


Abstract
RNA cleavage by bacterial RNA polymerase (RNAP) has been implicated in transcriptional proofreading and reactivation of arrested transcription elongation complexes but its molecular mechanism is less understood than the mechanism of nucleotide addition, despite both reactions taking place in the same active site. RNAP from the radioresistant bacterium Deinococcus radiodurans is characterized by highly efficient intrinsic RNA cleavage in comparison with Escherichia coli RNAP. We find that the enhanced RNA cleavage activity largely derives from amino acid substitutions in the trigger loop (TL), a mobile element of the active site involved in various RNAP activities. The differences in RNA cleavage between these RNAPs disappear when the TL is deleted, or in the presence of GreA cleavage factors, which replace the TL in the active site. We propose that the TL substitutions modulate the RNA cleavage activity by altering the TL folding and its contacts with substrate RNA and that the resulting differences in transcriptional proofreading may play a role in bacterial stress adaptation.
INTRODUCTION
Transcription errors can be detrimental to cells even though they are not genetically heritable like replication errors. Recent studies revealed the in vivo transcription error rates of about 10−4–10−5 and demonstrated that changes in transcription fidelity can significantly affect gene expression, cell growth and genomic stability in bacteria (1–5) and eukaryotes (6–8).
Transcription fidelity primarily depends on the ability of RNA polymerase (RNAP) to select correct nucleotides during RNA synthesis. The catalytic cycle of nucleotide addition starts with its initial pairing with the DNA template nucleotide, followed by the folding of a mobile trigger loop (TL) that encloses the nucleotide substrate within the i + 1 site of the active center. Two divalent metal ions (usually, Mg2+) then catalyze nucleotidyl transfer, followed by TL unfolding and RNAP translocation (9–13). TL closure contributes up to 10 000-fold to the rate of phosphodiester bond formation and fidelity of transcription (10,14,15). Conformational changes of the TL during different steps of the nucleotide addition cycle are coupled with changes in adjacent elements of the active site, including the bridge helix (BH) and the F-loop (FL) (Figure 1 and Supplementary Figure S1) (9,16–18). Folding and unfolding of the TL also modulates RNAP movement along the DNA because the intimate interactions of the folded TL with the 3′-end of the nascent RNA impede forward translocation (19,20).
Figure 1.

Structure of the RNAP active site in the backtracked TEC. (A) Backtracked TEC of Tth RNAP (4WQS, (28)). RNAP subunits are indicated. (B) Enlarged view of the RNAP active site. The DNA/RNA hybrid and the 3′-end RNA nucleotide bound in the proofreading site are indicated. Also shown are the TL, BH and FL in the β′ subunit and the β subunit segment forming a part of the proofreading site; individual amino acids are labeled (Eco numbering). Position of the SI3 insertion in the TL is shown with a gray sphere; position of the TL deletions analyzed in this study is shown with a black line. (C) Alignment of the TL sequences in Eco, Dra and Tth RNAPs. The alpha-helical and unfolded segments in the open and closed TL conformations are shown below the alignment by thick and thin lines, respectively. The TL deletions in Eco and Dra RNAPs are underlined.
The second major mechanism that contributes to transcription fidelity is endonucleolytic cleavage of the nascent RNA transcript in the RNAP active site, which results in removal of misincorporated nucleotides (21–23). Intrinsic RNA cleavage is catalyzed by the same pair of catalytic divalent ions as nucleotide addition; however, the binding of the second of these ions during cleavage is relatively week and is a subject to regulation (13,24,25). RNA cleavage is preceded by RNAP backtracking along the DNA template, which is stimulated by nucleotide misincorporation (23,26,27). Bacterial RNAP predominantly removes dinucleotide fragments from the RNA 3′-end, and the 3′-terminal RNA nucleotide was shown to directly participate in the reaction, thus suggesting an ancient role for RNA in catalysis (22,23). Recent structural analysis of a backtracked transcription elongation complex (TEC) of Thermus thermophilus (Tth) RNAP revealed that the 3′-nt is bound in a specific proofreading site in the secondary channel; mutations in this site were shown to impair RNA cleavage (Figure 1 and Supplementary Figure S1A) (28).
The role of the TL in RNA cleavage remains controversial. Biochemical studies demonstrated that it is essential for cleavage and likely directly contacts the RNA substrate in thermophilic Thermus aquaticus (Taq) RNAP (27), but may be less important for cleavage in mesophilic Eco RNAP (15,22,27). In particular, substitution of catalytic residue H1242 in the TL in Taq RNAP much more severely affected catalysis than substitution of corresponding residue H936 in Eco RNAP (Figure 1) (27). However, in the published backtracked TEC structure of Thermus RNAP, the TL is removed away from the RNA substrate suggesting that the complex is in an inactive conformation (Figure 1B) (28).
Most bacterial cells contain specialized Gre factors (GreA and GreB in Escherichia coli) that stimulate RNA cleavage by RNAP in vitro and are likely important for transcriptional proofreading in vivo. Gre proteins bind within the secondary channel of RNAP and reach the active site, which requires unfolding of the TL and is accompanied by conformational changes in major RNAP domains and opening of the main channel holding nucleic acids (28). Gre factors were proposed to coordinate the second metal ion in the active site by acidic amino acid residues located at a tip of their effector domain, and to directly participate in catalysis through contacts with the RNA substrate and activation of the attacking water molecule (22,24,25,29). Gre factors were shown to play crucial role in reactivation of arrested TECs resulting from irreversible RNAP backtracking (30,31). In vitro and in vivo studies suggested that such stalled TECs can block DNA replication, resulting in increased level of mutagenesis and genome instability (32–34).
Deinococcus radiodurans (Dra) is a unique mesophilic bacterium, phylogenetically related to thermophilic Taq and Tth, that is highly resistant to ionizing radiation, ultraviolet light and chemical mutagens, thus suggesting the existence of specific mechanisms of stress resistance (35,36). In particular, D. radiodurans cells were shown to accumulate manganese ions under stress conditions, resulting in activation of several manganese-dependent enzymes and protective pathways (37). Previously, we found that Dra RNAP highly efficiently catalyzes RNA cleavage in comparison with E. coli (Eco) RNAP (38). The enhanced RNA cleavage activity may play an essential role under stress conditions, by decreasing transcriptional mutagenesis and minimizing transcription-replication conflicts. In this work, we performed comparative analysis of Dra and Eco RNAPs and identified factors that modulate their RNA cleavage activities.
MATERIALS AND METHODS
DNA, proteins and reagents
Mutant variants of the Eco rpoC and rpoB genes were obtained by site-directed polymerase chain reaction-mutagenesis of the pET29-rpoC and pIA545 plasmids and recloned into the pVS10 and pIA679 expression vectors containing His6-tags in the C-terminus of the β′ subunit and in the N-terminus of the β subunit, respectively (39). Wild-type and mutant Eco core RNAPs were expressed and purified from Eco BL21(DE3) cells as described in (39). Core Dra RNAP was purified either from Dra cells as described previously (40) or from E. coli BL21(DE3) cells expressing all core RNAP subunits from plasmid pET28-rpoACBZ-Dra. The plasmid was obtained by cloning of the rpoA, rpoB, rpoC and rpoZ genes of Dra into the pET28 vector under the control of T7 RNAP promoters. Mutant variants of the Dra rpoC gene were obtained by site-directed mutagenesis and cloned into the same plasmid. Eco cells containing pET28-rpoACBZ-Dra were grown for 16 h at 22°C in the presence of 0.1 mM IPTG. The cells were disrupted by sonication; RNAP was purified by Polymin P precipitation, followed by chromatography on Heparin-Sepharose (Heparin-HiTrap), Ni2+-affinity (HiTrap Chelating) and anionic-exchange (MonoQ) columns (GE Healthcare) (39,41). Eco and Dra GreA proteins with C-terminal His6-tags were expressed from plasmids pET28-Eco-GreA and pET28-Dra-GreA and purified by Polymin P precipitation (from flow-through), followed by Ni2+-affinity chromatography.
