pipeline capable of identifying both intergenic and intragenic NCL transcripts with high sensitivity and precision from RNA-seq data.
To address these issues, we developed a new method, NCLscan, which achieves the goal through a series of knowledge-based processes and integrates different mapping algorithms to enable the stepwise elimination of spurious events without losing sensitivity. NCLscan was found to be superior to 18 other currently available tools in terms of sensitivity and precision, when used to detect intragenic or intergenic NCL events in several benchmark datasets, including real and simulated datasets with different strategy choices for generating simulated dataset, depths of coverage, read lengths and expression levels of NCL transcripts. With the high accuracy, NCLscan was applied to distinguishing between different types of NCL transcripts (i.e., trans-spliced, circular or fusion transcripts) and then investigating these three types of NCL transcripts in terms of the prevalence, expression level and expression breadth by comparing the composition and populations of NCL transcripts between RNA samples with different treatments (i.e., poly(A)- and nonpoly(A)-selected RNA-seq data) from diverse human cell types. This comparative analysis revealed some endogenous features of NCL transcripts, which allowed us to distinguish between circular RNAs, trans-splicing events and fusion transcripts. NCLscan promises to facilitate the comprehensive characterization of various types of NCL transcripts on a transcriptome-wide scale.
MATERIALS AND METHODS
Data retrieval and availability
The human genomic sequences (hg19/GRCh37 assembly) and annotated transcripts were downloaded from the GENCODE project (version 19) at http://www.gencodegenes.org/releases/19.html. Datasets A and C (see Table 1) were downloaded from the website of the FusionMap group at http://www.arrayserver.com/wiki/index.php?title=FusionMap (60) and from the NCBI Sequence Read Archive at http://www.ncbi.nlm.nih.gov/sra under the accession number SRP003186 (61), respectively. Dataset B (negative dataset; Table 1) was provided from the author of (57). The links and the used parameters of the tools examined in this study were listed in Supplemental Table S1. The simulated datasets generated in this study are available from our ftp site at ftp://treeslab1.genomics.sinica.edu.tw/NCLscan. The poly(A)-/nonpoly(A)-selected RNA-seq (see Table 2) and SOLiD-based DNA-PET (1, 10 and 20 kb libraries) data were downloaded from the ENCODE consortium (62) at https://www.encodeproject.org/. NCLscan was implemented under the Bio-Linux (Ubuntu 14.04) or Mac operating systems on a 64-bit machine with ≥10 GB RAM. The NCLscan program, document and test dataset are publicly accessible from GitHub at https://github.com/TreesLab/NCLscan or our FTP site at ftp://treeslab1.genomics.sinica.edu.tw/NCLscan.
Table 1. RNA-seq datasets for evaluating the accuracy of fusion-identification tools.
| Dataset | Dataset type | Read type | Number of reads | Reference | |
|---|---|---|---|---|---|
| A | Simulated data | 75-bp paired-end | 57 209 | (60) | |
| B | Simulated data | 100-bp paired-end | 70 000 000 | (57,58) | |
| Ca | Real data | 50-bp paired-end | 8 412 431 | (MCF-7) | (61) |
| 6 800 166 | (KPL-4) | ||||
| 13 515 132 | (BT-474(1)) | ||||
| 7 915 382 | (BT-474(2)) | ||||
| 9 048 352 | (SK-BR-3(1)) | ||||
| 9 097 152 | (SK-BR-3(2)) | ||||
aThe dataset includes RNA-seq reads from four cell lines: MCF-7, KPL-4, BT-474 and SK-BR-3.
Table 2. Poly(A)- and nonpoly(A)-selected RNA-seq data used in this study.
| Sample | Description | Biosample type | Sex (life stage) | Poly(A)-selected dataa | Nonpoly(A)-selected dataa |
|---|---|---|---|---|---|
| H1 | Human embryonic stem cell | stem cell | Male (embryonic) | SRR307911, | SRR307923, |
| SRR307912 | SRR307924 | ||||
| K562 | Chronic myelogenous leukemia | immortalized cell | Female (adult) | SRR315336, | SRR307930, |
| SRR315337 | SRR307931 | ||||
| BJ | Skin fibroblast | immortalized cell | Male (newborn) | SRR307903, SRR307904 | SRR317065 |
| HepG2 | hepatocellular carcinoma | immortalized cell | Male (child) | SRR307926, | SRR307913, |
| SRR307927 | SRR307914 | ||||
| HUVEC | Umbilical vein endothelial cell | primary cell | Male (newborn) | SRR307905, SRR307906 | SRR317067 |
| NHEK | Epidermal keratinocytes | primary cell | Female (unknown) | SRR315327 | SRR315321, SRR315322 |
aThe RNA-seq data were downloaded from the ENCODE project (62).
The NCLscan pipeline
As shown in Figure 2A, NCLscan first aligned RNA-seq reads against the human reference genome (GRCh37) and annotated transcripts (GENCODE version 19) using BWA (63) with default parameters. The unmapped reads were aligned against the genome and transcripts using Novoalign (Novocraft Technologies) with the parameters: -r A 1 -n 30. Both ends of the unmapped paired-end reads were then joined end to end, to form concatenated sequences. These concatenated sequences were aligned against the reference genome using BLAT (64) with default parameters. The concatenated sequences with two split sequence segments that were linearly inconsistent with the reference genome were retained. The pipeline did not consider read-through gene fusions. To this end, concatenated sequences in which the distance between two split segments on the same strand of the same chromosome was <2 Mbp were discarded. In addition, a concatenated sequence satisfied one of the two criteria was not considered: (i) the concatenated sequence mapped to an unplaced (undetermined) contig; and (ii) the donor (or acceptor) side of the concatenated sequence mapped to more than one positions with similar BLAT mapping scores (the score difference between matches <5). Subsequently, all possible ‘putative NCL references’ with putative NCL junction sites were made according to the information of BLAT alignment output and GENCODE annotation. In a retained sequence, the NCL junction site had to locate at the splicing junction of an annotated exon and the corresponding putative NCL reference had to be no longer than the read length with insert sizes (e.g., 1000 bp in this study; Figure 2B and C). Reads that could not be mapped to the genome or annotated transcripts were aligned against these putative NCL references using Novoalign with the following parameters: -r A 1 -g 99 -x 99. There are two types of read that match to a putative NCL reference: encompassing reads and junction reads. A putative NCL reference was retained if it satisfied all of the following criteria: (i) all reads that match to the putative NCL reference must not have an alternative co-linear explanation within a single gene or between two close genes; (ii) at least one junction read supports the putative NCL junction of the putative NCL reference, which must span the NCL junction boundary by ≥10 bp on both sides of the junction site; and (iii) the collection of supporting reads (including encompassing and junction reads) must span the NCL junction boundary by ≥50 bp (default value; the span range is a user-assignable parameter) on both sides of the junction site (Figure 2D). Of note, the default value was set because the read lengths of most currently-available RNA-seq reads were >50 bp. Finally, since different BLAT parameters may generate different alignment results, the retained NCL references were aligned against the reference genome using BLAT with a different set of parameters (-titleSize = 9 -stepSize = 9 -repMatch = 32768). Only the NCL transcript candidates that did not contain alternative co-linear explanations were reported in the final output.
Figure 2.
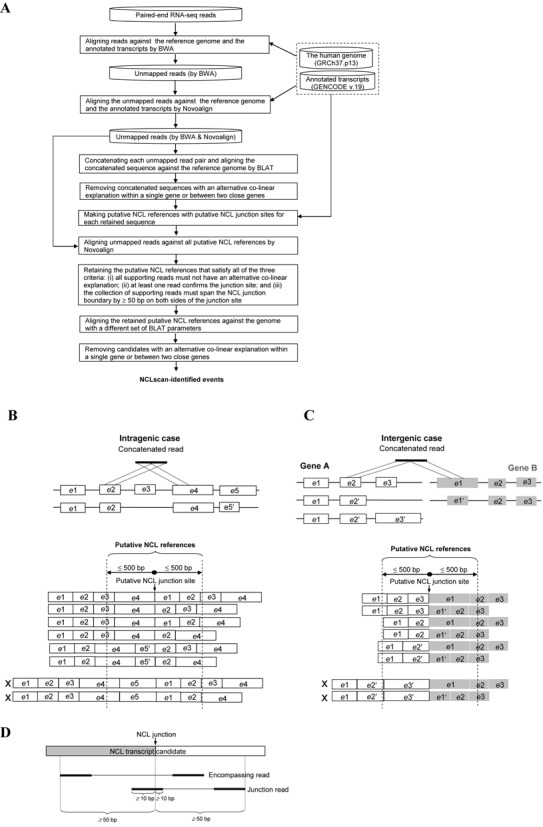
Identification of NCL transcripts. (A) Flowchart depicting the NCLscan pipeline. (B and C) Schematic illustrations of possible ‘putative NCL references’ with putative NCL junction sites (based on BLAT alignment output and GENCODE annotation): (B) intragenic case and (C) intergenic case. The characters ‘e’ and ‘X’ represent ‘exon’ and the putative NCL reference that is not considered, respectively. (D) Schematic illustration of a retained putative NCL reference, which satisfies all of the criteria listed in (A).
