


Abstract
Next-generation sequencing (NGS) technologies have transformed genomic research and have the potential to revolutionize clinical medicine. However, the background error rates of sequencing instruments and limitations in targeted read coverage have precluded the detection of rare DNA sequence variants by NGS. Here we describe a method, termed CypherSeq, which combines double-stranded barcoding error correction and rolling circle amplification (RCA)-based target enrichment to vastly improve NGS-based rare variant detection. The CypherSeq methodology involves the ligation of sample DNA into circular vectors, which contain double-stranded barcodes for computational error correction and adapters for library preparation and sequencing. CypherSeq is capable of detecting rare mutations genome-wide as well as those within specific target genes via RCA-based enrichment. We demonstrate that CypherSeq is capable of correcting errors incurred during library preparation and sequencing to reproducibly detect mutations down to a frequency of 2.4 × 10−7 per base pair, and report the frequency and spectra of spontaneous and ethyl methanesulfonate-induced mutations across the Saccharomyces cerevisiae genome.
INTRODUCTION
Next-generation sequencing (NGS) is transforming biomedical research, enabling high-throughput and low-cost sequencing of hundreds of billions of DNA base pairs (1). Traditional NGS approaches generate a consensus sequence, which averages the sequence heterogeneity within any given sample. As sequencing has advanced, new techniques have been developed to accurately measure a limited degree of sample heterogeneity (2,3). These techniques have been applied to a myriad of research applications including metagenomic sequencing of gut microbiomes (4,5), tracking in vitro genetic evolution (6,7), and identifying genetic drivers in human diseases (8–10). Efforts have been made to develop NGS-based rare variant detection for new clinical tools such as early cancer diagnosis, identification of optimal therapeutic approaches and monitoring of treatment response (3,11) by discovering oncogenic mutations present in low abundance cell populations. The utility of these applications is dependent on their power to identify mutant populations of smaller and smaller size, particularly in cancer, where genetic heterogeneity is common and minuscule sub-clonal populations can have disproportional effects on clinical outcomes (12). There exist three main barriers that limit NGS-based rare variant detection: (i) the intrinsic error frequency of high-throughput sequencing, (ii) the number of reads a sequencing platform can produce and (iii) the amount of input DNA available.
NGS sequencing is subject to 5 × 10−4 (13,14) to 10−2 (15,16) substitution errors per nucleotide, primarily due to polymerase errors that occur during library preparation, cluster formation and sequencing by synthesis. Variant detection below this frequency remains indistinguishable from experimental error. Many groups have worked to improve the error rate of NGS with both computational (3) and molecular approaches (17–19). The CAPP-Seq system use statistical models to parse error from real variants, which permits a mutation to be detected among a background of 5000 nucleotides or 2 × 10 −4 substitutions errors per nucleotide (3). Computational approaches such as CAPP-Seq are useful for the detection of known variants, which have been independently characterized. To date, the most accurate molecular approaches for error correction are based on DNA barcoding technologies in which each read is assigned a unique identifier and amplified. Multiple copies of each read are then sequenced, and a consensus is created. Errors that are introduced by library preparation polymerase chain reaction (PCR) and instrumentation are eliminated when the consensus sequence is made because they are present in only a fraction of the reads with the same barcode. Utilizing 12–14 base pair single stranded barcodes, the Safe-Sequencing System improves mutation detection down to roughly 10−5 mutations per base pair (17). Several other groups have also described similar molecular barcode-based error-reducing methodologies (19–21). One notable example is the Duplex Sequencing method in which each double-stranded template molecule is tagged with a double-stranded barcode (19). The double stranded barcode tracks copies created from both strands of the original template utilizing family consensus information from both strands to eliminate library preparation- and sequencing-errors and correct for DNA damage sites. The use of double-stranded barcodes permitted the detection of 1 mutation in 4 × 105 wild-type base pairs, though, theoretically, double-stranded barcoding should permit the resolution of <1 mutant base among 109 wild-type nucleotides (19).
The number of reads produced from an NGS instrument is an important factor for rare variant detection. The coverage depth required at a site in order to detect a variant is inversely proportional to its frequency within a sample, requiring ever greater depth to detect rarer variants. For example, detecting a variant in ‘gene X’ present in 1 out of every 105 genomes would require at least 105 coverage of ‘gene X’. 105 reads is not difficult to achieve, however with conventional approaches the rest of the genome, roughly 3 × 109 bp, would also be sequenced at a depth of 105, requiring 2.4 × 1012 (2.4 trillion) 125 bp reads or the equivalent of 1200 HiSeq lanes, which is cost prohibitive. This problem is compounded when combined with error correcting sequencing technologies which, due to the need for redundant barcoded reads, reduce the number of unique reads produced (17–19). As there are practical constraints on the read yield available from current sequencing platforms, detection of extremely rare variants cannot be performed quantitatively for each site genome-wide and must be limited to specific genomic targets of interest. In order to ensure adequate read depth, target sequences must be enriched within the heterogeneous input sample to limit off-target sequence reads.
Two primary forms of enrichment have been widely used: affinity purification (22) and PCR amplification (11). Affinity purification, which relies on hybridization probes to preferentially bind targeted sequences, is the most common enrichment method used in conjunction with NGS and is the basis for whole exome sequencing approaches (22). As with all purification protocols, there is considerable target loss (23). Target loss also limits the sensitivity of variant detection as the detection limit is bounded by the number of target copies present. Many of the most clinically relevant samples such as tissues, sera or biopsies are finite and precious, with only a small sample provided for testing. Loss of DNA during sample preparation or enrichment cannot be compensated for by scaling up inputs and thus further reduces detection sensitivity for rare variants. To avoid high sample loss, PCR amplification of targeted sequences can be used to enrich, as no input copies are lost to wash steps. PCR-based enrichment has been used in conjunction with the Safe-SeqS system to achieve higher depth but at a cost to accuracy (11,17). In that study however, sensitivity was limited to 1 substitution in 104 bases, roughly an order of magnitude lower than previously achieved by the Safe-Sequencing System, because the PCR was performed prior to barcoding, and polymerase mistakes made during amplification become indistinguishable from true variants (Supplementary Table SI3 from ref. (11)). The combination of powerful error correction and limited sample loss will be required to enhance the detection limit further.
Here we present a new NGS-based method, termed CypherSeq, designed to overcome the three main barriers to rare variant detection: (i) error correction, (ii) read depth and (iii) enrichment. CypherSeq employs double-stranded molecular barcoding to achieve high sensitivity basecalling. Additionally, we exploit the circular nature of the plasmid-based sequencing library to enrich for specific targets using rolling circle amplification (RCA) based enrichment to reduce off-target reads and maximize read depth. CypherSeq's combination of accuracy and enrichment will enable the full potential of personalized, sequencing-based clinical applications to be realized.