Schematics of nucleic-acid scaffolds used in the analysis of nucleotide addition and RNA cleavage are shown in Supplementary Figures S2, S3 and S4. DNA and RNA oligonucleotides used in the analysis were purchased from IBA Biotech (Göttingen, Germany), Fidelity Systems (Gaithersburg, MD, USA), Eurofins Genomics (Ebersberg, Germany), DNA Synthesis and Syntol (Moscow, Russia). Cytidine-5′-[(α,β)-methyleno]triphosphate (CMPCPP) was from Jena Bioscience (Jena, Germany). DNA template containing a fragment of the Eco rpoB gene fused to the λPR promoter for measurements of elongation rates was obtained as described in (41).
Analysis of nucleotide addition and RNA elongation
TECs for analysis of nucleotide addition were assembled on a minimal nucleic acid scaffold containing 5′-P32-labeled RNA (Supplementary Figure S3) or a complete scaffold containing fluorescently labeled RNA or DNA oligonucleotides (Supplementary Figure S2) as described in (42) and (43), respectively. The catalytic rates were measured after mixing the samples with nucleotides (1 mM uridine triphosphate [UTP] or cytidine triphosphate [CTP] in the case of the minimal scaffold and 200 μM guanosine triphosphate [GTP] or 2′-dGTP in the case of the complete scaffold) in a rapid quench-flow instrument (QFM-400, BioLogic and RQF 3, KinTek Corporation, respectively) or by manual mixing. Most measurements were performed at 25°C, unless otherwise indicated. The reactions were quenched with 0.5 M HCl, the RNA products were separated by denaturing Polyacrylamide gelelectrophoresis (PAGE) and analyzed by phosphorimaging or fluorimetry. The data on nucleotide addition obtained in the minimal scaffold system were fitted to the single-exponential equation. The experiments on nucleotide addition and translocation on the complete scaffold are described in detail in Supplementary Methods. Measurements of average elongation rates was performed on the λPR-rpoB template as previously described (41) (see Supplementary Figure S8 legend for details).
RNA cleavage assays
RNA cleavage was performed in TECs assembled on synthetic nucleic-acid scaffolds as previously described (41,43). RNA primer was 5′-labeled with γ-[32P]-ATP and T4 polynucleotide kinase and mixed with the template DNA oligonucleotide (600 nM and 1.5 μM final concentrations, respectively) in buffer containing 40 mM Tris–HCl, pH 7.9, 40 mM NaCl and 0.1 mM ethylenediaminetetraacetic acid (EDTA). The samples were heated to 65°C and cooled down to 20°C at 1°C/min. One microliter of the RNA–DNA hybrid solution (0.6 pmol RNA/1.5 pmol template DNA) was then mixed with core RNAP solution (5 pmol) in 10 μl of buffer containing 40 mM Tris–HCl, pH 7.9 and 40 mM NaCl and incubated for 20 min at 30°C. A total of 10 pmol of the non-template DNA oligonucleotide was added to the mixture and the samples were incubated for 20 min at 30°C. The samples were diluted 5–10× with transcription buffer containing 40 mM Tris–HCl, pH 7.9, 40 mM NaCl and RNA cleavage was initiated by the addition of MgCl2 or MnCl2 to indicated concentrations. GreA-dependent RNA cleavage was analyzed in a similar way. Gre proteins were added to preformed TECs of Eco or Dra RNAPs (to 5 or 1 μM final concentrations in Figure 7B and C, respectively) and the samples were incubated for 3 min at 30 or 37°C. RNA cleavage was initiated by the addition of MgCl2 to 10 mM. Gre-containing reactions with wild-type RNAPs were performed on a KinTek Chemical Quench Flow Model RQF-3 mixer (KinTek, Austin, USA). The reactions were stopped after various time intervals by the addition of equal volume of stop-solution containing 8M urea and 20 mM EDTA, and RNA products were analyzed by 15% denaturing PAGE. The data were fitted to the equation:
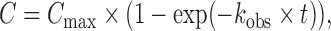 |
where C is the fraction of cleaved RNA, Cmax is the maximal cleavage level, kobs is the observed first-order rate constant and t is the reaction time.
Figure 7.

Effects of TL deletions and GreA factors on RNA cleavage by Eco and Dra RNAPs. (A) Structure of the RNAP active site in complex with GreA (28). Position of the analyzed TL deletions is shown with a line. (B) Kinetics of RNA cleavage in the rA-dG TEC by wild-type and ΔTL RNAP variants (measured at 37°C) and by wild-type RNAPs in the presence of GreA (measured at 30°C). (C) The effects of the Eco GreA factor on RNA cleavage measured at 37°C in the rA-dT TECs assembled with Eco-ΔSI3 and Eco-G1136M(ΔSI3) RNAPs.
Fe2+-induced RNA cleavage
The TECs were assembled in the same way as described above but the non-template DNA oligonucleotide contained a biotin residue in its 5′-end. The complexes were bound to magnetic avidin agarose beads (Dynabeads MyOne Streptavidin C1 Invitrogen), pre-washed in high salt buffer (40 mM Tris–HCl, pH 7.9, 1 M NaCl) and equilibrated with reaction buffer (40 mM Tris–HCl, pH 7.9, 40 mM NaCl), for 25 min at 25°C with occasional shaking. Unbound TECs were removed by washing the beads three times with 1 ml of the reaction buffer. Reaction was initiated by the addition of DTT to 10 mM and (NH4)2FeSO4 to 20 or 200 μM and terminated after 20 min by the addition of the stop-solution. The RNA products were resolved by 29% denaturing PAGE.
RESULTS
RNA synthesis by Eco and Dra RNAPs
Dra and Eco RNAPs transcribe genomes of very divergent bacterial phyla but evolved to operate in similarly mesophilic environments. Therefore, before starting the analysis of RNA cleavage it was imperative to compare RNA synthesis by these two enzymes. To assess possible differences in nucleotide addition and translocation states of the TECs formed by these RNAPs, we employed a set of kinetics and equilibrium assays previously developed in our work (17,44).
We first performed parallel, time-resolved measurements of nucleotide incorporation and post-catalytic relaxation (the combined rate of pyrophosphate release and translocation) in Eco and Dra TECs. To allow accurate comparisons of the catalytic rates, most experiments on nucleotide addition were performed at 25°C since at this temperature the reactions can be resolved in time by rapid quench-flow methods. Both Eco and Dra can actively grow and divide at this temperature and both RNAPs were shown to have comparable elongation rates in the range between 0 and 37°C (40) suggesting that measurements at 25°C allow adequate comparison of RNAP properties. The experiments were performed using a complete synthetic nucleic-acid scaffold containing fluorescent labels at the RNA 5′-end and in the template DNA (Figure 2 and Supplementary Figure S2). RNA extension was monitored by a rapid chemical quench-flow method, whereas translocation was monitored by measuring the increase in fluorescence of the 6-methyl-isoxanthopterin base (6-MI) incorporated in the template DNA strand in a stopped-flow instrument (44). Both Eco and Dra RNAPs fully (>80%, Supplementary Figure S2) extended the RNA transcript after the addition of cognate GTP, followed by rapid TEC translocation. Combined kinetic analysis of the data revealed that Dra RNAP is 2-fold slower in the nucleotide addition but translocates forward with similar or up to twofold faster rate than Eco RNAP (Figure 2B). Although the rate of the post-catalytic relaxation is faster in Dra RNAP, it constitutes only a small fraction of the total half-life of the nucleotide addition cycle (25–30% for Eco RNAP and 10–15% for Dra RNAP).