Generation of simulated datasets
Simulated NCL transcripts were selected from the annotated genes (GENCODE version 19). The genes would be considered to be the source of simulated NCL transcripts only when they satisfied both of the following criteria: (i) they must not be in the blacklist provided by FusionMap (e.g., pseudogenes, mitochondrial or ribosomal genes) (60); (ii) for intergenic NCL events, the gene pairs must not belong to the same gene families; and (iii) the distance between paired genes on the same strand of the same chromosome must not be <2 Mbp. The junction site in each simulated NCL transcript was random and located at the boundary of an annotated exon. In addition, the length of simulated NCL transcripts should be ≥500 bp and the upstream and downstream sequences should be longer than 100 bp (59). After the simulation process, we obtained 100 intragenic NCL transcripts and 100 intergenic NCL transcripts. The co-linear transcripts were obtained from the annotated protein-coding genes which met two criteria: (i) possessed a status of ‘known’; and (ii) ≥300 bp in length. Based on the co-linear transcripts and the 200 simulated intragenic/intergenic NCL transcripts, we used Mason v.0.1.2 (65) to generate paired-end RNA-seq reads (INS = 170 bp, SD = 20) with different depths of coverage (5-, 10-, 20- and 50-fold) and different read lengths (2 × 50, 2 × 100 and 2 × 150 bp). In addition, to evaluate the performance and accuracy of NCL transcript identification tools under different expression levels, we also generated paired-end RNA-seq reads from the 200 intragenic/intergenic NCL transcripts with 5-, 10-, 20-, 50-, 100-, 150- and 200-fold expression levels, and then mixed these simulated reads with a RNA-seq dataset (used as background data) generated from the co-linear transcripts. Of note, the Mason parameters were the same as those used in the Bowtie2 paper (66).
Accuracy measurement of NCL-transcript identification tools
Sensitivity (Sn), precision (Sp) and F1 values are defined as follows:
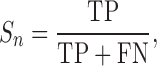 |
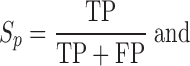 |
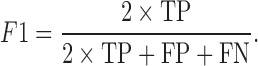 |
TP (true positive), FP (false positive) and FN (false negative) represent the number of correctly identified NCL events, the number of incorrectly identified NCL events and the number of missing NCL events, respectively. Sn, Sp, and F1 values all range from 0 to 1; higher value indicates higher level of accuracy. In consideration of mapping criteria of each tool, for each simulated or validated fusion transcript, a true positive hit was recorded when the distance between the correct NCL junction site and junction site identified by the tested tool was <10 bp (59).
Detection of genomic structure variations
To examine whether certain identified circRNAs were potentially derived from SVs, we used SVDetect (67), which can identify SVs from paired-end/mate-pair NGS data generated by the SOLiD platform, to detect SVs based on the K562 DNA-PET data from the ENCODE project. If an identified circRNA overlapped any breakpoint regions ±1000 bp of a detected SV, such an intragenic event was regarded as a potential SV-derived NCL event. Of the 8915 circRNA candidates detected in K562 cells, 594 were identified as potential SV-derived NCL events (Supplemental Table S2).
RESULTS
Identification of NCL transcripts
NCLscan identifies NCL transcripts from paired-end RNA-seq data. The overall schematic of NCLscan is depicted in Figure 2A. Firstly, co-linearly matched reads are eliminated by mapping the RNA-seq reads against the reference genome and well-known transcripts (i.e., annotated coding and non-coding transcripts in GENCODE) using a stepwise alignment strategy, which integrates two read-mapping algorithms (‘Materials and Methods’ section; Figure 2A). Next, the two ends of each unmapped read are concatenated to generate a continuous sequence. Each concatenated sequence is then aligned against the reference genome using BLAT (64). Only concatenated sequences that contain NCL segments are retained after this step (‘Materials and Methods’ section). For each retained sequence, we make ‘putative NCL references’ with putative NCL junction sites (Figure 2B and C) on the basis of the corresponding BLAT alignment result and GENCODE annotation. Of note, we only consider candidates in which splice junctions agree to well-known junction sites, because such candidates are more reliable than those with junction sites not matching exon boundaries (22,32,43,52). All possible combinations of transcript isoforms are considered while making the putative NCL references. To avoid the putative NCL references inferred from reads with an abnormal inner size, we limit the putative NCL references to 1000 bp in length (Figure 2B and C). Subsequently, reads that cannot be mapped to the genome or transcriptome (i.e., unmapped reads in Figure 2A) are aligned against the putative NCL references. There are two types of reads that can be mapped to a putative NCL reference: ‘encompassing read’, which connects two parental transcript segments but doesn't support the NCL junction site, and ‘junction read’, which overlaps the junction site. A putative NCL reference will be retained if it satisfies all of the following criteria: (i) all reads that match to the putative NCL reference must not have an alternative co-linear explanation, (ii) at least one junction read supports the putative NCL reference and (iii) the NCL junction boundary must be supported by read evidence (including encompassing and junction reads) spanning across 50 bp on both sides of the junction site (Figure 2D). Of note, the third rule is used to eliminate skew mapping between reads and the corresponding putative NCL reference. Since the use of BLAT-alignments with different sets of parameters could be more effective at detecting possible co-linear explanations of an expressed sequence than single operation with default parameters (32), the retained references are aligned against the reference genome using a different set of BLAT parameters (‘Materials and Methods’ section) to further remove potential false calls. Only the candidates that meet all the requirements are considered as high-confidence NCL transcripts, which are used in the following analyses.
Sensitivity and precision of NCLscan for intergenic NCL transcript detection
We first assess the accuracy of NCLscan at detecting intergenic NCL transcripts based on two simulated datasets (i.e., Datasets A (60) and B (57,58)), which were generated by different methods. Dataset A, which is a semi-artificial dataset generated by the FusionMap group (60), contains a set of artificial 75-bp paired-end reads generated according to 50 simulated fusion transcripts and a background set of real RNA-seq reads that is not expected to harbor any fusion events and has been applied to evaluation of fusion-identification tools (57–60). Dataset B is an artificial dataset provided by Carrara et al. (57,58), in which 100-bp paired-end reads were built by BEERS (68) based on well-annotated co-linear transcripts. Dataset B is regarded as a negative dataset, as it does not contain any fusion events. Thus, it is particularly useful for assessing the number of false calls reported by fusion-identification tools (57,58). With respect to the size of each dataset, Dataset A is a small collection of 0.057 million reads (Table 1), which can be used to efficiently evaluate the accuracy of fusion-detecting tools. In contrast, Dataset B, which contains 70 million reads, is much closer to real datasets (Table 1).