MATERIALS AND METHODS
CypherSeq design and generation of empty library stocks
To create the CypherSeq library construct, two 195 base PAGE Ultramer DNA oligonucleotides (Integrated DNA Technologies, Coralville, IA, USA) were designed (Supplementary Table SI1). These oligonucleotides contain EcoRI and BamHI restriction enzyme cut sites, Illumina adapter sequences (Nextera v 1.0 from the original Epicentre product literature; Epicentre Biotechnologies, Madison, WI, USA), Illumina identifying indices (N501 and N701 or N702), and two random 7-nt barcodes flanking a SmaI restriction enzyme cut site (Figure 1A). To create a double-stranded product from the single-stranded DNA oligonucleotide, two cycles of PCR were performed using PfuUltra High-Fidelity DNA Polymerase (Agilent Technologies, Santa Clara, CA, USA) and Nextera adapter-specific primers (Supplementary Table SI2) as per the manufacturer's instructions. The following cycling conditions were used: 95°C for 2 min, followed by two cycles of 95°C for 1 min and 64°C for 10 min. The double-stranded nature of the product was verified using a SmaI (New England BioLabs, Ipswich, MA, USA) restriction digest and gel electrophoresis. The double-stranded product was then purified using the Zymo Research DNA Clean & Concentrator-5 kit (Zymo Research, Irvine, CA, USA) and subjected to EcoRI/BamHI restriction digest using BamHI-HF (New England Biolabs) and EcoRI-HF (New England Biolabs) to prepare the construct for ligation into an EcoRI/BamHI-digested pUC19 backbone. Digested vector and construct were run on a 1.5% UltraPure Low-Melting Point Agarose (Invitrogen) electrophoresis gel with 1× SYBR Safe (Life Technologies, Grand Island, NY, USA), and the appropriate bands were manually excised. The DNA in the gel fragments was then purified using a Zymoclean Gel DNA Recovery kit (Zymo Research) and DNA was quantified using a spectrophotometer (Nanophotometer, Implen, Inc., Munich, Germany). Ligation reactions were performed with high-concentration T4 DNA ligase (Life Technologies) with a 1:6 vector to insert ratio, followed by overnight incubation at 16°C. The following day, the ligation reactions were ethanol precipitated and resuspended in water. The purified ligation DNA was electroporated into ElectroMAX DH10B T1 Phage Resistant Cells (Life Technologies). The transformed cells were plated on LB agar media containing 100 μg/ml carbenicillin and grown overnight at 37°C. Colonies were scraped off plates with a glass cell spreader in LB media. DNA was purified using the QIAquick Spin Miniprep Kit (Qiagen, Hilden, Germany) and DNA quantities and qualities were evaluated by spectrophotometry (Nanophotometer, Implen). Barcode diversity was determined by sequencing (Supplementary Figure S1).
Figure 1.
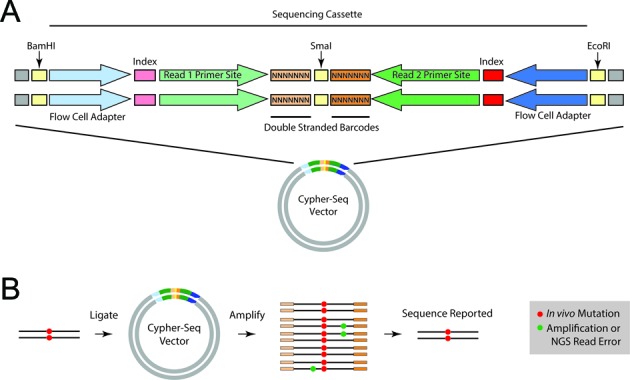
Diagram of CypherSeq construct and sequencing workflow. (A) The vector consists of a pUC19 plasmid backbone into which the sequencing cassette has been ligated. The sequencing cassette consists of the Nextera adaptors (flow cell sequences, indexes and read primer sequences) and two 7-nt double-stranded, random barcodes flanking a blunt-ended restriction site (SmaI). (B) Sheared DNA containing wild-type (black line) or mutated (red) bases are ligated into the sequencing library vector at the Smal site. This ligation can be performed with blunted DNA fragments, or alternatively, A-tailed DNA fragments and T-tailed vectors, each prepared with extended incubation with Taq polymerase and dATP or dTTP, respectively. The resultant sequencing library is amplified to generate a family for each barcode, either via PCR with primers matching the ends of Nextera adaptors or by transformation and growth in bacteria followed by restriction digest to isolate the sequencing cassettes, and then sequenced on an Illumina flow cell. The reads are grouped together using their associated barcodes into barcode families. True mutations (red) will be observed in all or most (>90%) of the barcode family, while mutations arising from PCR-introduced errors or sequencing error (green) will be present in a small fraction of the barcode family (<90%). Thus, the error-corrected sequence is generated by filtering for sites with >90% consensus within each barcode family.
Cell culture
SKOV-3 cells were grown in McCoy's 5a Medium (Life Technologies) supplemented with 10% fetal bovine serum (Hyclone), 1.5 mM L-glutamine, 2200 mg/l sodium bicarbonate and Penicillin/Streptomycin (Hyclone). CaOV3 cells were grown in Dulbecco's Modified Eagle's Medium (Invitrogen) supplemented with 10% fetal bovine serum, 4 mM L-glutamine, 4500 mg/l glucose, 1 mM sodium pyruvate (Hyclone), 1500 mg/l sodium bicarbonate and penicillin-streptomycin.