Figure 2.

Comparative analysis of nucleotide addition and translocation by Eco and Dra RNAPs. (A) Schematics of the experiment. RNA was extended with GTP (200 μM) or 2′-dGTP, followed by TEC translocation. Upon translocation the 6-MI base migrates to the upstream edge of the RNA–DNA hybrid with concomitant increase in fluorescence intensity. (B) Time courses of GMP addition (discrete black time points) and translocation (continuous traces) by Eco (top) and Dra (bottom) RNAPs. The schematics of the analyses and the lower and upper bounds of parameters (calculated at a 10% increase in Chi2) are depicted below the graphs. Best fit curves were drawn using best fit values of parameters described in Supplementary Methods. (C) Fluorescent levels of the Eco (top) and Dra (bottom) TECs after assembly and following the extension by GTP or 2′-dGTP in the presence and absence of CMPCPP. Data were averaged over two to three independent experiments. Error bars are standard deviations.
To assess the completeness of translocation, we forward-biased the GMP-extended TEC with CMPCPP (a non-hydrolyzable analog of CTP), corresponding to the next incoming nucleoside triphosphate (NTP) in our system. For both Eco and Dra TECs, the addition of CMPCPP did not result in a further increase in fluorescence intensities (Figure 2C). Furthermore, similar changes in TEC fluorescence were observed after the incorporation of 2′-dGMP, which promotes forward translocation (because the 2′-OH group at the RNA 3′-end is essential for stabilizing the pre-translocated state), and the addition of CMPCPP to such complexes (20). These observations suggest that the fractions of the pre-translocated state are below the detection threshold (∼10%) in both Eco and Dra TECs.
Overall, the analysis of nucleotide addition and translocation by Eco and Dra RNAP revealed only marginal differences in the rates of the individual steps in the nucleotide addition cycle between the two enzymes. While it is not certain at present if the observed differences have any physiological and/or mechanistic significance, they cannot explain a significantly higher intrinsic RNA cleavage activity of Dra RNAP.
Stimulation of nucleotide misincorporation by manganese ions
We then analyzed the catalytic rates in TECs assembled on a minimal nucleic acid scaffold stabilized in the post-translocated state (Figure 3A, Supplementary Figure S3A), which allows comparison of incorporation of correct and incorrect nucleotides in the absence of RNA cleavage (42). Similarly to the complete scaffold described above, both RNAPs had comparable rates of the correct nucleotide incorporation (UTP), although Dra RNAP was faster than Eco RNAP in this case (Supplementary Figure S3B).
Figure 3.

Effects of manganese on nucleotide incorporation and transcription fidelity. (A) Scheme of the reaction with the minimal nucleic acid scaffold. (B) Kinetics of cognate UTP addition (1 mM, 10°C) by Eco and Dra RNAPs in the presence of magnesium or manganese ions. The plot shows accumulation of the 9 nt RNA product over the reaction time (normalized to the maximal extension for each reaction). (C) Kinetics of non-cognate CTP addition (1 mM, 25°C).
Manganese ions are known to increase misincorporation of non-complementary nucleotides by RNA and DNA polymerases (e.g., (45)). We therefore compared transcription fidelity of Dra and Eco RNAPs in the presence of magnesium and manganese ions, by analyzing cognate UTP and non-cognate CTP addition on the minimal scaffold template. Due to the very fast correct nucleotide incorporation in the presence of manganese, the experiment with UTP was performed at 10°C. Manganese ions stimulated uridine monophosphate (UMP) incorporation, resulting in about 4- and 2.5-fold increase in the catalytic rates for Eco and Dra RNAPs, respectively (Figure 3B, C and Supplementary Figure S3C). At the same time, the rates of the non-cognate cytidine monophosphate (CMP) incorporation were much stronger stimulated by Mn2+ resulting in an overall decrease in the fidelity of RNA synthesis. This effect was larger for Dra than for Eco RNAP (420- versus 75-fold stimulation), leading to a larger decrease in the transcription fidelity (Figure 3B, C and Supplementary Figure S3C).
RNA cleavage by Eco and Dra RNAPs in correct and mismatched TECs
Previous studies revealed enhanced RNA cleavage by Dra RNAP in a model TEC containing fully complementary RNA (38). In this work, analysis of the cleavage reaction was performed in several types of TECs assembled on synthetic nucleic-acid scaffolds. Most experiments were performed with mismatched TEC rA-dG, containing a non-complementary adenine at the RNA 3′-end located opposite guanine in the template DNA, and correct TEC rA-dT, in which the 3′-adenine was complementary to a template thymine (Supplementary Figure S4) (modified from Ref. (23)). The rA–dG complex corresponds to the TEC that is formed after misincorporation of an incorrect nucleotide in the RNA transcript, which facilitates RNAP backtracking and RNA cleavage (23). In addition, we analyzed mismatched TEC rC–dT, which contained a non-complementary cytosine at the RNA 3′-end opposite template thymine, and another variant of correct TEC with the rA–dT pair at the RNA 3′-end (rA-dT2) (Supplementary Figure S4). Most RNA cleavage experiments were performed at 37°C, the temperature optimum for both RNAPs.
RNA cleavage in all analyzed TECs resulted in removal of a dinucleotide from the RNA 3′-end (Supplementary Figure S5). Dra RNAP cleaved RNA dramatically faster than Eco RNAP (Figure 4, Supplementary Figure S5, Table 1 and Supplementary Table S1). In particular, we observed 20-, 18- and 56-fold differences in the cleavage rates in the rA–dG, rA–dT and rC–dT TECs, respectively (at 10 mM MgCl2, 37°C) (Table 1). For each RNAP, the rates of intrinsic RNA cleavage were the highest in the case of the mismatched complex rA–dG containing unpaired adenine residue at the RNA 3′-end (kobs = 0.39 and 7.9 min−1 for Eco and Dra RNAPs, respectively). Similar differences were observed when the experiment was repeated at 25°C, although the absolute rates of the reaction were ∼2.5× lower for both RNAPs (Supplementary Figure S6). The rates of the cleavage in the correct complex rA–dT were more than 30× lower for both RNAPs, likely because base-pairing of the RNA 3′-end prevents RNAP backtracking. The RNA cleavage rates in the mismatched complex rC–dT were also lower in comparison with rA–dG, likely because the unpaired adenine in the latter complex is directly involved in the cleavage reaction (23). Overall, our data demonstrate that Dra RNAP efficiently cleaves RNA in TECs of various structures.
Figure 4.

Kinetics of RNA cleavage by Eco and Dra RNAPs. Schemes of the TECs used for analysis of the cleavage rates are shown above the plots (see Supplementary Figure S4 for scaffold structures). The amounts of cleaved RNA are normalized to the maximal cleavage observed in each reaction. Averages and standard deviations from 2–4 independent experiments are shown.