We used these two simulated datasets to compare NCLscan with 15 publicly-available tools, including BreakFusion (69), ChimeraScan (70), CRAC (71), deFuse (38), FusionAnalyser (41), FusionFinder (37), FusionHunter (72), FusionMap (60), FusionQ (73), MapSplice2 (74), STAR (75), segemehl (76), SOAPfuse (59), TopHat-Fusion (36) and TopHat-Fusion-post (36). All tools for evaluation were used with default parameters or the parameters suggested by the authors (Supplemental Table S1). We then evaluated the performance of each tool based on sensitivity (Sn), precision (Sp) and F1 score (‘Materials and Methods’ section). For Dataset A, NCLscan exhibited the greatest precision and F1 score, and the third highest sensitivity (Sn = 0.86) among 16 tools (Figure 3A). It is worth noting that NCLscan did not identify any false positives (Sp = 1); furthermore, although STAR and TopHat-Fusion had better sensitivity than NCLscan, their advances in sensitivity were subject to the trade-off in precision (Sp = 0.79 for STAR; Sp = 0.2 for TopHat-Fusion) (Figure 3A). Moreover, although FusionHunter and ChimeraScan also exhibited 100% precision (Sp = 1) on Dataset A, they were not satisfying with respect to sensitivity (both Sn < 0.4) (Figure 3A). Overall, NCLscan exhibited the highest F1 score (Figure 3A), indicating its superiority in terms of sensitivity and precision. For Dataset B, while most tools detected an enormous number of false NCL events (e.g., TopHat-Fusion detected >180 000 events; Figure 3B), NCLscan didn't report any false calls. Since Dataset B is a negative dataset, any NCL events detected in this dataset must be false positives. The result revealed that the existing fusion-detecting tools generally have severe problems with false positives, particularly when detecting fusion transcripts in a large dataset. In contrast, NCLscan still kept 100% precision on a large dataset, indicating the robustness of NCLscan for detecting NCL events with high accuracy. It should be noted that CRAC and FusionQ could not finish analyzing Dataset B after more than 1 week had elapsed, and so the results shown for the two tools were for only part of this dataset; even so, hundreds of false fusions were delivered by the two tools (Figure 3B).
Figure 3.
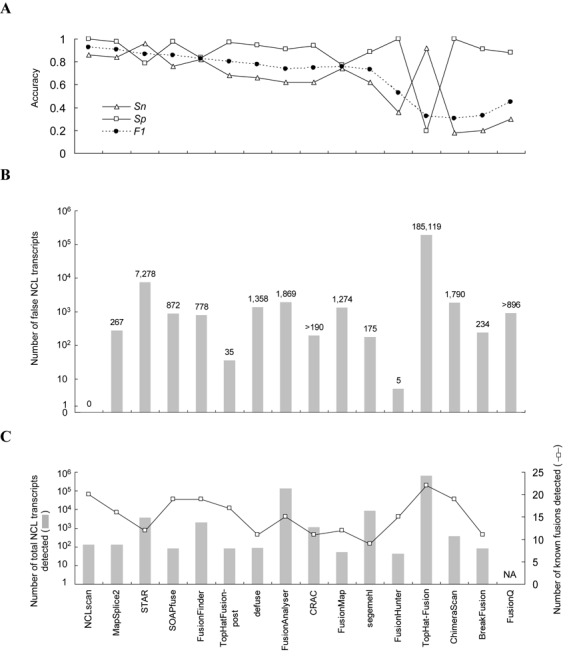
Comparison of the accuracy of 16 tools for detecting intergenic NCL transcripts based on (A) Dataset A, (B) Dataset B and (C) Dataset C. Datasets A and B are simulated datasets; Dataset C is a real dataset. The results for CRAC and FusionQ on Dataset B represent only one half and one fifth of the RNA-seq reads, respectively, for the reason that the two tools could not finish the calculation of the whole reads of Dataset B within 1 week. For the same reason, the result of FusionQ on Dataset C is not available. NA, not available.
Furthermore, we applied the 16 tools to a real RNA-seq dataset (Dataset C). This dataset contains 50-bp paired-end reads from four breast cancer cell lines (Table 1), in which 22 non-read-through fusion transcripts (e.g., the fusion events with a distance of ≤2 Mbp between paired genes on the same strand of the same chromosome were not considered) were experimentally validated by both RT-PCR and array comparative genomic hybridization (aCGH) (Supplemental Table S3) (61). Since the authenticity of the observed NCL events were often hampered by in vitro artifacts (22,32,45,77,78), the identified fusion events that have passed multiple experimental validations were more likely to be genuine than those did not. We found that NCLscan achieved high sensitivity, which was able to detect 20 out of the 22 fusion events (Figure 3C). SKA2-MYO19 was not reported by NCLscan because of the ambiguous alignments with an alternative co-linear explanation or mapping to an unplaced (undetermined) contig. LAMP1-MCF2L was missed due to the skew mapping between reads and the corresponding putative NCL reference (Supplemental Figure S1A). Of note, if the smaller size of the span range (e.g., 30 bp) was set, LAMP1-MCF2L could be detected by NCLscan but the number of the identified NCL candidates would be increased, increasing the risk of accepting false positives (Supplemental Figure S1B). Although TopHat-Fusion detected all 22 known fusions, it should be noted that TopHat-Fusion reported as many as 630 812 candidates. In view of the high false-positive rates of TopHat-Fusion when applied to the simulated datasets (Datasets A and B; Figure 3A and B), users of this tool may need to make extra efforts to reduce potential false calls. NCLscan was also applied to another read RNA-seq data from the prostate cancer cell line VCaP and successfully detected all the three non-read-through fusion transcripts that were validated by both qRT-PCR and fluorescence in situ hybridization (FISH) (48) and collected in the ChiTARS database (79) (Supplemental Tables S3 and S4), further supporting the high level of sensitivity of NCLscan. On the other hand, some tools capable of detecting intergenic NCL transcripts also delivered a great number of intragenic NCL events from Dataset C (i.e., NCLscan, STAR, MapSplice2, CRAC, segemehl and TopHat-Fusion) (Supplemental Table S5). This suggests that there remains a considerable number of uncharacterized intragenic NCL transcripts (or circRNAs) awaiting further discovery within cancer samples.
Sensitivity and precision of NCLscan for simultaneously detecting intergenic and intragenic NCL transcripts
We emphasize that NCLscan is also capable of detecting intragenic NCL transcripts. To further evaluate the sensitivity and precision of NCLscan for simultaneously detecting intergenic and intragenic NCL transcripts, we applied NCLscan and eight other tools (STAR, MapSplice2, CRAC, segemehl, TopHat-Fusion, find_circ (29), CIRCexplorer (80) and CIRI (81)) to simulated RNA-seq datasets. Of note, find_circ, CIRCexplorer and CIRI were designed for detection of intragenic NCL transcripts (circRNAs) only. We used short-read simulator Mason (65) to generate artificial paired-end RNA-seq reads from the mix of the following transcripts: (i) 100 simulated intergenic NCL transcripts, (ii) 100 simulated intragenic NCL transcripts and (iii) well-annotated co-linear transcripts, and simulated a variety of data conditions of different read depths (5-, 10-, 20- and 50-fold) and of different read lengths (2 × 50, 2 × 100 and 2 × 150 bp) (‘Materials and Methods’ section). The identification results of these tools were reported in Supplemental Table S6. As expected, Sn values increased with increasing depth or length of reads, regardless of whether intragenic or intergenic NCL transcripts were detected (Figure 4A). In terms of sensitivity with shorter reads (e.g., 2 × 50 bp), NCLscan and TopHat-fusion were effective at detecting intragenic events, whereas MapSplice2 appeared to yield good sensitivity for detecting intergenic ones. For longer reads (e.g., 2 × 150 bp), NCLscan, STAR, TopHat-fusion, segemehl and the three circRNA-detecting tools (i.e., find_circ, CIRCexplorer and CIRI) represented comparable sensitivity for detecting intragenic events; MapSplice2 was more sensitive for detecting intergenic events than for detecting intragenic ones, whereas segemehl exhibited the reverse trend (Figure 4A). As for Sp, NCLscan reported zero false calls on all tested datasets under different simulated conditions (Supplemental Figure S2), yielding the highest precision (all Sp = 1) among the tested tools (Figure 4A). Obviously, NCLscan was much more precise than the tool (i.e., CIRCexplorer) that exhibited the second best precision (all Sp < 0.72; Figure 4A and Supplemental Figure S3A). With the exception of NCLscan, the Sp values appeared to be affected by read depth and length (Figure 4A), in which the number of false positives markedly increased with increasing depth of reads and marginally decreased with increasing length of reads (Supplemental Figure S2). In contrast to NCLscan with no false positives, all the other tested tools identified a considerable number of false positives (Supplemental Figure S2), which severely reduced the Sp values and then F1 scores under varied simulated conditions (Figure 4A). For example, although TopHat-fusion generally had higher Sn values than the other tools, it yielded the lowest Sp, resulting in a relatively poor balance between sensitivity and precision (all F1 scores < 0.1; Figure 4A). In general, NCLscan had comparable Sn values and the highest Sp values without compromising the false positive rate, exhibiting the best balance between sensitivity and precision on all tested datasets.