TP53 exon 5 dilution library preparation
SKOV-3 and CaOV3 cells were harvested and DNA was extracted as directed by the DNeasy Blood and Tissue Kit (Qiagen). Primers were designed to amplify exon 5 of human TP53 (Supplementary Table SI3), and 30 cycles of PCR were performed on SKOV-3 and CaOV3 DNA using 0.5 μM primers and GoTaq Hot Start Colorless Master Mix (Promega) with the following cycling conditions: 95°C for 2 min; 30 cycles of 95°C for 30 s, 63°C for 30 s, 72°C for 1 min; followed by 72°C for 5 min. Each PCR product was then cloned into pCR-4-TOPO vectors (Invitrogen) following the TOPO TA cloning kit's recommendations, transformed into One Shot TOP10 Chemically Competent Escherichia coli cells (Invitrogen), plated on LB agar media containing 100 μg/ml carbenicillin and incubated overnight at 37°C. Ten colonies were picked for each cell type and cultured overnight. The DNA from the overnight LB cultures was purified using the QIAquick Spin Miniprep Kit (Qiagen). Sequencing of the TOPO clones was performed by the Fred Hutchinson Cancer Research Center's Genomics core facility using capillary electrophoresis-based sequencing on an Applied Biosystems 3730xl DNA Analyzer. After sequence analysis, five TOPO clones were selected. One contained wild-type SKOV-3 TP53 exon 5 DNA (named WT), one contained CaOV3 TP53 exon 5 DNA (406 C > T, named MUT1) and three additional clones (MUT2, MUT3 and MUT4) containing distinct TP53 exon 5 mutations (455 C > T, 394 A > G and 431 A > G, respectively), likely introduced by PCR errors. Two TOPO clone DNA mixtures were prepared: (i) the four mutant clones were combined at the appropriate ratios to obtain a 10-fold dilution curve and (ii) the mutant mixture described in (i) was diluted into the wild-type background to render a 10−2 total mutant frequency. Both mixtures were subjected to double enzymatic digestion using AfeI (New England BioLabs) and ScaI-HF (New England BioLabs) and run on a 1.5% UltraPure Low-Melting Point Agarose (Invitrogen) electrophoresis gel with 1× SYBR Safe (Life Technologies, Grand Island, NY, USA) to separate the TP53 exon 5 insert from the TOPO vector backbone. The appropriate bands corresponding to the TP53 exon 5 insert were excised from the gel and purified using the Zymoclean Gel DNA Recovery kit (Zymo Research) and DNA was quantified via spectrophotometry.
In parallel, empty CypherSeq libraries were prepared for insertion of the p53 exon 5. DNA representing 3.6 million different barcoded CypherSeq vectors (as estimated by bacterial colony counts) containing the N701 Illumina index (hereafter named ‘N701-CypherSeq’) was digested with SmaI (New England BioLabs) and treated with Antarctic phosphatase (New England BioLabs) before electrophoresis on a 1.5% low melting-point agarose gel containing 1× SYBR Safe (Life Technologies). The digested vector was then excised from the gel, purified using the Zymo-Clean gel DNA recovery kit (Zymo Research) and quantified by spectrophotometry. The same steps were taken to prepare an estimated 3.8 million different barcoded CypherSeq vectors containing the N702 Illumina index (hereafter named ‘N702-CypherSeq’).
Ligation reactions were set up for each library using high-concentration T4 DNA ligase (Life Technologies) and a 1:6 vector-to-insert ratio for overnight incubation at 16°C. N701-CypherSeq was used in the preparation of the wild-type + 1% mutant library whereas N702-CypherSeq was used in the preparation of the 100% mutant library. The 100% mutant library was sequenced to determine the relative ratios of each mutant within the mutant mix that was diluted 1:100 into the wild-type background. The following day, the ligation reactions were ethanol precipitated and resuspended in water. The purified ligation DNA was electroporated into ElectroMAX DH10B T1 Phage Resistant Cells (Life Technologies). The transformed cells were plated on LB agar media containing 100 μg/ml carbenicillin and grown overnight at 37°C. Colonies were carefully scraped off plates with a glass cell spreader in LB media. Each plate contained ∼300 000 colonies. DNA was purified from each plate separately using the QIAquick Spin Miniprep Kit (Qiagen, Hilden, Germany) and DNA quantities and qualities were evaluated by spectrophotometry. Equal quantities of DNA were combined from three separate minipreps for each index to create N701-p53 and N702-p53 libraries with an estimated 900 000 different barcoded CypherSeq vectors containing p53 exon 5 inserts.
Alternatively, libraries can also be prepared by PCR amplification in place of transformation and bacterial amplification when the vector and DNA inserts are ligated using a T/A cloning method. In this protocol the SmaI digested vectors were not dephosphorylated and instead incubated with Klenow fragment and dTTP for 2 h. The blunted DNA, after inactivation of the blunting enzyme used to repair the sheared DNA ends, is incubated with the Klenow fragment and dATP for 2 h. The vector with T overhangs and DNA fragments with A overhangs are ligated at a ratio of 1:2 with T4 DNA ligase overnight.
5-cycle polymerase chain reactions were then set up using GoTaq Polymerase and primers (Supplementary Table SI4) designed against the Illumina adapter ends to prepare the two libraries for sequencing. The PCR conditions were as follows: 95°C for 2 min, followed by 5 cycles of 95°C for 30 s, 63°C for 30 s and 72°C for 1 min, followed by 72°C for 5 min. The PCR products were run on a 1.5% low melting-point agarose gel containing 1× SYBR Safe (Life Technologies). The cut vector material was then excised from the gel, purified using the Zymoclean Gel DNA Recovery kit (Zymo Research) and the resulting DNA was quantified utilizing the Quantisize methodology as described previously (24) (see Supplementary Table SI5). The samples were then diluted and denatured before loading on the Illumina MiSeq sequencing instrument as per the manufacturer's instructions.
Droplet digital PCR
Samples were prepared for droplet digital PCR (ddPCR) in 25 μl reactions containing 2× ddPCR Master Mix (Bio-Rad), 250 nM TaqMan probe, 900 nM of each of the above primers and ∼10 000 copies of the target DNA. A total of 20 μl of each reaction mixture were added to the sample wells of a droplet generator DG8 cartridge (Bio-Rad) and 70 μl ddPCR Droplet Generation Oil (Bio-Rad) to the oil wells of the cartridge for use in the QX100 Droplet Generator (Bio-Rad) and up to 20 000 emulsified droplets were then generated for each sample. Forty microliters of the generated droplet emulsions were transferred to Twin.tec semi-skirted 96-well PCR plates (Eppendorf, Hamburg, Germany), which were then heat-sealed with pierceable foil sheets using a PX1 PCR plate sealer (Bio-Rad). The reactions were thermally cycled using the following protocol: 95°C for 10 min, followed by 50 cycles of 94°C for 30 s and 60°C for 1 min. The fluorescence of each droplet was measured using the QX100 Droplet Reader (Bio-Rad) as described by the manufacturer. All reactions were performed with at least two replicates to measure experimental error in the assay.