Table 1. The rates of RNA cleavage by Eco, Dra RNAPs and their mutant variants in different TECs.
| Cleavage rate (kobs, min−1) | |||
|---|---|---|---|
| RNAP | rA-dG | rA-dT | rC-dT |
| Eco | 0.39 ± 0.07 | 0.012 ± 0.003 | 0.014 ± 0.001 |
| 1 | 1 | 1 | |
| Dra | 7.9 ± 1.0 | 0.22 ± 0.05 | 0.78 ± 0.08 |
| 20.1 | 18.2 | 55.7 | |
| Eco ΔSI3 | 0.49 ± 0.16 | 0.016 ± 0.004 | 0.045 ± 0.007 |
| 1.2 | 1.3 | 3.2 | |
| Eco-TL-Dra | 3.4 ± 1.7 | 0.155 ± 0.039 | |
| 8.7 | 13.1 | ||
| Eco-β′I937T(ΔSI3) | 0.78 | 0.018 ± 0.006 | |
| 2.0 | 1.5 | ||
| Eco-β′G1136M (ΔSI3) | 2.0 ± 0.2 | 0.144 ± 0.070 | 0.298 ± 0.055 |
| 5.0 | 12.2 | 21.3 | |
| Dra-β′M1271G | 5.3 ± 0.2 | 0.095 | 0.42 ± 0.12 |
| 13.6 | 8.0 | 29.7 | |
| Eco ΔTL | 0.0050 ± 0.0004 | 0.00088 ± 0.00051 | |
| 0.013 | 0.074 | ||
| Dra ΔTL | 0.0024 ± 0.0003 | 0.0011 ± 0.0007 | |
| 0.006 | 0.09 | ||
| Eco + GreA | 890 ± 175 | ||
| 2300 | |||
| Dra + GreA | 860 ± 50 | ||
| 2200 | |||
The rates were measured at 37°C and 10 mM MgCl2 for most reactions and at 30°C for reactions containing GreA factors. Averages and standard deviations from 2–4 independent experiments are shown. The numbers in bold indicate the cleavage rates relative to the rate of wild-type E. coli RNAP in the same TEC.
Catalytic metal binding by Eco and Dra RNAPs during RNA cleavage
To reveal possible differences between Eco and Dra RNAPs in the binding of the second catalytic metal ion during RNA cleavage, the reactions in the rA–dG and rA–dT complexes were performed at different divalent ion concentrations. At all Mg2+ concentrations, RNA cleavage by Dra RNAP was much faster than in the case of Eco RNAP and the maximal rates of the reaction at saturation were 14.5× higher for Dra RNAP in both TECs (Figure 5, Table 2). However, the apparent dissociation constants (Kd,app) for the binding of the catalytic ions in both complexes were comparable for Eco and Dra RNAPs, with only ∼1.5-fold better binding of Mg2+ to Dra RNAP in the rA–dG complex (Kd,app = 13.9 and 8.3 mM, respectively).
Figure 5.

Dependencies of the RNA cleavage rates by Eco and Dra RNAPs on magnesium concentration for rA-dG (left) and rA-dT (right) TECs. For each TEC, the cleavage rates for Dra and Eco RNAPs are shown on the left and right y-axes, respectively.
Table 2. Kinetic parameters of RNA cleavage by Eco and Dra RNAPs.
| TEC, Me2+, RNAP | Kd,app Me2+ (mM) | kobsmax (min−1) |
|---|---|---|
| rA-dT, Mg2+ | ||
| Eco | 53.8 ± 13.2 | 0.11 ± 0.013 |
| 1 | 1 | |
| Dra | 41.9 ± 14.4 | 1.6 ± 0.6 |
| 0.8 | 14.5 | |
| rA-dG, Mg2+ | ||
| Eco | 13.9 ± 2.8 | 0.87 ± 0.21 |
| 1 | 1 | |
| Dra | 8.3 ± 1.4 | 12.6 ± 0.2 |
| 0.6 | 14.5 | |
| Eco-β′A455E | 3.9 ± 1.7 | 1.5 ± 0.2 |
| 0.3 | 1.7 | |
| Eco-TL-Dra | 11.6 ± 3.1 | 4.1 ± 0.3 |
| 0.8 | 4.7 | |
| rA-dG, Mn2+ | ||
| Eco | 2.8 ± 1.6 | 1.1 ± 0.2 |
| 1 | 1 | |
| Dra | 1.6 ± 0.1 | 13.7 ± 2.4 |
| 0.6 | 12.5 | |
The Kd,app and kobs,max values were determined from measurements of RNA cleavage rates at different metal concentrations (see Figure 5). The numbers in bold indicate the values relative to the wild-type E. coli RNAP in the same TEC.
The apparent Kd values for Mg2+ binding by both RNAPs were lower for the mismatched rA–dG TEC (about 4–5×; Table 2), likely because the translocation equilibrium in this complex is shifted toward backtracked state, in which the unpaired RNA 3′-end facilitates metal binding. This contrasts previously reported observations for Taq RNAP for which the apparent Kd values for Mg2+ did not depend on the complementarity of the 3′-terminal RNA nucleotide (23).
Previously, Dra-specific substitution A455E near the metal-binding NADFDGD-motif of the β′ subunit was shown to slightly stimulate RNA cleavage by Eco RNAP (38). We therefore measured the apparent Kd value for Mg2+ in the cleavage reaction for the Eco-β′A455E RNAP. The substitution slightly increased the maximal rate of catalysis and the apparent affinity of magnesium in the rA-dG TEC (Table 2). Therefore, this substitution may account for the somewhat greater affinity of Dra RNAP to magnesium ions, probably by changing the conformation of the adjacent NADFDGD-motif. However, it cannot fully explain the large differences in the RNA cleavage rates between the Eco and Dra RNAPs.
We then analyzed RNA cleavage in the presence of Mn2+ ions and compared their apparent affinities in the cleavage reaction for Eco and Dra RNAPs. Both RNAPs bound manganese ions about 5-fold better than magnesium ions (Kd,app = 2.8 and 1.6 mM, respectively, in the rA–dG TEC), with no changes in the maximal rates of the reaction (Table 2). Therefore, manganese does not significantly change the intrinsic RNA proofreading activity of Dra RNAP. Overall, the observed differences in the RNA cleavage rates cannot be explained by different efficiencies of divalent ion binding in the active sites of Eco and Dra RNAPs.
Effects of Dra-specific amino acid substitutions on RNA cleavage
To identify specific elements of the active site of Dra RNAP that might be responsible for the increased RNA cleavage rate, we obtained a series of mosaic Eco RNAP variants with substitutions of individual elements that contact the RNA 3′-end with corresponding Dra sequences. In particular, we introduced substitutions present in the TL, FL, BH in the β′ subunit and several substitutions in the RNA proofreading site in the β subunit, including P567A and substitution M681A of a conserved methionine residue that was previously shown to be critically important for cleavage in Tth RNAP (28) (Figure 1, Supplementary Figures S1 and S7). In addition, we analyzed substitution of the β′ lid domain (three amino acid substitutions in total) that contacts the upstream part of the RNA-DNA hybrid. All mutant core RNAP variants were overexpressed in Eco cells and purified to homogeneity.
Most analyzed Dra-specific substitutions, including substitutions of the whole FL-BH segment, the lid domain, and point amino acid substitutions β′L783G in the BH and βP567A in the β proofreading site did not significantly affect RNA cleavage by Eco RNAP (Supplementary Table S1). Alanine substitution of βM681 also did not affect RNA cleavage (kobs = 0.38 min−1 in comparison with 0.39 min−1 for wild-type Eco RNAP in the rA–dG complex), unlike to previously reported tryptophan substitution of the corresponding residue in Tth RNAP (M560W) that dramatically impaired RNA cleavage (28).
In contrast, substitution of the whole TL (in Eco-TL-Dra RNAP) significantly activated RNA cleavage by the mosaic RNAP (8.7- and 13.1-fold in the rA–dG and rA–dT TECs, respectively, at 10 mM MgCl2; Table 1). This substitution also increased the maximal cleavage rate at saturating Mg2+ concentrations but did not significantly change the apparent Kd value for Mg2+ binding (Table 2). Thus, the TL may be the main determinant of the observed differences in RNA cleavage.