Figure 4.
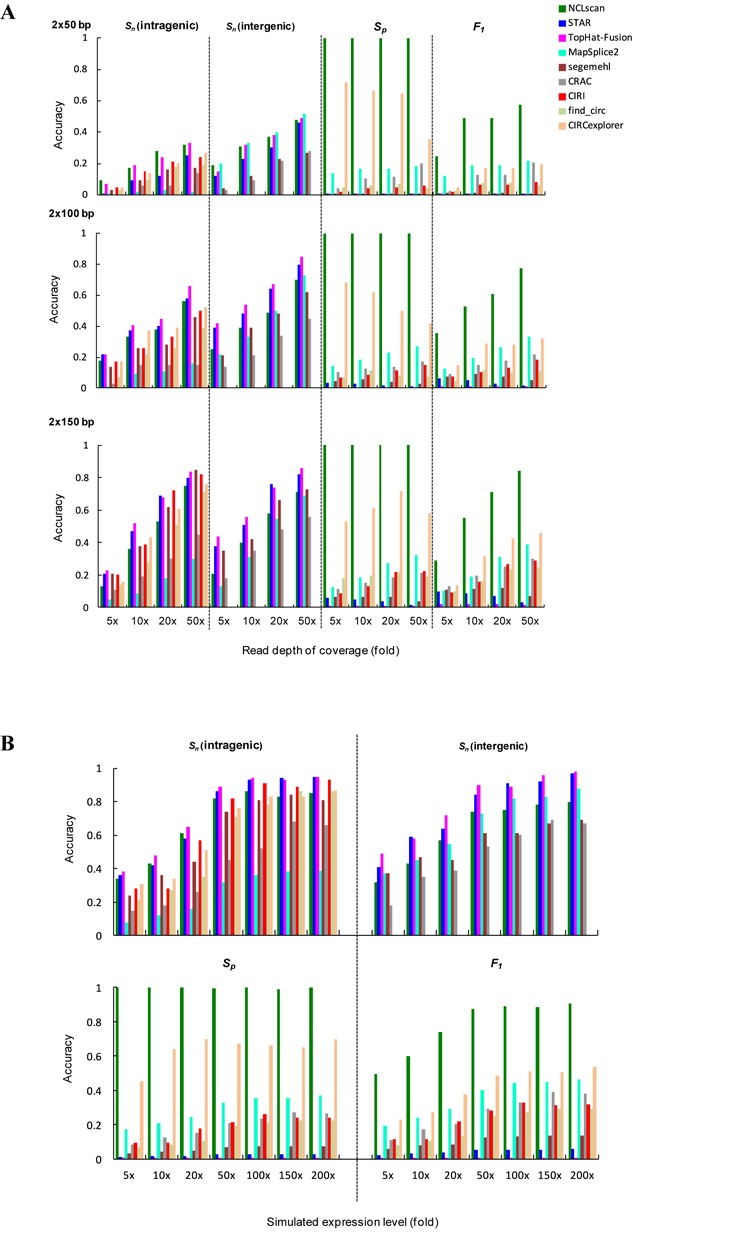
Evaluation of the sensitivity, precision and F1 score of NCLscan for simultaneous detection of intergenic and intragenic NCL transcripts based on simulated datasets of (A) different depths and lengths of reads, and (B) different expression levels (5- to 200-fold) of NCL transcripts.
We further explored the effect of expression level on the sensitivity and precision of NCL transcript identification. We utilized Mason to generate paired-end reads from the aforementioned 100 simulated intragenic and 100 simulated intergenic NCL transcripts with different expression levels (5- to 200-fold), and then mixed these datasets with the same background dataset generated from co-linear transcripts (‘Materials and Methods’ section). The identification results of the examined tools were listed in Supplemental Table S6. For all examined tools, sensitivity generally increased with increasing expression level of NCL transcript; however, this tendency weakened when NCL transcript expression level reached 100-fold or greater (Figure 4B and Supplemental Figure S3B). In general, TopHat-fusion exhibited the highest Sn values, but it still possessed the lowest Sp and F1 values for all tested datasets. With the exception of TopHat-fusion, for intragenic events, NCLscan was more sensitive at lower expression levels, whereas NCLscan, STAR, segemehl and the three circRNA-detecting tools yielded comparable sensitivity at higher expression levels (Figure 4B). For intergenic event, while NCLscan, STAR, MapSplice2 and segemehl exhibited the comparable sensitivity for detecting events at lower expression levels, NCLscan, STAR and MapSplice2 appeared to be effective for detecting ones at higher expression levels (Figure 4B). These results indicated that our method exhibited comparative sensitivity at all simulated expression levels. Importantly, while the other tools suffered poor precision, we emphasized that NCLscan maintained the highest Sp values and F1 scores on the tested datasets, regardless of the expression level of NCL transcripts (Figure 4B and Supplemental Figure S3B).
Regarding the NCLscan identification on the 19 simulated datasets (see Supplemental Table S6), the true events were not reported by NCLscan because of the following reasons (which were not mutually exclusive): (i) a low level of read depth or NCL transcript expression (ii) skew mapping (see also Supplemental Figure S1A); and (iii) ambiguous alignments with very close mapping scores between matches (see ‘Materials and Methods’ section; an example was given in Supplemental Table S7). On the other hand, NCLscan yielded three false positives on two of these simulated datasets (Supplemental Table S6). The reason was that the simulator (i.e., Mason) had randomly added a reasonable proportion of mismatches into the generated reads for simulating real reads (65). It was possible that a generated read matched to a false position with a higher mapping score than all the other possible alignments (‘Materials and Methods’ section), thus yielding false positives (Supplemental Table S7). We emphasized that NCLscan yielded only three false positives on these 19 simulated datasets, supporting its high level of precision.
Taken together, our results reveal that NCL transcript-identification tools are often susceptible to false-positive detections. In contrast, NCLscan, while retaining comparable sensitivity, significantly outperforms the other tools in terms of precision (all P-values < 10−15 by the two-tailed Fisher's exact test), regardless of read depth, read length or NCL transcript expression level. NCLscan thus achieves the highest F1 scores on all simulated datasets, indicating its superior performance with a good balance between sensitivity and precision.
Discrimination between circRNAs, trans-spliced RNAs and fusion transcripts
We next applied NCLscan to NCL transcript identification using six real datasets, each of which contained both poly(A)- and nonpoly(A)-selected RNA-seq data (Table 2), from the ENCODE project (82). These datasets comprised of multiple types of cultured cell lines (including primary, immortalized and stem cells; Table 2). The poly(A)- and nonpoly(A)-selected RNA samples were generated by the same laboratory (82). The nonpoly(A)-selected samples were derived from ribo-and poly(A)-depleted RNA. It has been shown that most trans-spliced RNA products are polyadenylated, but circRNAs are not (25,28,32,78,80). Therefore, it is possible to classify the detected NCL transcript candidates into the following four groups based on their presence status in poly(A)- and nonpoly(A)-selected data (see also Supplemental Table S8).
Group I: intragenic events detected in poly(A)-selected data, which were expected to be intragenic trans-spliced RNAs.
Group II: intergenic events detected in poly(A)-selected data, which were expected to be intergenic trans-spliced RNAs or fusion transcripts.
Group III: intragenic events detected in nonpoly(A)-selected data, which were expected to be circRNAs.
Group IV: intergenic events detected in nonpoly(A)-selected data, which were expected to be fusion transcripts.
As shown in Figure 5A, we observed that NCL transcripts in Group III (predicted circRNA products) were more common than those in the other groups; this trend was independent of cell type and read depth of RNA-seq data. This suggests that circRNAs might be the predominant type of NCL RNA products. However, one might consider that some of the events in Group III could in fact be trans-spliced rather than circRNAs, due to the incomplete depletion of poly(A)-tailed RNAs. To address this, we calculated the ratio of the number of intergenic events to that of intragenic ones in each sample. Based on the facts that events in Groups II and IV were definitely not circRNAs and that both Groups I and II were highly probable to be trans-splicing events, we speculated that if trans-splicing events contributed highly to the candidates detected in the nonpoly(A)-selected samples, the quantity ratio of intragenic to intergenic events in nonpoly(A)-selected sample (i.e., Group III versus Group IV) should be comparable to the ratio in poly(A)-selected sample (i.e., Group I versus Group II). However, this analysis showed that the quantity ratio of intragenic-to-intergenic events was much higher in nonpoly(A)-selected samples than in poly(A)-selected ones (all P-values < 10−15 by the two-tailed Fisher's exact test; Figure 5B). In addition, we found that only a small fraction (0.9–2.9%) of events in Group III were detected in poly(A)-selected sample (Figure 5C), in agreement with a previous report (28). These results suggested that most events in Group III should belong to circRNAs.