Rolling circle amplification and affinity purification
Genomic DNA from CaOV3 cells was sheared into 150 bp fragments with a Covaris S220 focused ultrasonicator, followed by gel isolation and end repair with a Quick Blunting Kit (NEB). The DNA was then cloned into the CypherSeq vector to generate a library of random CaOV3 genomic fragments. The above-described SKOV-3 p53 exon 5 library (WT) was diluted into a library of CaOV3 genomic DNA to render one p53 copy per 10 000 genomic targets. The DNA was then denatured in preparation for RCA in 20 μl reactions containing 1 μM p53-targeted biotinylated primer (Supplementary Table SI6) and 1× AccuTaq LA PCR Buffer (Sigma-Aldrich). The reactions were heated to 95°C for 4 min then cooled gradually in a step-wise fashion down to 63°C. During the cooling period, 16 μl reactions containing 120 units of Bst 2.0 WarmStart DNA Polymerase (New England BioLabs), 1× Bst 2.0 WarmStart DNA Polymerase master mix, 2 mM dGTP, 2 mM dATP, 2 mM dCTP, 2 mM dTTP and 200 μg/ml BSA were prepared and heated to 63°C before 4 μl of the denatured DNA was added. The RCA reactions were then maintained in a Bio-Rad MyCycler thermal cycler at 63°C for 96 h. Affinity purification of the biotin-labeled RCA products was performed using the Dynabeads kilobaseBINDER Kit (Life Technologies), following the manufacturer's instructions. Prior to quantification, RCA samples were sheared into 1 kb fragments with the Covaris S220 to separate concatemerized copies. p53-containing and total CypherSeq constructs were measured via ddPCR as described above with the p53ex5 (Supplementary Table SI7) and Quantisize (Supplementary Table SI5) ddPCR assays, respectively. Finally, the sample was prepared by limited PCR with the library primers (Supplementary Table SI4), sequenced with an Illumina MiSeq and aligned to the human genome sequence or p53 exon 5 sequence to measure enrichment.
Yeast culture and mutagenesis
Saccharomyces cerevisiae strain S288C was cultured from a single colony in standard YPD media overnight for a total of 28.3 generations (calculated by cell count), washed into water and counted with a hemocytometer. The culture was split into two 5 ml cultures containing ∼1.7 × 108 cells each and incubated at 30°C in the presence or absence of 100 μl ethyl methanesulfonate (EMS) (Sigma-Aldrich), a common laboratory yeast mutagen. After 1 h, both the treated and untreated cultures were pelleted, washed twice with 5% sodium thiosulfate to inactivate any remaining EMS and resuspended in water. The yeast cultures were plated on media containing both YPD and 5-fluoroorotic acid (5-FOA), a selective agent (25). Yeast growth on 5-FOA plates requires a loss of function mutation of the URA3 gene, which can be used to monitor mutation induction after mutagen exposure (25). Mutation induction of EMS was qualitatively confirmed by growth on 5-FOA selective YPD plates (Supplementary Figure S3). The yeast cultures were allowed to recover in YPD for 3 h, roughly two doubling times, to convert the EMS damage into mutation, followed by DNA extraction via the ‘Rather Rapid Genomic Prep’, described previously (26). Finally, the DNA was sheared into 150 bp fragments with a Covaris 220 and prepared as a CypherSeq sequencing library as described above.
Sequencing specific S288C laboratory strain
In order to establish a highly accurate reference sequence for variant calling, the specific strain of S288C used in this study was sequenced using the Nextera XT kit (Illumina). Input DNA was taken from the untreated yeast grown on the same day as the mutagenesis experiment in order to ensure all spontaneous mutations occurring during culture were sampled. Sequencing was performed with the Illumina MiSeq with a 300 cycle MiSeq v2 Reagent Kit (Illumina).
Illumina MiSeq sequencing
Sequencing runs were performed as instructed by the manufacturer's protocol, with the exception that Nextera v. 1.0 sequencing primers (Supplementary Table SI8) were spiked into the sequencing primer mixes contained in the MiSeq sequencing cartridges. A total of 6.8 μl of a 50 uM stock of the Nextera v.1.0 Read 1, Index and Read 2 primers were added to positions 12, 13 and 14, respectively, of the MiSeq cartridge.
Computational analysis
The CypherSeq algorithm for barcode family sequence correction was performed in R using the ShortRead (27) package. About 151 nt, raw sequence reads consisted of the following sequence elements: 7 nt barcode sequence (NNNNNNN) + 3 nt linker sequence (e.g. CCC) + insert sequence (variable length) + (potentially) up to 85 nt adaptor sequence (Figure 1A). Initial processing of raw sequence reads includes family barcode trimming, adaptor trimming and quality filtering (Supplementary Figure S2). First, a family identifier for each read pair was saved, consisting of the barcode and linker sequences plus the first 13 nt of the insert sequence from each read pair. The approach combines the diversity of the molecular barcodes (Supplementary Figure S1A) with the diversity of DNA fragment break-points to reach a theoretical maximum of 4(14 + 13) or 1.8 × 1016 unique identifiers. Reads with Ns anywhere in this family identifier sequence were discarded. The barcode and linker sequences were then removed. In order to recognize the adaptor sequence on the 3′ end of the read for adaptor trimming a minimum overlap of 10 nt at a maximum mismatch rate of 0.05 (i.e. 4 mismatches in 80 nt) is required. Trimmed reads <50 nt were discarded.
Trimmed reads and quality scores were exported into new FASTQ files which were aligned using BWA to the full reference genome, either human or yeast for our experiments (28). The yeast sequences were aligned to the SGD S288C genome (‘sacCer3’), not the strain-specific sequence, to ensure reads mapping to regions with poor coverage in the strain-specific sequence were correctly mapped (29). Following alignment, paired reads were further filtered based on the following criteria: (i) all reads were required to be paired; (ii) if a target locus was specified (i.e. p53 exon 5), both reads in a pair were required to overlap the target locus; (iii) each read in a pair was required to have a minimum aligned sequence length of 50 nt; (iv) no Ns were allowed in either pair; (v) nucleotide positions with a quality score <30 were recorded as missing data; (vi) no more than 20% of the sequence in either pair was allowed to have a quality score lower than 30, or the entire read pair was discarded; and finally, (vii) reads aligning to genomic regions containing low complexity or short-period tandem repeats, as identified by the repeat masking program ‘tantan’, were discarded (30) (Supplementary Figure S2).
Following quality filtering, three bases on the 5′ and 3′ read ends were masked, as substitutions introduced after genomic shearing by end-repair prior to barcode ligation cannot be computationally corrected by family consensus building. Reads were then ‘expanded’ by overlaying the read sequence on the reference using the CIGAR string, allowing family members to align properly in a consensus matrix. Read pairs were next re-associated with their family IDs and sorted into their respective families. Families with fewer than 10 read-pair members were discarded.
Error correction was performed on each family as follows. A consensus matrix of the family was made, and the consensus sequence taken at the 90% level. Positions with <90% consensus were recorded as missing data. Read positions with a family read depth <10 were also encoded as missing data (i.e. if a family consisted of 20 reads [10 read pairs] and 11 reads had missing data at position 5, the family consensus for position 5 was set to missing).
Finally, the global site-specific mutational frequency was calculated by considering a consensus matrix of all family consensus sequences.
For whole genome analysis of the yeast data, a further filter step was performed to remove mutations called due to reads which mapped to large repeats not flagged by the ‘tantan’ program, using the YeastMine tool of the Saccharomyces Genome Database to identify repetitive regions which consisted of long tandom repeats, pseudogenes, retrotransposons, rRNA repeats and transposable gene elements (31). Mutation calls were then made based on the strain-specific reference sequence.