Dra RNAP, similarly to related Taq/Tth RNAPs, naturally lacks large insertion (‘SI3’) present in the TL in Eco RNAP (Figure 1C). It was hypothesized that the presence of the SI3 insertion might explain the low efficiency of RNA cleavage by Eco RNAP in comparison with Taq RNAP (27). All studied Eco RNAP variants with Dra-specific substitutions in the TL also lacked the SI3 domain and were therefore compared with a control RNAP lacking this domain. The SI3 deletion in Eco RNAP (Eco ΔSI3) by itself did not significantly change the RNA cleavage rate in most TECs except rC–dA where it activated the reaction about 3-fold (in comparison with 55-fold difference in the rates between Eco and Dra RNAPs in this TEC) (Table 1). Thus, the absence of the SI3 insertion cannot explain the increased RNA cleavage rate by Dra and Eco-TL-Dra RNAPs.
To identify individual amino acid residues in the TL that might modulate RNA cleavage, we obtained additional Eco RNAP variants with Dra-specific single amino acids substitutions in the TL. In particular, we introduced substitution β′I937T(ΔSI3) in the N-terminal part of the TL near residue H936 that was shown to be directly involved in RNA cleavage (27), and substitution G1136M(ΔSI3) in the C-terminal part of the TL. The latter substitution is located at the border of the unfolded TL segment in the backtracked complex that becomes alpha-helical during nucleotide addition (G1136 corresponds to M1271 in Dra RNAP and Q1254 in Thermus RNAP; Figure 1B and Supplementary Figure S1). Previously, the G1136S substitution at the same position was shown to significantly affect catalytic properties of Eco RNAP (see ‘Discussion’ section; (46,47)).
Substitutions I937T(ΔSI3) and G1136M(ΔSI3) increased the RNA cleavage rates in different TEC variants 1.5–2-fold and 5–21-fold, respectively (Figure 4, Supplementary Figure S6 and Table 1). We therefore conclude that the G1136M substitution likely makes a major contribution to the increased cleavage rate of the Eco-TL-Dra and wild-type Dra RNAPs. At the same time, this substitution only marginally increased the average rate of transcription elongation measured on the λPR-rpoB DNA template (Supplementary Figure S8) and the rate of single-nucleotide addition measured on the minimal scaffold template (Supplementary Figure S3B), suggesting that it has a specific effect on RNA cleavage.
To test whether substitutions in the TL may also affect RNA cleavage by Dra RNAP, we obtained its mutant variant with a reciprocal substitution M1271G, located at the same position as G1136M in Eco RNAP. The M1271G substitution reduced the RNA cleavage rate by Dra RNAP in all analyzed TEC variants (about 2-fold, Table 1), although the effect was much less dramatic than the stimulatory effect of the Eco-G1136M(ΔSI3) substitution. The M1271G substitution also slightly decreased the nucleotide addition rate in comparison with Dra RNAP (Supplementary Figure S3B). Therefore, substitutions at this TL position can modulate catalytic properties of RNAPs from various bacteria.
No effects of the TL mutations on the translocation bias of the mismatched TEC
Dra-specific substitutions in the TL might stimulate RNA cleavage by (i) facilitating Mg2+ binding, (ii) affecting the translocation state of the RNAP active site and (iii) changing the TL conformation and its contacts with the RNA substrate. In addition, they might also indirectly affect the conformation of other elements of the active site. As was shown in metal titration experiments, substitutions in the TL do not affect Mg2+ affinity (see above, Table 2). Dra RNAP also does not differ from Eco RNAP in the translocation state of the active site in the TEC used for analysis of nucleotide addition (Figure 2). To reveal whether the TL substitutions might have any effect on the TEC translocation during the cleavage reaction, we analyzed RNA cleavage by hydroxyl radicals generated by Fe2+ ions bound in the RNAP active site in the rA–dG complex. Although the unpaired 3′-end likely stabilizes this complex in the backtracked state, the lower cleavage rate by Eco RNAP could potentially result from transient forward translocation of the complex. However, the Fe2+-induced RNA cleavage patterns were identical for wild-type Eco and Dra RNAPs and for the Eco-G1136M(ΔSI3) mutant. In particular, the cleavage resulted in the appearance of RNA products shortened by 1–3 nt from the 3′-end, likely corresponding to the backtracked TEC conformation (Figure 6). Thus, these RNAPs do not differ in the translocation states of the active site in this TEC, despite having very different RNA cleavage rates. We therefore propose that Dra-specific substitutions may modulate catalysis through local changes in the active site conformation during RNA cleavage (see ‘Discussion’ section).
Figure 6.

Comparison of the translocation states of the 3′-mismatched TECs assembled with Eco and Dra RNAPs. Fe2+-induced RNA cleavage by wild-type Eco, Dra and Eco-G1136M(ΔSI3) RNAPs in the rA-dG TEC. Positions of the starting 15 nt RNA and cleaved transcripts (−1, −2, −3) are indicated at the left. A control reaction with Eco GreA factor shows positions of RNA products resulting from endonucleolytic cleavage. Fe2+ cleavage positions are shown with arrows on the scaffold scheme on the right; the i and i + 1 sites of the active center are indicated.
Effects of TL deletions and GreA factors on RNA cleavage by Eco and Dra RNAPs
To confirm that the TL directly contributes to the differences in RNA cleavage between Eco and Dra RNAPs, we analyzed the effects of TL deletions on the cleavage rates. Deletion of the TL dramatically impaired RNA cleavage by Dra RNAP, but had less severe effect on Eco RNAP (Figure 7B and Table 1). Both deletions more strongly affected the rates of RNA cleavage in the mismatched than in the correct TEC. In particular, the rates for ΔTL Eco and Dra RNAPs were decreased 80- and 3300-fold, respectively, in the rA–dG TEC and 13- and 200-fold in the rA–dT TEC. Presumably, the weaker effect of the TL deletions on RNA cleavage in the latter complex results from superimposition of potentially opposite effects of these deletions on RNA cleavage and RNAP backtracking in the fully matched TEC. As a result, the ΔTL Eco and Dra RNAPs had comparable rates of RNA cleavage in both types of TECs (Table 1).
We then measured RNA cleavage in the presence of GreA cleavage factors, which were shown to substitute the TL in the RNAP active site (Figure 7A) (28). Eco RNAP was more strongly activated by Eco GreA than Dra RNAP by its cognate GreA factor (2300 and 110-fold stimulation, respectively, in the rA–dG TEC; Table 1). As a result, the rates of RNA cleavage by both RNAP became identical in the presence of corresponding GreA factors.
Finally, we analyzed the effects of Eco Gre factors on RNA cleavage by Eco ΔSI3 and Eco-G1136M(ΔSI3) RNAPs. In accordance with published data (15), GreB could not stimulate RNA cleavage by these RNAP variants, likely because the SI3 domain, which is absent in both RNAPs, is essential for GreB binding. In contrast, GreA significantly stimulated RNA cleavage by both RNAPs, indicating that the SI3 domain is not absolutely required for the GreA action (Figure 7C). Notably, the RNA cleavage rates by the Eco ΔSI3 and Eco-G1136M(ΔSI3) RNAPs became identical in the presence of GreA. Overall, these results further suggest that the TL determines differences in RNA cleavage between Eco and Dra RNAPs, which disappear in the absence of the TL or in the presence of Gre factors.