Figure 5.
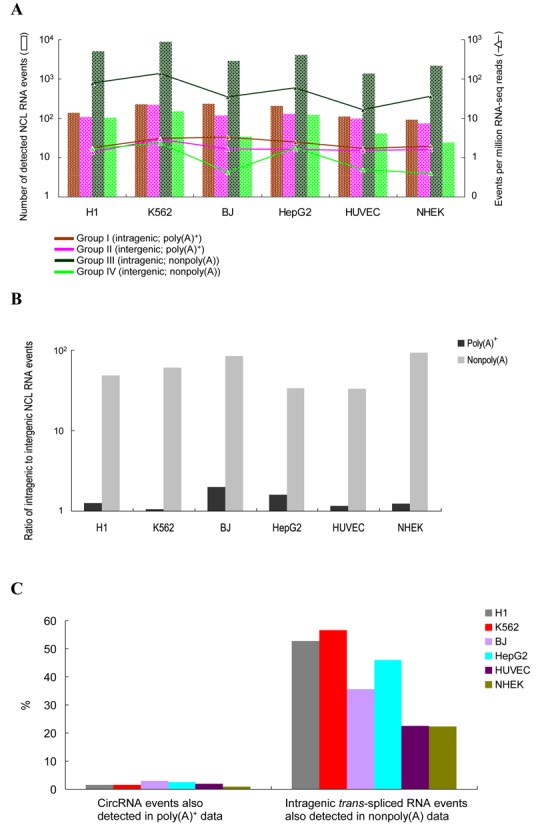
Distinctions between trans-splicing events, circular RNAs and fusion transcripts based on poly(A)- and nonpoly(A)-selected RNA-seq data. (A) Numbers of the identified Groups I to IV NCL events (see the text) in the six cultured cell lines (Table 2). (B) Quantity ratio of intragenic to intergenic NCL RNA events. (C) Percentages of circular RNA candidates (Group III) observed in poly(A)-selected samples and those of intragenic trans-spliced transcript candidates (Group I) observed in nonpoly(A)-selected samples.
Intriguingly, in contrast to the low percentage of events in Group III detected in poly(A)-selected sample, a considerable fraction (22.2–56.6%) of events in Group I were also detected in nonpoly(A)-selected sample (Figure 5C). Since trans-spliced transcripts and circRNAs have been shown to be able to share the same junctions (32), the events detected in both Groups I and III may be attributed to both trans-splicing and circRNA products. Our results thus suggest that a considerable proportion of intragenic trans-spliced RNAs (i.e., Group I) may share the same NCL junctions with circRNAs (i.e., Group III).
Moreover, genomic structural variations (e.g., insertions, duplications and translocations) can also create scrambled exons (54,83,84), which may contribute false positives in the identified circRNA candidates. To address this possibility, we detected potential structure variations (SVs) by analyzing DNA paired-end tags (DNA-PETs) from K562 cells (62) using SVDetect (67). We examined the circRNA candidates (i.e., the Group III candidates; Supplemental Table S4), which were detected from the same sample (K562 cells), and found that only 6.7% of them (594 out of 8915 events) might be formed by the detected SVs (Supplemental Table S2; ‘Materials and Methods’ section). This suggested that most identified intragenic events in nonpoly(A)-selected samples might not be subject to the consequences of SVs.
We next examined the cell-type specificities and expression levels of the four groups of NCL transcripts. Generally, the majority (>70%) of NCL transcripts were detected in only one cell type (Figure 6A), and a considerable percentage of them were supported by a small number of reads (Figure 6B and Supplemental Figure S4). Comparisons of NCL transcripts in the four groups revealed two observations: (i) intragenic NCL transcripts (Groups I and III) tended to present in more cell types and supported by more reads than intergenic ones (Groups II and IV); and (ii) for intragenic NCL events, circRNAs (Group III) tended to exhibit less cell type-specificity (Figure 6A) and higher expression level (Figure 6B and Supplemental Figure S4) than intragenic trans-spliced transcripts (Group I). Of particular importance, more than a quarter of circRNAs (27.8%; 4468 out of 16 063 events) could be detected in multiple cell types (Figure 6A). Our results suggest that circRNAs exhibit a greater expression breadth and a higher expression level than other groups of NCL transcripts, also reflecting that the number of the detected circRNA events is much greater than those of the detected trans-splicing and fusion transcript events (Figure 5A). Moreover, 17.7% (2849 out of 16 063 events) of the circRNAs identified by NCLscan were also detected in different cell types by Guo et al. (28), and over half (51%, 2527 out of 4975 events) of the detected circRNAs in H1 human embryonic stem cells were also detected from H9 human embryonic stem cells by Zhang et al. (80) (Supplemental Table S6). These observations further suggest that certain circRNAs are broadly expressed in a variety of human cells, also reflecting previous reports that circRNAs are abundant in transcriptomes (25,26).
Figure 6.
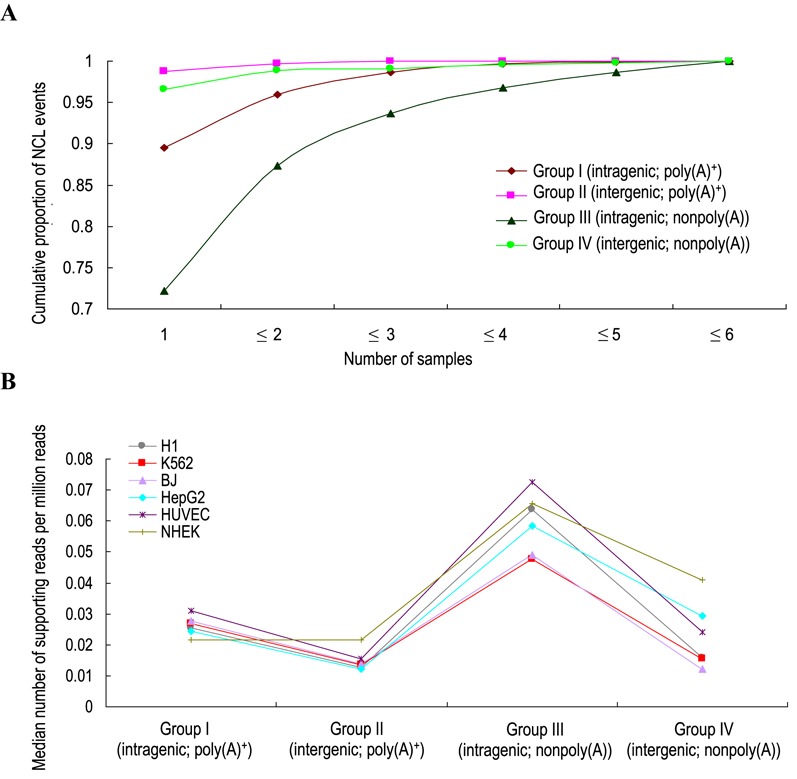
Comparison of (A) cell-type specificity and (B) expression level of the four groups of NCL transcripts.