RESULTS
The CypherSeq vector
CypherSeq method is designed to use a circular, double-stranded, dual-barcoded bacterial vector that contains the adapter sequences required for sequencing on Illumina platforms and two 7-nt double-stranded random barcodes flanking a blunt-ended restriction site (SmaI) (Figure 1A). The sample DNA is ligated into CypherSeq vectors at the SmaI site and prepared for sequencing with limited PCR of the sequencing cassette region or bacteria amplification followed by restriction digest to isolate the cassette (Figure 1B). After sequencing, reads with identical barcode pairs are grouped into barcode families and consensus sequences are created for each family, thereby eliminating errors introduced during library preparation and sequencing. Mutations occurring in a minority of reads from a given barcode family are excluded, whereas mutations present in at least 90% of all reads from a given barcode pair and its reverse complement are counted as true (Figure 1B).
Titration of barcode diversity
A unique advantage of the CypherSeq vector is that it was engineered into a plasmid backbone that can be transformed, amplified and maintained in bacteria. As the CypherSeq vector contains a beta-lactamase gene, we were able to transform E. coli with CypherSeq vectors and select for resistant colonies on media containing carbenicillin. To estimate the total number of unique reads or barcodes per experiment, dilutions of the transformed bacteria are plated in parallel and individual colonies are counted. Conveniently, by combining aliquots of CypherSeq DNA preparations with known barcode/read counts, the number of barcodes can easily be titrated to adapt to the requirements of each application. For example, the barcode/read number can be easily reduced to scale an experiment down to a smaller sequencing platform and ensure adequate redundant sequencing for error correction. In addition, libraries with titrated barcode numbers can be stored long-term as DNA preparations or glycerol stocks.
Error correction and rare mutation detection
The sensitivity and specificity with which CypherSeq can identify and recover rare mutants was resolved in reconstruction experiments, where four different p53 mutant sequences were diluted in known ratios into a background of wild-type p53 DNA, cloned into a library of CypherSeq vectors that harbor ∼4 million unique barcodes and prepared for sequencing by PCR. This resulted in sequencing reads that captured a highly diverse array of barcode pairs, with family sizes ranging from 1 to 348 members, and where 287 410 barcodes were represented by 10 or more family members. When target region reads that passed Q30 filters were compared to a wild-type sequence, the substitution frequency averaged 8.9 × 10−4 errors per base pair and ranged from 1.4 × 10−4 to 6.21 × 10−2, thus masking the true concentrations of all spiked-in p53 mutant molecules (Figure 2A). However, when we compiled reads with identical barcodes into families (Figure 1B) and applied the CypherSeq error correction algorithm (Supplementary Figure S2A), no unexpected base substitutions were reported (Figure 2B). Additionally, the four spiked-in mutant molecules were each captured at the expected ratio across several orders of magnitude, with a single mutant gene copy being resolved among more than 105 wild-type copies, or a frequency of 2.4 × 10−7 mutations per base pair (Figure 2C).
Figure 2.
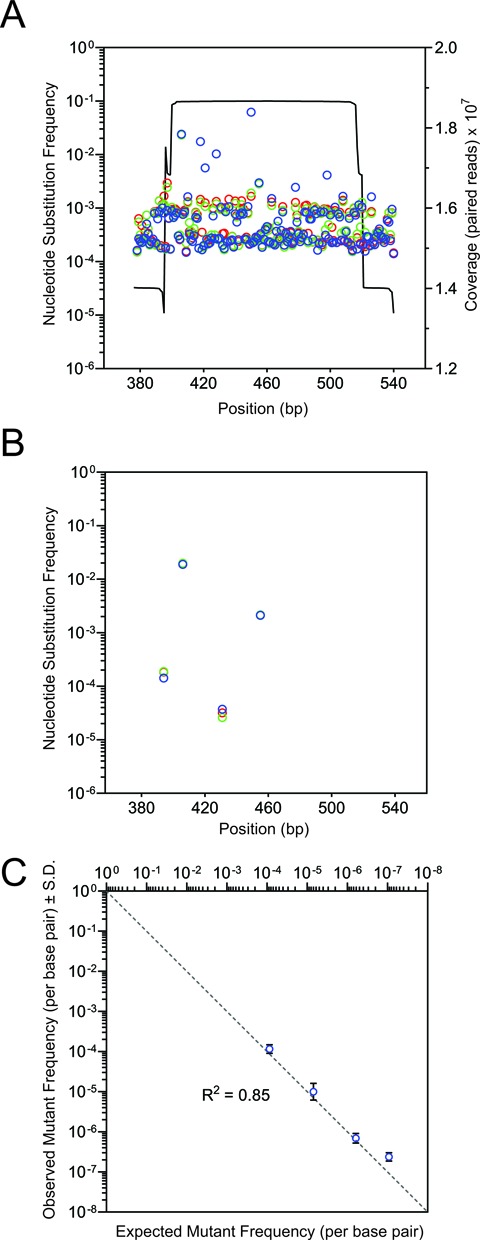
CypherSeq error correction and detection of rare mutations. A CypherSeq library composed of wild-type p53 exon 5 sequences and four mutant sequences spiked in at known ratios (1 in 102, 103, 104, 105) was prepared for sequencing in triplicate (red, blue and green) by limited PCR and sequenced with >107-fold coverage. (A) Base substitutions called in the p53 exon 5 library using a standard Q30 quality filter prevent accurate detection of the known spiked-in mutant molecules due to the high background of false positive mutation calls. (B) Application of the CypherSeq computational workflow eliminated false positive mutation calls and (C) enabled accurate identification of spiked-in mutations.