DISCUSSION
RNA cleavage by bacterial RNAP was proposed to play essential roles in transcriptional proofreading and resolution of transcription-replication conflicts in bacterial cells but the detailed mechanism of the cleavage reaction is under debate. Based on comparison of Eco and Dra RNAPs, we made several important observations on the catalytic mechanism of RNAP. In particular, analysis of Dra RNAP helped us to elucidate specific details of RNA cleavage, which is highly efficient in this RNAP, and to identify RNAP elements involved in this reaction. Our findings are briefly discussed below.
Phylogenetically distant Eco and Dra RNAPs revealed similar parameters of nucleotide addition and TEC translocation, suggesting that the basic catalytic mechanism of RNA synthesis is highly conserved in bacteria. Both Eco and Dra RNAPs adopt a predominantly post-translocated conformation following the cycle of nucleotide addition in the TEC used for our analysis. Previous kinetic analyses of nucleotide addition by Eco RNAP suggested that forward translocation of the TEC is likely controlled by the unfolding kinetics of the TL (19,20,48). In particular, it was shown that stabilization of the closed active site with the folded TL by tagetitoxin (20) or amino acid substitutions (49) leads to significantly elevated fractions of the pre-translocated state. Therefore, based on the low occupancy of the pre-translocation state in Dra and Eco TECs we suggest that the closed conformation of the TL during nucleotide addition is not more stable in Dra RNAP than in Eco RNAP. At the same time, Dra RNAP, similarly to extensively characterized Thermus RNAP, differs from Eco RNAP in RNA cleavage and its response to transcription factors, probably as a result of altered TL dynamics (see below; (38,50,51)).
Dra RNAP is highly efficient in RNA cleavage in TECs of various structures, including mismatched complexes formed after nucleotide misincorporation. Recently published data suggest that RNA cleavage by RNAP plays an important role in RNA proofreading in vivo, and any changes in this activity may significantly affect transcriptional mutagenesis. In particular, deletions of Gre factors increased the transcription error rate in Eco (26). Similarly, mutations in Saccharomyces cerevisiae RNAPII located in the TL, BH and in the sites interacting with the RNA cleavage factor TFIIS were shown to increase transcription errors, by decreasing the fidelity of nucleotide incorporation and by impairing TFIIS-dependent transcript cleavage (7). The RNA cleavage activity has also been implicated in reactivation of backtracked TECs during transcription-replication encounters and at DNA damage sites (34,52,53). In particular, DNA lesions were shown to block transcription by S. cerevisiae RNAPII in vitro, and the stalled TECs were susceptible to the action of TFIIS (54–56) suggesting that they are prone to backtracking. Furthermore, RNAP backtracking promoted by the UvrD helicase is an essential step of nucleotide excision repair, followed by RNA cleavage and TEC reactivation (57). We therefore propose that the enhanced RNA cleavage activity of Dra RNAP may be important for RNA proofreading and/or in reactivation of arrested TECs whose formation is likely greatly stimulated by genome damage.
Dra RNAP can efficiently utilize both magnesium and manganese ions for the cleavage reaction, which may be physiologically significant since Dra cells were shown to accumulate manganese under stress conditions (37). The increased cleavage rate of this RNAP is not explained by improved binding of the catalytic ions or stimulation of catalysis by manganese in comparison with Eco RNAP. However, manganese ions do stimulate RNA synthesis and dramatically increase nucleotide misincorporation by Dra RNAP. Similarly, manganese ions were shown to stimulate several Dra enzymes involved in stress resistance, including DNA polymerases I and X whose translesion activity is increased in the presence of Mn2+ (58,59). Previously, Mn2+ was shown to increase nucleotide misincorporation by S. cerevisiae RNAPII, probably by sequestering nucleotide substrates within the active site and stabilizing the folded TL conformation (45). However, nucleotide misincorporation by Dra RNAP is increased even when the reactions are immediately quenched with hydrochloric acid, thus suggesting that Mn2+ may stimulate catalysis independently of the TL folding. The decreased fidelity of Mn2+-dependent RNA synthesis may potentially explain the need for the higher level of the RNA cleavage activity displayed by Dra RNAP.
Species-specific variations in the TL modulate the RNA cleavage activity of bacterial RNAPs. In contrast to previously analyzed thermophilic Taq RNAP (27), the use of mesophilic Dra RNAP allowed its direct comparison with Eco RNAP under identical conditions. We showed that the contribution of the TL to catalysis is different in these RNAPs, as a result of non-conserved amino acid substitutions, thus explaining previous controversies on the role of the TL in RNA cleavage (15,27). A single Dra-specific amino acid substitution in the TL greatly stimulated RNA cleavage by Eco RNAP and may therefore be largely responsible for the highly efficient catalysis in Dra RNAP. The G1136M substitution is located at the C-terminus of the unfolded TL segment that becomes α-helical after TL folding (Figure 1 and Supplementary Figure S1). Substitutions in the C-terminal helix of the TL and adjacent RNAP segment, including E1103G in S. cerevisiae RNAPII (60) and G1136S in Eco RNAP (47) were previously shown to significantly affect RNAP catalysis. In particular, the G1136S substitution was proposed to stimulate RNAP translocation and suppress transcription pausing and termination, likely by stimulating TL folding (46,47). The latter substitution was also shown to increase pH-stimulated RNA hydrolysis by Eco RNAP but this phenomenon has not been studied in detail (47). We propose that the G1136M(ΔSI3) and, probably, G1136S substitutions may affect TL folding and its contacts with the RNA 3′-end during cleavage, for example, by stabilizing a specific TL conformation involved in RNA cleavage. In the backtracked complex structure of Tth RNAP, residue Q1254 in the unfolded TL (corresponds to G1136 in Eco RNAPs) directly contacts the N-terminal part of the TL, in particular, conserved residue R933 (R1239 in Eco RNAP) that has been implicated in nucleotide addition and pausing (14,15) (Supplementary Figure S1A). Phyla-specific substitutions may therefore modulate RNAP catalysis through changes in this and other contacts. Interestingly, analysis of the TL sequences from various bacterial lineages reveals that the presence of a glycine residue at positions corresponding to Eco G1136 correlates with the presence of the SI3 insertion in the TL (Supplementary Figure S9). Furthermore, most RNAPs lacking this insertion contain either glutamine or, less frequently, histidine or methionine residues, similarly to the Thermus/Deinococcus lineage. Since Gre proteins and other secondary channel factors are sensitive to the SI3 presence (15,61), it will be interesting to establish whether these correlations are important for regulation of RNA cleavage in these RNAPs.
Combination of the effects of the G1136M substitution and two other substitutions, I937T in the TL and A455E in the Mg2+-binding site can likely fully explain the large difference in the RNA cleavage rate between Eco and Dra RNAPs. Thus, complex adaptive changes in RNAP properties can result from only a few substitutions in key elements involved in catalysis.
Deletions of the TL impair RNA cleavage by both Eco and Dra RNAPs but the effect is much stronger for the Dra RNAP; as a result, the RNA cleavage rates become comparable for both ΔTL RNAPs. Eco and Dra GreA factors significantly stimulate RNA cleavage by their cognate RNAPs and also eliminate the TL-dependent differences in the rates of the reaction. Eco GreA also activates RNA cleavage by the Eco-G1136M(ΔSI3) RNAP and a control Eco ΔSI3 RNAP to the same level. This agrees with the idea that the TL is not involved in Gre-dependent RNA cleavage (28,29). Overall, the data suggest that the TL plays an important role in intrinsic RNA cleavage in various RNAPs and is likely to determine interspecies variations in this reaction by a direct mechanism, rather than through indirect conformational changes of adjacent RNAP elements.