DISCUSSION
In this study, we developed a new pipeline, NCLscan, which is rather advantageous in the identification of both intragenic and intergenic NCL transcripts from paired-end RNA-seq data. We showed that the accuracy performance of NCLscan was superior over 18 currently available tools, including 11 fusion-detecting tools, 3 circRNA-detecting tools and 5 tools that are capable of detecting both intragenic and intergenic NCL transcripts, in terms of sensitivity and precision (Figures 3 and 4). Interestingly, by evaluating accuracy of these tools, we observed striking differences in accuracy between different versions or parameter settings of the same tool. For example, for Dataset A, TopHat-Fusion was advantageous in sensitivity (Sn = 0.92), but exhibited poor precision (Sp = 0.2) as trade-off. In contrast, TopHat-Fusion-post, an updated version of TopHat-Fusion for detecting intergenic NCL events, achieved excellent precision (Sp = 0.97), but it was worse in terms of sensitivity (Sn = 0.68) (Figure 3A). Another example is CIRCexplorer, which is a TopHat-Fusion-based tool for detecting intragenic NCL events (80). Our results revealed that CIRCexplorer was more precise but less sensitive than TopHat-Fusion (Figure 4). A similar scenario was observed for the updated version of segemehl (v. 0.2.0) and its previous release (v. 0.1.9) (Supplemental Table S9). In addition, many more false calls were reported in Dataset B by FusionAnalyzer without filters than by the same tool with filters (1869 versus 59); moreover, the use of its filters would even rule out all candidates in Dataset A (Supplemental Table S9). These results suggested that certain NCL RNA-identification strategies achieved better precision by sacrificing sensitivity, emphasizing the difficulty in reaching a balance between sensitivity and precision. It was noteworthy that although some tools, such as MapSplice2, SOAPfuse, defuse, FusionAnalyser, CRAC, ChimeraScan and BreakFusion, exhibited good precision (Sp > 0.9) on a small dataset (e.g., Dataset A; Figure 3A), they reported hundreds to thousands of false calls on a larger one (e.g., Dataset B; Figure 3B). This reveals that it is more challenging to remove false positives from a large dataset than from a smaller one. It seems that the larger the dataset is, the more ambiguous alignments there exist, resulting in a larger number of false calls. Here, we confirmed that NCLscan consistently achieved both the highest Sp and F1 values on a variety of simulated datasets, regardless of strategy choice for generating simulated dataset (i.e., Datasets A and B and the simulated datasets generated in this study), read depth, read length, or NCL transcript expression level (Figures 3A, B and 4). It is important to note that NCLscan achieved near 100% precision on all the simulated datasets examined (all Sp > 0.98). We also applied NCLscan to two real RNA-seq datasets (Supplemental Table S3), for which it demonstrated a high level of sensitivity (Figure 3C and Supplemental Table S4). These results indicated that NCLscan can effectively minimize false discovery rate, while maintain a good balance between sensitivity and precision.
Of note, since several studies have suggested that in vitro artifacts are the source of most detected sense-antisense fusion candidates (22,77,85), the current version of NCLscan does not consider this type of chimeric RNAs. However, a few sense-antisense fusions have been confirmed to be associated with the response to chemotherapy in cancer patients (86,87). Some sense-antisense fusion candidates were also provided in a prominent chimera database (i.e., ChiTARS (79)). It is worthwhile to add the capability of detecting sense-antisense chimeras to NCLscan in the future.
We further analyzed poly(A)- and nonpoly(A)-selected RNA-seq data from the ENCODE project to categorize the NCL transcripts into four groups: intragenic and intergenic events in poly(A)-selected samples, and intragenic and intergenic events in nonpoly(A)-selected samples; which enables us to distinguish between trans-splicing events, circRNAs and fusion transcripts. Our results revealed that the number of circRNAs was >10× larger than the numbers of other groups of NCL transcripts (Figure 5A). This trend was generally observed in diverse cell types, suggesting that circRNAs predominated in NCL RNAs. Recent studies have demonstrated that exon circularization is closely associated with Alu-based complementary sequences in flanking introns (26,80). Thus, widespread Alu elements in human introns may make a considerable contribution to the biogenesis of circRNAs (80), accounting for the wide expression of such RNAs in transcriptomes. In addition, we found that a considerable proportion (22.2–55.8%) of intragenic trans-splicing events were also observed in nonpoly(A)-selected samples (Figure 5C). By performing RNase R treatment and RT-PCR/qRT-PCR experiments in human embryonic stem cells (ESCs, line H1), we showed that the selected intragenic trans-splicing events were also resistant to RNase R degradation (Supplemental Note). These results suggest that certain intragenic trans-spliced RNAs may share the same NCL junction with circRNAs, although we cannot completely eliminate the possibility that poly(A)-selected RNA-seq data may still remain a few circRNA products (25). The above result is consistent with our previous study that an observed PtNCL splicing junction may result from trans-splicing RNA, circRNA or both (32).
Moreover, comparison of these four groups of NCL transcripts further revealed two interesting findings. First, intragenic events (Groups I and III) appeared to be less cell type-specific and expressed at a higher level than intergenic ones (Groups II and IV) (Figure 6A and B). Intragenic events may be more abundant than intergenic ones because intragenic splicing within the same gene tends to cause a higher local concentration of transcripts than intergenic splicing between different genes. On the basis of comparison of different reverse transcription products (22,32,77,78) in human H1 ESCs, our experimental validations also exhibited that the tested Groups I and III events had a higher proportion of authentic NCL transcripts than the tested Groups II and IV ones (Supplemental Note), in accord with the previous observations that intergenic events were less common than intragenic ones (22,32,45). Second, circRNAs (Group III) tended to be expressed more ubiquitously, less cell type-specifically and more abundantly than other types of NCL transcript (Figures 5A, 6A and B). This implies that circRNAs may play more diverse cellular roles than expected. For example, some circRNAs have been demonstrated to act as microRNA sponges or play a role in other non-catalytic cellular functions (29,33,88), and as such, these circRNAs are expressed at substantial levels within cells.
Interestingly, in addition to BCR-ABL1, a classic example of gene fusion, we also detected a known fusion event IMMP2L-DOCK4 in K562 cells (Supplemental Table S4). These two intergenic events were detected in both poly(A)- and nonpoly(A)-selected samples, supporting the hypothesis that fusion transcripts can simultaneously contribute to both Groups II and IV. The IMMP2L-DOCK4 fusion event was previously shown to be associated with autism and dyslexia (89,90), and might play an important role in neurite differentiation (91). This is the first time to detect the IMMP2L-DOCK4 fusion event in K562 cells, providing a hint of future studies to investigate the role of this fusion event in chronic myeloid leukemia.
In conclusion, we presented a high accurate method, NCLscan, for detecting intragenic and intergenic NCL transcripts. We showed that NCLscan achieved a better accuracy in terms of sensitivity and precision than 18 other tools, minimizing long and costly experimental validations. Applying NCLscan to different types of RNA-seq data, such as poly(A)- and nonpoly(A)-selected RNA-seq data, was found to be a feasible way of distinguishing between trans-splicing events, circRNAs and fusion transcripts. We suggest that circRNAs exhibit a higher level of prevalence, expression level and expression breadth than other types of NCL transcripts. With different biological and mechanical roles, NCL transcripts provide an alternative means of increasing transcriptome complexity. Our study thus provides an efficient methodology for the design of future experimental studies to functionally probe different types of NCL event in transcriptome. Accumulating evidence has indicated the biological significance of NCL events (1,22,92–99), and as such, these largely uncharted classes of transcripts should not be overlooked in biomedical studies, especially in those seeking to develop therapeutics.
Supplementary Material
Acknowledgments
We thank Chuang-Chieh Lin for operating FusionFinder, MapSplice2 and segemehl, and Shuo-Huang Chen for operating STAR. The authors are especially grateful to Dr. Raffaele A. Calogero for providing Dataset B (negative dataset), Dr. Steve Hoffmann (segemehl group) and Dr. Nicolas Philippe (CRAC group) for helpful suggestions on operating software, Dr. Heng Li for helpful comments on generating simulated datasets and Prof. Hung-Chih Kuo and his group members for providing human H1 ESC samples and experimental facilities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Genomics Research Center, Academia Sinica, Taiwan (internal funding); Ministry of Science and Technology (MOST), Taiwan [MOST 103-2628-B-001-001-MY4, MOST 103-2911-I-001-507]. Funding for open access charge: Genomics Research Center, Academia Sinica, Taiwan; Ministry of Science and Technology (MOST), Taiwan [MOST 103-2628-B-001-001-MY4, MOST 103-2911-I-001-507].
Conflict of interest statement. None declared.