Genome-wide quantification and characterization of spontaneous and induced mutations
The high sensitivity of the CypherSeq methodology not only permits high coverage targeted resolution of rare variant detection, but also allows for the characterization of genome-wide mutagenesis. To highlight this aspect, we enumerated the frequency of spontaneous and mutagen-induced mutations in S. cerevisiae. Specifically, a liquid culture of S. cerevisiae was established from a single yeast cell, expanded for 16 h, and divided in half. One-half of the cells were treated with EMS, a potent alkylating agent and mutagen, for 1 h, while the remaining half was left untreated. Next, both EMS-treated and untreated cells were allowed to undergo 3 h of continuous growth, followed by plating on 5-FOA restrictive agar plates, which select for a non-functional URA3 gene (32). Cultures that were treated with EMS exhibited a greater frequency of 5-FOA resistant colonies than untreated cells (Supplementary Figure S3), as was expected following the induction of mutation by EMS. To characterize genome-wide mutagenesis, DNA was extracted from the remaining cells, sheared and processed for CypherSeq analysis. We first generated a strain-specific consensus sequence to distinguish mutations from single nucleotide polymorphisms (SNPs) by performing traditional NGS on the untreated culture. Bases that passed a Phred quality score filter of 30 and differed from this strain-specific consensus sequence in our CypherSeq analysis were denoted as de novo mutations. Without the CypherSeq error correction algorithm applied, overall error frequencies of 4.3 × 10−4 and 3.9 × 10−4 mutations per base were measured for untreated and EMS-treated yeast, respectively. Moreover, the spectrum of substitutions called in the untreated and EMS-treated samples runs were similar (Figure 3A). However, when the double-stranded barcode error correction algorithm was applied to the same dataset, the base substitution frequencies decreased to 1.4 × 10−6 and 4.6 × 10−6 mutations per base, for DNA extracted from untreated and EMS-treated yeast, respectively (Figure 3B). Moreover, in accord with previous studies that have demonstrated that EMS treatment nearly exclusively (>99%) results in the induction of G to A transitions (33), our observed increase in mutation frequency in the EMS-treated yeast was almost exclusively the result of a significant increase in the frequency of G to A transitions (P < 1 × 10−15, two-sample test for equality of proportions with Yates continuity correction, n = 26 174 632 and 25 954 910).
Figure 3.
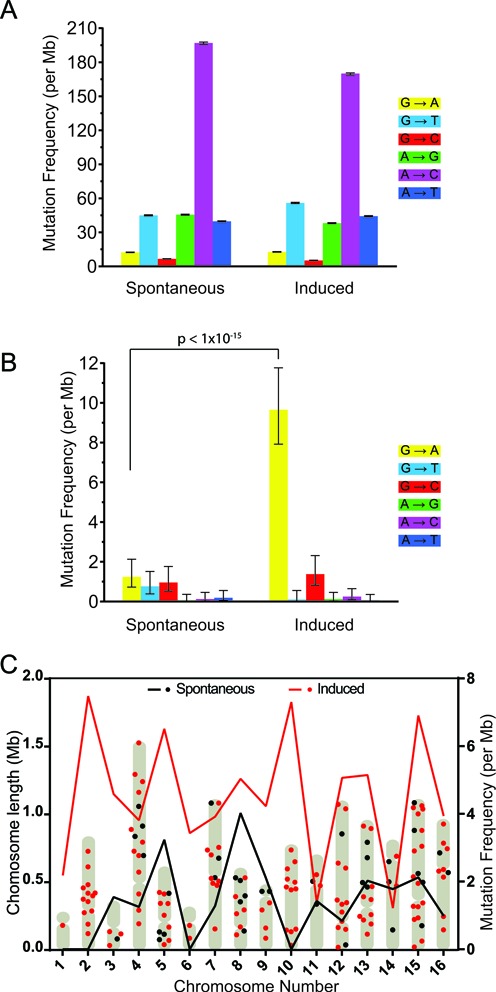
Characterization of spontaneous and induced mutations in Saccharomyces cerevisiae. (A) Whole genome mutational spectrum observed in untreated and EMS-treated yeast using standard Q30 quality filters revealed no significant difference between the samples. (B) CypherSeq error correction removed a large number of false positive mutation calls, and revealed an increase in G:C to A:T transitions in EMS-treated yeast. About 95% confidence intervals were calculated using the Wilson score interval method and p values were calculated using a two-sample test for probabilities of success with continuity correction. (C) The chromosomal position (left axis) and mutational frequency per megabase for each chromosome (right axis) is shown for the untreated (black) and EMS-treated (red) samples across the yeast genome following CypherSeq error correction.
The mutations detected in DNA extracted from untreated and EMS-treated yeast were distributed throughout the genome with an average distance between mutations of 73.0 ± 6.4 kb and 53.1 ± 3.9 kb, respectively (Figure 3C). The number of mutations detected on each chromosome is correlated to the chromosome length in both datasets (P < 0.05, t-test, n = 17), and no chromosomal bias with respect to mutational density was observed (Supplementary Figure S4).
Target-specific enrichment
Error-correction allows unprecedented sensitivity in detecting rare mutations, but to be both robust and financially viable in the enumeration of site-specific mutations high coverage depth restricted to target sequences is required. Thus, in a heterogeneous library, target enrichment is necessary to increase targeted coverage and reduce the number of superfluous off-target DNA sequencing reads. The circular nature of CypherSeq libraries enables target-specific enrichment and amplification via RCA (Figure 4). Briefly, enrichment is performed by annealing a 5′-biotinylated, target-specific primer for replication of the circular vectors containing the targeted region of interest in an isothermal RCA reaction. This creates single-stranded DNA concatemers, each containing multiple copies of the construct and target of interest. To employ double-stranded molecular barcoded error correction, however, RCA must be performed with at least two primers, each of which must target complementary DNA strands (Figure 4). A 5′-biotinylation on the RCA primer allows further enrichment by isolating the RCA product from unamplified templates via streptavidin affinity purification. To test the efficiency of CypherSeq target-specific enrichment, we targeted CypherSeq vectors that harbored sequences of p53 exon 5 in a background of vectors that contained randomly sheared genomic DNA. Prior to amplification, the ratio of p53 target to total genomic library was 1 : 5 × 103 (Figure 5). Following the use of a p53 exon 5-targeted biotinylated RCA primer, the p53 target was amplified and resulted in a 2.1-fold increase over background. Subsequent streptavidin magnetic separation resulted in a 977-fold enrichment (Figure 5) and 17.9% alignment of all the NGS reads to p53.
Figure 4.