Contributions of the TL to RNA cleavage likely vary in RNAPs from different domains of life. Intrinsic RNA cleavage by eukaryotic RNAPII from S. cerevisiae is much less efficient in comparison with Eco RNAP (62), and unfolded TL in backtracked RNAPII structures is located far away from the RNA substrate (63,64). Thus, the low efficiency of RNA cleavage by RNAPII may result from the absence of stimulatory effect of the TL on cleavage. Furthermore, the TL was shown to be unimportant for RNA cleavage in archaeal RNAP (65). It is therefore likely that in these RNAPs the reaction mainly depends on the action of accessory cleavage factors, TFIIS and TFS. In contrast, highly efficient intrinsic RNA cleavage by Dra RNAP may serve as an alternative, factor-independent pathway for reactivation of stalled TECs. Multiple partially redundant systems for dealing with genome damage and transcription stalling may provide evolutionary advantage to Deinococcus and other extremophilic bacterial lineages. For example, the presence of multiple DNA repair systems with overlapping specificities is well documented in Dra (35,36). In addition, Dra cells contain two ‘anti-Gre’ Gfh factors which may outcompete GreA under certain conditions (66). Investigation of the in vivo role of RNA cleavage and its regulation in transcription fidelity and stress response in Dra cells is therefore an important goal of future studies.
Supplementary Material
Acknowledgments
We thank I. Artsimovitch for plasmids, Arkady Mustaev and Sergei Borukhov for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Science Foundation [14-14-01074] (analysis of RNA cleavage); Academy of Finland [286205] (analysis of RNA synthesis). Funding for open access charge: Russian Science Foundation [14-14-01074].
Conflict of interest statement. None declared.
REFERENCES
- 1.Gordon A.J., Satory D., Halliday J.A., Herman C. Heritable change caused by transient transcription errors. PLoS Genet. 2013;9:e1003595. doi: 10.1371/journal.pgen.1003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imashimizu M., Oshima T., Lubkowska L., Kashlev M. Direct assessment of transcription fidelity by high-resolution RNA sequencing. Nucleic Acids Res. 2013;41:9090–9104. doi: 10.1093/nar/gkt698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roghanian M., Zenkin N., Yuzenkova Y. Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Res. 2015;43:1529–1536. doi: 10.1093/nar/gkv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satory D., Gordon A.J., Wang M., Halliday J.A., Golding I., Herman C. DksA involvement in transcription fidelity buffers stochastic epigenetic change. Nucleic Acids Res. 2015;43:10190–10199. doi: 10.1093/nar/gkv839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y.N., Lubkowska L., Hui M., Court C., Chen S., Court D.L., Strathern J., Jin D.J., Kashlev M. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. J. Biol. Chem. 2013;288:2700–2710. doi: 10.1074/jbc.M112.429464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gout J.F., Thomas W.K., Smith Z., Okamoto K., Lynch M. Large-scale detection of in vivo transcription errors. Proc. Natl. Acad. Sci. U.S.A. 2013;110:18584–18589. doi: 10.1073/pnas.1309843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irvin J.D., Kireeva M.L., Gotte D.R., Shafer B.K., Huang I., Kashlev M., Strathern J.N. A genetic assay for transcription errors reveals multilayer control of RNA polymerase II fidelity. PLoS Genet. 2014;10:e1004532. doi: 10.1371/journal.pgen.1004532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strathern J., Malagon F., Irvin J., Gotte D., Shafer B., Kireeva M., Lubkowska L., Jin D.J., Kashlev M. The fidelity of transcription: RPB1 (RPO21) mutations that increase transcriptional slippage in S. cerevisiae. J. Biol. Chem. 2013;288:2689–2699. doi: 10.1074/jbc.M112.429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vassylyev D.G., Vassylyeva M.N., Zhang J., Palangat M., Artsimovitch I., Landick R. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 10.Wang D., Bushnell D.A., Westover K.D., Kaplan C.D., Kornberg R.D. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belogurov G.A., Artsimovitch I. Regulation of transcript elongation. Annu. Rev. Microbiol. 2015;69:49–69. doi: 10.1146/annurev-micro-091014-104047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu. Rev. Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sosunov V., Sosunova E., Mustaev A., Bass I., Nikiforov V., Goldfarb A. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 2003;22:2234–2244. doi: 10.1093/emboj/cdg193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuzenkova Y., Bochkareva A., Tadigotla V.R., Roghanian M., Zorov S., Severinov K., Zenkin N. Stepwise mechanism for transcription fidelity. BMC Biol. 2010;8:54. doi: 10.1186/1741-7007-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J., Palangat M., Landick R. Role of the RNA polymerase trigger loop in catalysis and pausing. Nat. Struct. Mol. Biol. 2010;17:99–104. doi: 10.1038/nsmb.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brueckner F., Cramer P. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat. Struct. Mol. Biol. 2008;15:811–818. doi: 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- 17.Miropolskaya N., Esyunina D., Klimasauskas S., Nikiforov V., Artsimovitch I., Kulbachinskiy A. Interplay between the trigger loop and the F loop during RNA polymerase catalysis. Nucleic Acids Res. 2014;42:544–552. doi: 10.1093/nar/gkt877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miropolskaya N., Nikiforov V., Klimasauskas S., Artsimovitch I., Kulbachinskiy A. Modulation of RNA polymerase activity through trigger loop folding. Transcription. 2010;1:89–94. doi: 10.4161/trns.1.2.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feig M., Burton Z.F. RNA polymerase II with open and closed trigger loops: active site dynamics and nucleic acid translocation. Biophys. J. 2010;99:2577–2586. doi: 10.1016/j.bpj.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malinen A.M., Turtola M., Parthiban M., Vainonen L., Johnson M.S., Belogurov G.A. Active site opening and closure control translocation of multisubunit RNA polymerase. Nucleic Acids Res. 2012;40:7442–7451. doi: 10.1093/nar/gks383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orlova M., Newlands J., Das A., Goldfarb A., Borukhov S. Intrinsic transcript cleavage activity of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1995;92:4596–4600. doi: 10.1073/pnas.92.10.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sosunova E., Sosunov V., Epshtein V., Nikiforov V., Mustaev A. Control of transcriptional fidelity by active center tuning as derived from RNA polymerase endonuclease reaction. J. Biol. Chem. 2013;288:6688–6703. doi: 10.1074/jbc.M112.424002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zenkin N., Yuzenkova Y., Severinov K. Transcript-assisted transcriptional proofreading. Science. 2006;313:518–520. doi: 10.1126/science.1127422. [DOI] [PubMed] [Google Scholar]