REFERENCES
- 1.Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shtivelman E., Lifshitz B., Gale R.P., Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 3.Mitelman F., Johansson B., Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat. Genet. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- 4.Mitelman F., Johansson B., Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 5.Frohling S., Dohner H. Chromosomal abnormalities in cancer. N. Engl. J. Med. 2008;359:722–734. doi: 10.1056/NEJMra0803109. [DOI] [PubMed] [Google Scholar]
- 6.Westbrook C.A., Hooberman A.L., Spino C., Dodge R.K., Larson R.A., Davey F., Wurster-Hill D.H., Sobol R.E., Schiffer C., Bloomfield C.D. Clinical significance of the BCR-ABL fusion gene in adult acute lymphoblastic leukemia: a Cancer and Leukemia Group B Study (8762) Blood. 1992;80:2983–2990. [PubMed] [Google Scholar]
- 7.O'Brien S.G., Guilhot F., Larson R.A., Gathmann I., Baccarani M., Cervantes F., Cornelissen J.J., Fischer T., Hochhaus A., Hughes T., et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 8.Druker B.J., Guilhot F., O'Brien S.G., Gathmann I., Kantarjian H., Gattermann N., Deininger M.W., Silver R.T., Goldman J.M., Stone R.M., et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 9.Tkachuk D.C., Westbrook C.A., Andreeff M., Donlon T.A., Cleary M.L., Suryanarayan K., Homge M., Redner A., Gray J., Pinkel D. Detection of bcr-abl fusion in chronic myelogeneous leukemia by in situ hybridization. Science. 1990;250:559–562. doi: 10.1126/science.2237408. [DOI] [PubMed] [Google Scholar]
- 10.Tognon C., Knezevich S.R., Huntsman D., Roskelley C.D., Melnyk N., Mathers J.A., Becker L., Carneiro F., MacPherson N., Horsman D., et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 11.Barlund M., Monni O., Weaver J.D., Kauraniemi P., Sauter G., Heiskanen M., Kallioniemi O.P., Kallioniemi A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–317. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- 12.Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.W., Varambally S., Cao X., Tchinda J., Kuefer R., et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 13.Kumar-Sinha C., Tomlins S.A., Chinnaiyan A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H., et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 15.Persson M., Andren Y., Mark J., Horlings H.M., Persson F., Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bass A.J., Lawrence M.S., Brace L.E., Ramos A.H., Drier Y., Cibulskis K., Sougnez C., Voet D., Saksena G., Sivachenko A., et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konarska M.M., Padgett R.A., Sharp P.A. Trans splicing of mRNA precursors in vitro. Cell. 1985;42:165–171. doi: 10.1016/s0092-8674(85)80112-4. [DOI] [PubMed] [Google Scholar]
- 18.Solnick D. Trans splicing of mRNA precursors. Cell. 1985;42:157–164. doi: 10.1016/s0092-8674(85)80111-2. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Wang J., Mor G., Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 20.Schoenfelder S., Clay I., Fraser P. The transcriptional interactome: gene expression in 3D. Curr. Opin. Genet. Dev. 2010;20:127–133. doi: 10.1016/j.gde.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Rickman D.S., Pflueger D., Moss B., VanDoren V.E., Chen C.X., de la Taille A., Kuefer R., Tewari A.K., Setlur S.R., Demichelis F., et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C.S., Yu C.Y., Chuang C.Y., Hsiao M., Kao C.F., Kuo H.C., Chuang T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24:25–36. doi: 10.1101/gr.159483.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu M.T., Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280:339–340. doi: 10.1038/280339a0. [DOI] [PubMed] [Google Scholar]
- 24.Nigro J.M., Cho K.R., Fearon E.R., Kern S.E., Ruppert J.M., Oliner J.D., Kinzler K.W., Vogelstein B. Scrambled exons. Cell. 1991;64:607–613. doi: 10.1016/0092-8674(91)90244-s. [DOI] [PubMed] [Google Scholar]
- 25.Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeck W.R., Sorrentino J.A., Wang K., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P.L., Bao Y., Yee M.C., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Salzman J. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014;9:e90859. doi: 10.1371/journal.pone.0090859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J.U., Agarwal V., Guo H., Bartel D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M., et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 30.Ivanov A., Memczak S., Wyler E., Torti F., Porath H.T., Orejuela M.R., Piechotta M., Levanon E.Y., Landthaler M., Dieterich C., et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–177. doi: 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 31.Westholm J.O., Miura P., Olson S., Shenker S., Joseph B., Sanfilippo P., Celniker S.E., Graveley B.R., Lai E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9:1966–1980. doi: 10.1016/j.celrep.2014.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu C.Y., Liu H.J., Hung L.Y., Kuo H.C., Chuang T.J. Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro? Nucleic Acids Res. 2014;42:9410–9423. doi: 10.1093/nar/gku643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 34.Bachmayr-Heyda A., Reiner A.T., Auer K., Sukhbaatar N., Aust S., Bachleitner-Hofmann T., Mesteri I., Grunt T.W., Zeillinger R., Pils D. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 2015;5:8057. doi: 10.1038/srep08057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hentze M.W., Preiss T. Circular RNAs: splicing's enigma variations. EMBO J. 2013;32:923–925. doi: 10.1038/emboj.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim D., Salzberg S.L. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Francis R.W., Thompson-Wicking K., Carter K.W., Anderson D., Kees U.R., Beesley A.H. FusionFinder: a software tool to identify expressed gene fusion candidates from RNA-Seq data. PLoS One. 2012;7:e39987. doi: 10.1371/journal.pone.0039987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McPherson A., Hormozdiari F., Zayed A., Giuliany R., Ha G., Sun M.G., Griffith M., Heravi Moussavi A., Senz J., Melnyk N., et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakarya O., Breu H., Radovich M., Chen Y., Wang Y.N., Barbacioru C., Utiramerur S., Whitley P.P., Brockman J.P., Vatta P., et al. RNA-Seq mapping and detection of gene fusions with a suffix array algorithm. PLoS Comput. Biol. 2012;8:e1002464. doi: 10.1371/journal.pcbi.1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abate F., Acquaviva A., Paciello G., Foti C., Ficarra E., Ferrarini A., Delledonne M., Iacobucci I., Soverini S., Martinelli G., et al. Bellerophontes: an RNA-Seq data analysis framework for chimeric transcripts discovery based on accurate fusion model. Bioinformatics. 2012;28:2114–2121. doi: 10.1093/bioinformatics/bts334. [DOI] [PubMed] [Google Scholar]
- 41.Piazza R., Pirola A., Spinelli R., Valletta S., Redaelli S., Magistroni V., Gambacorti-Passerini C. FusionAnalyser: a new graphical, event-driven tool for fusion rearrangements discovery. Nucleic Acids Res. 2012;40:e123. doi: 10.1093/nar/gks394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffmann S., Otto C., Doose G., Tanzer A., Langenberger D., Christ S., Kunz M., Holdt L., Teupser D., Hackermueller J., et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol. 2014;15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim P., Yoon S., Kim N., Lee S., Ko M., Lee H., Kang H., Kim J. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res. 2010;38:D81–D85. doi: 10.1093/nar/gkp982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ha K.C., Lalonde E., Li L., Cavallone L., Natrajan R., Lambros M.B., Mitsopoulos C., Hakas J., Kozarewa I., Fenwick K., et al. Identification of gene fusion transcripts by transcriptome sequencing in BRCA1-mutated breast cancers and cell lines. BMC Med. Genomics. 2011;4:75. doi: 10.1186/1755-8794-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McManus C.J., Duff M.O., Eipper-Mains J., Graveley B.R. Global analysis of trans-splicing in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12975–12979. doi: 10.1073/pnas.1007586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang G., Guo G., Hu X., Zhang Y., Li Q., Li R., Zhuang R., Lu Z., He Z., Fang X., et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20:646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Q., Caballero O.L., Levy S., Stevenson B.J., Iseli C., de Souza S.J., Galante P.A., Busam D., Leversha M.A., Chadalavada K., et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1886–1891. doi: 10.1073/pnas.0812945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J., et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma L., Yang S., Zhao W., Tang Z., Zhang T., Li K. Identification and analysis of pig chimeric mRNAs using RNA sequencing data. BMC Genomics. 2012;13:429. doi: 10.1186/1471-2164-13-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inaki K., Hillmer A.M., Ukil L., Yao F., Woo X.Y., Vardy L.A., Zawack K.F., Lee C.W., Ariyaratne P.N., Chan Y.S., et al. Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res. 2011;21:676–687. doi: 10.1101/gr.113225.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Balool H.H., Weber D., Liu Y., Wade M., Guleria K., Nam P.L., Clayton J., Rowe W., Coxhead J., Irving J., et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011;21:1788–1799. doi: 10.1101/gr.116442.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glazar P., Papavasileiou P., Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–1670. doi: 10.1261/rna.043687.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao X., Shepelev V., Fedorov A. Bioinformatic analysis of exon repetition, exon scrambling and trans-splicing in humans. Bioinformatics. 2006;22:692–698. doi: 10.1093/bioinformatics/bti795. [DOI] [PubMed] [Google Scholar]