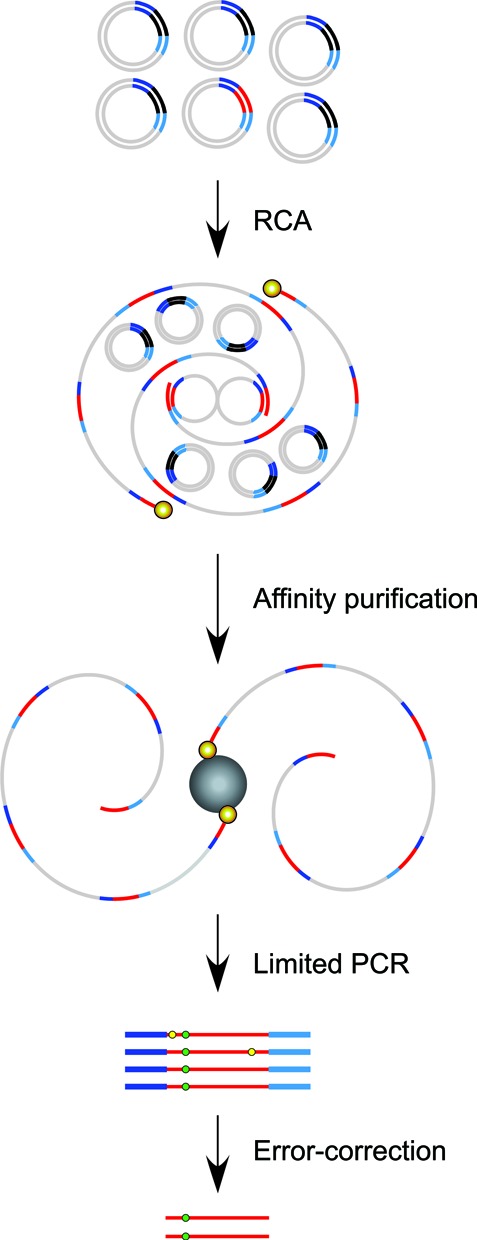
Overview of rolling circle amplification (RCA) enrichment from CypherSeq libraries. A CypherSeq vector library is amplified by extension of biotinylated, target-specific primers using the strand displacement synthesis-proficient polymerase Bst. Two primers, one targeting each of the complementary strands, must be used to achieve double-strand molecular barcoded error correction. Template CypherSeq vectors containing non-target sequences remain unamplified while templates containing the target sequence are amplified via RCA into long single-stranded products containing redundant copies of the target sequence and sequencing cassette. Unlike conventional PCR, each redundant copy of the target sequence is copied directly from the original DNA fragment. Thus, errors occurring in early rounds of amplification are not reproduced in later duplications, preventing exponential amplification of error. The RCA products are purified using magnetic streptavidin-coated beads, subjected to limited PCR with the library preparation primers (Supplementary Table SI4), and sequenced. The error correction methodology is performed identically to samples not subjected to enrichment. Namely, sequencing reads are compiled by barcode and a consensus is made for each barcode family independently. Substitutions occurring in <90% of the reads within a family are rejected as artifacts, while substitutions present in all or nearly all (>90%) of a family are accepted as true mutations.
Figure 5.
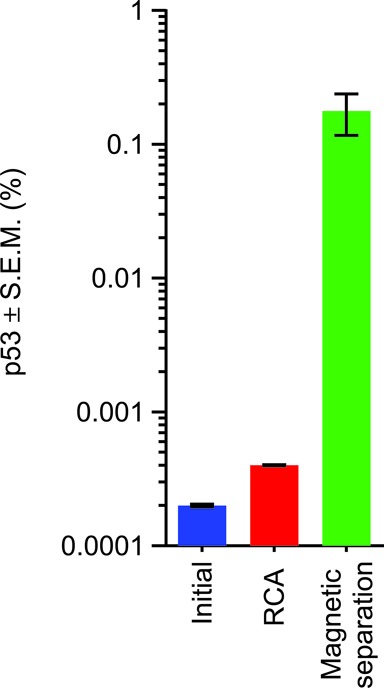
Targeted enrichment of p53 with RCA. A CypherSeq library was generated by ligating sheared ovarian cancer cell line genomic DNA, which also harbored the target gene of interest (p53) in a diluted minority (initial). The library was subjected to RCA with a biotinylated p53-specific primer as described in Figure 4, leading to an increase in the number of p53-specific targets (RCA). Isolation of the RCA products by affinity purification followed by limited PCR (magnetic separation), and subsequent NGS, resulted in 17.9% of all reads mapping to p53.
DISCUSSION
The CypherSeq methodology incorporates the error-correcting capabilities of double-stranded barcodes into a circular construct that carries all the components required for NGS. The sequencing construct is cloned into a bacterial plasmid, and thus permits the replication and storage of the barcoded CypherSeq vectors in bacteria, whereas its circular nature allows for enrichment and amplification of specific targets via RCA. The CypherSeq workflow is compatible across many NGS platforms including the Illumina, Ion Torrent, Pacific Bio, 454 and SMRT systems, and is also capable of large scale multiplexing using conventional indexes.
We demonstrate that CypherSeq corrects errors inherent in NGS sequencing outputs allowing detection of mutations down to a frequency of 2.4 × 10−7 per base pair. However, the sensitivity of the CypherSeq methodology is likely even greater, as double-stranded barcoding-based error correction can theoretically permit the resolution of mutation frequencies as low as 10−9–10−10 per nucleotide (19), and depends upon the number of unique reads generated. Direct comparisons of the sensitivity reported here with those published for other sequencing methodologies are approximate, as the number of rare mutations present in the input DNA were not empirically determined for these other methods prior to sequencing (17–19). In the absence of a reconstruction experiment in which a mixture of DNA sequence variants with known frequencies is used, as performed here, the possibility of false negatives cannot be ascertained. Nevertheless, with the CypherSeq methodology, we demonstrate a level of sensitivity for the detection of rare mutations of at least an order of magnitude greater than has been reported for any other NGS-based method, of which we are aware.
Similarly to other molecular barcode strategies, in order to achieve accurate error correction, multiple copies of the same read must be generated to create barcode families, reducing the coverage attainable from a single sequencing run by a factor of the average family size. However, unlike in traditional consensus generation, which requires high coverage depth from multiple input molecules, the increased accuracy afforded by barcode-based error reduction technologies permits accurate base calls for each read family without redundant coverage. Coverage depth in the CypherSeq context is not reflective of accuracy but rather a measure of the number of genomes sampled at a given location.
As such, it is possible to assess de novo mutation genome-wide, even when read coverage is low (Figure 3). A similar genome-wide assessment of mutagenesis has been reported previously using a single cell sequencing approach (34,35). These methods, however, require lineage expansion via cell proliferation and multiple single cell sequences to be determined in order to sample the population sufficiently and ensure that independent sub-clonal populations are sampled. Moreover, single cell sequencing requires whole genome amplification, which can introduce sequence errors and reduce accuracy. CypherSeq offers a simpler, more accurate method to enumerate sub-clonal mutations in diverse cell populations, without the requirement of cell proliferation, where each read family can be considered as originating from a single individual cell, yielding the same single cell resolution without multiple independent whole genome sequencing experiments.
Translation of robust rare variant detection methods, such as CypherSeq, to the clinic have the potential to dramatically transform disease diagnostics, monitoring and prognostication. Circulating tumor DNA (ctDNA) and circulating tumor cells (CTCs) are detectable in the blood of most patients with advanced cancer and in a significant percentage of patients in the early stages of cancer (36,37). Early cancer diagnosis is currently the most promising approach to reducing mortality, as early detection is associated with more favorable prognosis for nearly all cancer types (36). Reliable detection of early-stage cancer, by quantifying ctDNA or CTCs marked by cancer-specific mutations, will require the most highly sensitive and specific rare variant detection assays to enable screening in a vast background of wild-type normal cells. By exploiting CypherSeq's highly sensitive error correction abilities and by targeting the enrichment step to a panel of genes known to be mutated in cancers, we expect CypherSeq will be able to achieve the sensitivity and specificity required for the early detection of disease.