- 24.Laptenko O., Lee J., Lomakin I., Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sosunova E., Sosunov V., Kozlov M., Nikiforov V., Goldfarb A., Mustaev A. Donation of catalytic residues to RNA polymerase active center by transcription factor Gre. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15469–15474. doi: 10.1073/pnas.2536698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imashimizu M., Takahashi H., Oshima T., McIntosh C., Bubunenko M., Court D.L., Kashlev M. Visualizing translocation dynamics and nascent transcript errors in paused RNA polymerases in vivo. Genome Biol. 2015;16:98. doi: 10.1186/s13059-015-0666-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuzenkova Y., Zenkin N. Central role of the RNA polymerase trigger loop in intrinsic RNA hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10878–10883. doi: 10.1073/pnas.0914424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekine S., Murayama Y., Svetlov V., Nudler E., Yokoyama S. The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Mol. Cell. 2015;57:408–421. doi: 10.1016/j.molcel.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 29.Roghanian M., Yuzenkova Y., Zenkin N. Controlled interplay between trigger loop and Gre factor in the RNA polymerase active centre. Nucleic Acids Res. 2011;39:4352–4359. doi: 10.1093/nar/gkq1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komissarova N., Kashlev M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1755–1760. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nudler E., Mustaev A., Lukhtanov E., Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 32.Pomerantz R.T., O'Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762–766. doi: 10.1038/nature07527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pomerantz R.T., O'Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makarova K.S., Aravind L., Wolf Y.I., Tatusov R.L., Minton K.W., Koonin E.V., Daly M.J. Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol. Mol. Biol. Rev. 2001;65:44–79. doi: 10.1128/MMBR.65.1.44-79.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slade D., Radman M. Oxidative stress resistance in Deinococcus radiodurans. Microbiol. Mol. Biol. Rev. 2011;75:133–191. doi: 10.1128/MMBR.00015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daly M.J., Gaidamakova E.K., Matrosova V.Y., Vasilenko A., Zhai M., Venkateswaran A., Hess M., Omelchenko M.V., Kostandarithes H.M., Makarova K.S., et al. Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science. 2004;306:1025–1028. doi: 10.1126/science.1103185. [DOI] [PubMed] [Google Scholar]
- 38.Pupov D.V., Barinova N.A., Kulbachinskiy A.V. Analysis of RNA cleavage by RNA polymerases from Escherichia coli and Deinococcus radiodurans. Biochemistry (Mosc) 2008;73:725–729. doi: 10.1134/s000629790806014x. [DOI] [PubMed] [Google Scholar]
- 39.Svetlov V., Artsimovitch I. Purification of bacterial RNA polymerase: tools and protocols. Methods Mol. Biol. 2015;1276:13–29. doi: 10.1007/978-1-4939-2392-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulbachinskiy A., Bass I., Bogdanova E., Goldfarb A., Nikiforov V. Cold sensitivity of thermophilic and mesophilic RNA polymerases. J. Bacteriol. 2004;186:7818–7820. doi: 10.1128/JB.186.22.7818-7820.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esyunina D., Klimuk E., Severinov K., Kulbachinskiy A. Distinct pathways of RNA polymerase regulation by a phage-encoded factor. Proc. Natl. Acad. Sci. U.S.A. 2015;112:2017–2022. doi: 10.1073/pnas.1416330112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miropolskaya N., Artsimovitch I., Klimasauskas S., Nikiforov V., Kulbachinskiy A. Allosteric control of catalysis by the F loop of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18942–18947. doi: 10.1073/pnas.0905402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komissarova N., Kireeva M.L., Becker J., Sidorenkov I., Kashlev M. Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods Enzymol. 2003;371:233–251. doi: 10.1016/S0076-6879(03)71017-9. [DOI] [PubMed] [Google Scholar]
- 44.Malinen A.M., Turtola M., Belogurov G.A. Monitoring translocation of multisubunit RNA polymerase along the DNA with fluorescent base analogues. Methods Mol. Biol. 2015;1276:31–51. doi: 10.1007/978-1-4939-2392-2_3. [DOI] [PubMed] [Google Scholar]
- 45.Walmacq C., Kireeva M.L., Irvin J., Nedialkov Y., Lubkowska L., Malagon F., Strathern J.N., Kashlev M. Rpb9 subunit controls transcription fidelity by delaying NTP sequestration in RNA polymerase II. J. Biol. Chem. 2009;284:19601–19612. doi: 10.1074/jbc.M109.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mejia Y.X., Nudler E., Bustamante C. Trigger loop folding determines transcription rate of Escherichia coli's RNA polymerase. Proc.Natl. Acad. Sci. U.S.A. 2015;112:743–748. doi: 10.1073/pnas.1421067112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bar-Nahum G., Epshtein V., Ruckenstein A.E., Rafikov R., Mustaev A., Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 48.Larson M.H., Zhou J., Kaplan C.D., Palangat M., Kornberg R.D., Landick R., Block S.M. Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6555–6560. doi: 10.1073/pnas.1200939109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malinen A.M., Nandymazumdar M., Turtola M., Malmi H., Grocholski T., Artsimovitch I., Belogurov G.A. CBR antimicrobials alter coupling between the bridge helix and the beta subunit in RNA polymerase. Nat. Commun. 2014;5:3408. doi: 10.1038/ncomms4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sevostyanova A., Artsimovitch I. Functional analysis of Thermus thermophilus transcription factor NusG. Nucleic Acids Res. 2010;38:7432–7445. doi: 10.1093/nar/gkq623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minakhin L., Nechaev S., Campbell E.A., Severinov K. Recombinant Thermus aquaticus RNA polymerase, a new tool for structure- based analysis of transcription. J. Bacteriol. 2001;183:71–76. doi: 10.1128/JB.183.1.71-76.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGlynn P., Savery N.J., Dillingham M.S. The conflict between DNA replication and transcription. Mol. Microbiol. 2012;85:12–20. doi: 10.1111/j.1365-2958.2012.08102.x. [DOI] [PubMed] [Google Scholar]
- 53.Nudler E. RNA polymerase backtracking in gene regulation and genome instability. Cell. 2012;149:1438–1445. doi: 10.1016/j.cell.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brueckner F., Hennecke U., Carell T., Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- 55.Damsma G.E., Alt A., Brueckner F., Carell T., Cramer P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat. Struct. Mol. Biol. 2007;14:1127–1133. doi: 10.1038/nsmb1314. [DOI] [PubMed] [Google Scholar]
- 56.Damsma G.E., Cramer P. Molecular basis of transcriptional mutagenesis at 8-oxoguanine. J. Biol. Chem. 2009;284:31658–31663. doi: 10.1074/jbc.M109.022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Epshtein V., Kamarthapu V., McGary K., Svetlov V., Ueberheide B., Proshkin S., Mironov A., Nudler E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinz K., Marx A. Lesion bypass activity of DNA polymerase A from the extremely radioresistant organism Deinococcus radiodurans. J. Biol. Chem. 2007;282:10908–10914. doi: 10.1074/jbc.M611404200. [DOI] [PubMed] [Google Scholar]
- 59.Khairnar N.P., Misra H.S. DNA polymerase X from Deinococcus radiodurans implicated in bacterial tolerance to DNA damage is characterized as a short patch base excision repair polymerase. Microbiology. 2009;155:3005–3014. doi: 10.1099/mic.0.029223-0. [DOI] [PubMed] [Google Scholar]
- 60.Kireeva M.L., Nedialkov Y.A., Cremona G.H., Purtov Y.A., Lubkowska L., Malagon F., Burton Z.F., Strathern J.N., Kashlev M. Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Mol. Cell. 2008;30:557–566. doi: 10.1016/j.molcel.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furman R., Tsodikov O.V., Wolf Y.I., Artsimovitch I. An insertion in the catalytic trigger loop gates the secondary channel of RNA polymerase. J. Mol. Biol. 2013;425:82–93. doi: 10.1016/j.jmb.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen S., Zenkin N. Transcript assisted phosphodiester bond hydrolysis by eukaryotic RNA polymerase II. Transcription. 2013;4:209–212. doi: 10.4161/trns.27062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheung A.C., Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- 64.Wang D., Bushnell D.A., Huang X., Westover K.D., Levitt M., Kornberg R.D. Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fouqueau T., Zeller M.E., Cheung A.C., Cramer P., Thomm M. The RNA polymerase trigger loop functions in all three phases of the transcription cycle. Nucleic Acids Res. 2013;41:7048–7059. doi: 10.1093/nar/gkt433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laptenko O., Kim S.S., Lee J., Starodubtseva M., Cava F., Berenguer J., Kong X.P., Borukhov S. pH-dependent conformational switch activates the inhibitor of transcription elongation. EMBO J. 2006;25:2131–2141. doi: 10.1038/sj.emboj.7601094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.