- 55.Ozsolak F., Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nacu S., Yuan W., Kan Z., Bhatt D., Rivers C.S., Stinson J., Peters B.A., Modrusan Z., Jung K., Seshagiri S., et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carrara M., Beccuti M., Cavallo F., Donatelli S., Lazzarato F., Cordero F., Calogero R.A. State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues. BMC Bioinformatics. 2013;14(Suppl. 7):S2. doi: 10.1186/1471-2105-14-S7-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carrara M., Beccuti M., Lazzarato F., Cavallo F., Cordero F., Donatelli S., Calogero R.A. State-of-the-art fusion-finder algorithms sensitivity and specificity. Biomed. Res. Int. 2013:340620. doi: 10.1155/2013/340620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jia W., Qiu K., He M., Song P., Zhou Q., Zhou F., Yu Y., Zhu D., Nickerson M.L., Wan S., et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013;14:R12. doi: 10.1186/gb-2013-14-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ge H., Liu K., Juan T., Fang F., Newman M., Hoeck W. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–1928. doi: 10.1093/bioinformatics/btr310. [DOI] [PubMed] [Google Scholar]
- 61.Edgren H., Murumagi A., Kangaspeska S., Nicorici D., Hongisto V., Kleivi K., Rye I.H., Nyberg S., Wolf M., Borresen-Dale A.L., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011;12:R6. doi: 10.1186/gb-2011-12-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holtgrewe M., Emde A.K., Weese D., Reinert K. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011;12:210. doi: 10.1186/1471-2105-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeitouni B., Boeva V., Janoueix-Lerosey I., Loeillet S., Legoix-ne P., Nicolas A., Delattre O., Barillot E. SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics. 2010;26:1895–1896. doi: 10.1093/bioinformatics/btq293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grant G.R., Farkas M.H., Pizarro A.D., Lahens N.F., Schug J., Brunk B.P., Stoeckert C.J., Hogenesch J.B., Pierce E.A. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen K., Wallis J.W., Kandoth C., Kalicki-Veizer J.M., Mungall K.L., Mungall A.J., Jones S.J., Marra M.A., Ley T.J., Mardis E.R., et al. BreakFusion: targeted assembly-based identification of gene fusions in whole transcriptome paired-end sequencing data. Bioinformatics. 2012;28:1923–1924. doi: 10.1093/bioinformatics/bts272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iyer M.K., Chinnaiyan A.M., Maher C.A. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Philippe N., Salson M., Commes T., Rivals E. CRAC: an integrated approach to the analysis of RNA-seq reads. Genome Biol. 2013;14:R30. doi: 10.1186/gb-2013-14-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y., Chien J., Smith D.I., Ma J. FusionHunter: identifying fusion transcripts in cancer using paired-end RNA-seq. Bioinformatics. 2011;27:1708–1710. doi: 10.1093/bioinformatics/btr265. [DOI] [PubMed] [Google Scholar]
- 73.Liu C., Ma J., Chang C.J., Zhou X. FusionQ: a novel approach for gene fusion detection and quantification from paired-end RNA-Seq. BMC Bioinformatics. 2013;14:193. doi: 10.1186/1471-2105-14-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang K., Singh D., Zeng Z., Coleman S.J., Huang Y., Savich G.L., He X., Mieczkowski P., Grimm S.A., Perou C.M., et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoffmann S., Otto C., Doose G., Tanzer A., Langenberger D., Christ S., Kunz M., Holdt L.M., Teupser D., Hackermuller J., et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014;15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Houseley J., Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen I., Chen C.Y., Chuang T.J. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip. Rev. RNA. 2015;6:563–579. doi: 10.1002/wrna.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frenkel-Morgenstern M., Gorohovski A., Vucenovic D., Maestre L., Valencia A. ChiTaRS 2.1–an improved database of the chimeric transcripts and RNA-seq data with novel sense-antisense chimeric RNA transcripts. Nucleic Acids Res. 2015;43:D68–D75. doi: 10.1093/nar/gku1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang X.O., Wang H.B., Zhang Y., Lu X., Chen L.L., Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–147. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 81.Gao Y., Wang J., Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4. doi: 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patthy L. Genome evolution and the evolution of exon-shuffling–a review. Gene. 1999;238:103–114. doi: 10.1016/s0378-1119(99)00228-0. [DOI] [PubMed] [Google Scholar]
- 84.Zhang F., Gu W., Hurles M.E., Lupski J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kapranov P., Drenkow J., Cheng J., Long J., Helt G., Dike S., Gingeras T.R. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–997. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murga Penas E.M., Cools J., Algenstaedt P., Hinz K., Seeger D., Schafhausen P., Schilling G., Marynen P., Hossfeld D.K., Dierlamm J. A novel cryptic translocation t(12;17)(p13;p12-p13) in a secondary acute myeloid leukemia results in a fusion of the ETV6 gene and the antisense strand of the PER1 gene. Genes Chromosomes Cancer. 2003;37:79–83. doi: 10.1002/gcc.10175. [DOI] [PubMed] [Google Scholar]
- 87.Tang M., Foo J., Gonen M., Guilhot J., Mahon F.X., Michor F. Selection pressure exerted by imatinib therapy leads to disparate outcomes of imatinib discontinuation trials. Haematologica. 2012;97:1553–1561. doi: 10.3324/haematol.2012.062844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bahn J.H., Zhang Q., Li F., Chan T.M., Lin X., Kim Y., Wong D.T., Xiao X. The Landscape of MicroRNA, Piwi-Interacting RNA, and Circular RNA in Human Saliva. Clin. Chem. 2015;61:230–221. doi: 10.1373/clinchem.2014.230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pagnamenta A.T., Bacchelli E., de Jonge M.V., Mirza G., Scerri T.S., Minopoli F., Chiocchetti A., Ludwig K.U., Hoffmann P., Paracchini S., et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol. Psychiatry. 2010;68:320–328. doi: 10.1016/j.biopsych.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maestrini E., Pagnamenta A.T., Lamb J.A., Bacchelli E., Sykes N.H., Sousa I., Toma C., Barnby G., Butler H., Winchester L., et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol. Psychiatry. 2010;15:954–968. doi: 10.1038/mp.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xiao Y., Peng Y., Wan J., Tang G., Chen Y., Tang J., Ye W.C., Ip N.Y., Shi L. The atypical guanine nucleotide exchange factor Dock4 regulates neurite differentiation through modulation of Rac1 GTPase and actin dynamics. J. Biol. Chem. 2013;288:20034–20045. doi: 10.1074/jbc.M113.458612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parra G., Reymond A., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Akiva P., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. doi: 10.1101/gr.4137606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frenkel-Morgenstern M., Valencia A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 2012;28:i67–i74. doi: 10.1093/bioinformatics/bts216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frenkel-Morgenstern M., Lacroix V., Ezkurdia I., Levin Y., Gabashvili A., Prilusky J., Del Pozo A., Tress M., Johnson R., Guigo R., et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–1242. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou J., Liao J., Zheng X., Shen H. Chimeric RNAs as potential biomarkers for tumor diagnosis. BMB Rep. 2012;45:133–140. doi: 10.5483/BMBRep.2012.45.3.133. [DOI] [PubMed] [Google Scholar]
- 97.Conn S.J., Pillman K.A., Toubia J., Conn V.M., Salmanidis M., Phillips C.A., Roslan S., Schreiber A.W., Gregory P.A., Goodall G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–1134. doi: 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 98.Lasda E., Parker R. Circular RNAs: diversity of form and function. RNA. 2014;20:1829–1842. doi: 10.1261/rna.047126.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jeck W.R., Sharpless N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014;32:453–461. doi: 10.1038/nbt.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.