AVAILABILITY
Data processing code has been made freely available for download from https://github.com/mtgregory.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank the Gottschling laboratory for their kind gift of the S288C S. cerevisiae strain used in this work, Haley BrinJones for technical laboratory support and Chris Warth for assistance with computational algorithm optimization.
FUNDING
Canary Foundation (to J.H.B.); Ellison Medical Foundation New Scholar Award [AG-NS-0577-09 to J.H.B.]; Outstanding New Environmental Scientist Award (R01) from the National Institute of Environmental Health Sciences [R01ES019319 to J.H.B.]; Congressionally Directed Medical Research Programs/U.S. Department of Defense [W81XWH-10-1-0563 to J.H.B.]; Pacific Ovarian Cancer Research Consortium Ovarian Cancer SPORE Award [P50 CA083636]; Scientific Scholar Award from the Marsha Rivkin Center for Ovarian Cancer Research (to J.A.B.); National Institute of Environmental Health Sciences of the National Institutes of Health under award number [F32ES021703 to S.D.T.]. Funding for open access charge: Outstanding New Environmental Scientist Award (R01) from the National Institute of Environmental Health Sciences [R01ES019319].
Conflict of interest statement. None declared.
REFERENCES
- 1.Shendure J., Ji H. Next-generation DNA sequencing. Nat. Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 2.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman A.M., Bratman S.V., To J., Wynne J.F., Eclov N.C., Modlin L.A., Liu C.L., Neal J.W., Wakelee H.A., Merritt R.E., et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S., Gordon J.I., Relman D.A., Fraser-Liggett C.M., Nelson K.E. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlotterer C., Kofler R., Versace E., Tobler R., Franssen S.U. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity. 2014;114:431–440. doi: 10.1038/hdy.2014.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlotterer C., Tobler R., Kofler R., Nolte V. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- 8.Druley T.E., Vallania F.L., Wegner D.J., Varley K.E., Knowles O.L., Bonds J.A., Robison S.W., Doniger S.W., Hamvas A., Cole F.S., et al. Quantification of rare allelic variants from pooled genomic DNA. Nat. Methods. 2009;6:263–265. doi: 10.1038/nmeth.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos E., Levinson B.T., Chasnoff S., Hughes A., Young A.L., Thornton K., Li A., Vallania F.L., Province M., Druley T.E. Population-based rare variant detection via pooled exome or custom hybridization capture with or without individual indexing. BMC Genomics. 2012;13:683–697. doi: 10.1186/1471-2164-13-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Power P.M., Bentley S.D., Parkhill J., Moxon E.R., Hood D.W. Investigations into genome diversity of Haemophilus influenzae using whole genome sequencing of clinical isolates and laboratory transformants. BMC microbiology. 2012;12:273. doi: 10.1186/1471-2180-12-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinde I., Bettegowda C., Wang Y., Wu J., Agrawal N., Shih Ie M., Kurman R., Dao F., Levine D.A., Giuntoli R., et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci. Transl. Med. 2013;5:167ra164. doi: 10.1126/scitranslmed.3004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uramoto H., Tanaka F. Recurrence after surgery in patients with NSCLC. Translational Lung Cancer Res. 2014;3:242–249. doi: 10.3978/j.issn.2218-6751.2013.12.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y., Wu J., Dressman D.C., Iacobuzio-Donahue C., Markowitz S.D., Velculescu V.E., Diaz L.A., Jr, Kinzler K.W., Vogelstein B., Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E., et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nazarian R., Shi H., Wang Q., Kong X., Koya R.C., Lee H., Chen Z., Lee M.K., Attar N., Sazegar H., et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quail M.A., Kozarewa I., Smith F., Scally A., Stephens P.J., Durbin R., Swerdlow H., Turner D.J. A large genome center's improvements to the Illumina sequencing system. Nat. Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinde I., Wu J., Papadopoulos N., Kinzler K.W., Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lou D.I., Hussmann J.A., McBee R.M., Acevedo A., Andino R., Press W.H., Sawyer S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc.Natl. Acad. Sci. U.S.A. 2013;110:19872–19877. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitt M.W., Kennedy S.R., Salk J.J., Fox E.J., Hiatt J.B., Loeb L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casbon J.A., Osborne R.J., Brenner S., Lichtenstein C.P. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Res. 2011;39:e81. doi: 10.1093/nar/gkr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jabara C.B., Jones C.D., Roach J., Anderson J.A., Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mertes F., Elsharawy A., Sauer S., van Helvoort J.M., van der Zaag P.J., Franke A., Nilsson M., Lehrach H., Brookes A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genomics. 2011;10:374–386. doi: 10.1093/bfgp/elr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rykalina V.N., Shadrin A.A., Amstislavskiy V.S., Rogaev E.I., Lehrach H., Borodina T.A. Exome sequencing from nanogram amounts of starting DNA: comparing three approaches. PloS one. 2014;9:e101154. doi: 10.1371/journal.pone.0101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurie M.T., Bertout J.A., Taylor S.D., Burton J.N., Shendure J.A., Bielas J.H. Simultaneous digital quantification and fluorescence-based size characterization of massively parallel sequencing libraries. BioTechniques. 2013;55:61–67. doi: 10.2144/000114063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boeke J.D., Trueheart J., Natsoulis G., Fink G.R. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman C.S., Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 27.Morgan M., Anders S., Lawrence M., Aboyoun P., Pages H., Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engel S.R., Dietrich F.S., Fisk D.G., Binkley G., Balakrishnan R., Costanzo M.C., Dwight S.S., Hitz B.C., Karra K., Nash R.S., et al. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 2014;4:389–398. doi: 10.1534/g3.113.008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frith M.C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2011;39:e23. doi: 10.1093/nar/gkq1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R., et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Greene E.A., Codomo C.A., Taylor N.E., Henikoff J.G., Till B.J., Reynolds S.H., Enns L.C., Burtner C., Johnson J.E., Odden A.R., et al. Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics. 2003;164:731–740. doi: 10.1093/genetics/164.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y., Waters J., Leung M.L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H., et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gundry M., Li W., Maqbool S.B., Vijg J. Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res. 2012;40:2032–2040. doi: 10.1093/nar/gkr949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bettegowda C., Sausen M., Leary R.J., Kinde I., Wang Y., Agrawal N., Bartlett B.R., Wang H., Luber B., Alani R.M., et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Y.F., Yang X.R., Zhou J., Qiu S.J., Fan J., Xu Y. Circulating tumor cells: advances in detection methods, biological issues, and clinical relevance. J. Cancer Res. Clin. Oncol. 2011;137:1151–1173. doi: 10.1007/s00432-011-0988-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.