

Abstract
Background:
Mutations of transthyretin (TTR) cause the most common type of autosomal-dominant hereditary systemic amyloidosis, which occurs worldwide. To date, more and more mutations in the TTR gene have been reported. Some variations in the clinical presentation are often observed in patients with the same mutation or the patients in the same family. The purpose of this study was to find out the clinicopathologic and genetic features of Chinese patients with hereditary TTR amyloidosis.
Methods:
Clinical and necessary examination materials were collected from nine patients of eight families with hereditary TTR amyloidosis at Peking University First Hospital from January 2007 to November 2014. Sural nerve biopsies were taken for eight patients and skin biopsies were taken in the calf/upper arm for two patients, for light and electron microscopy examination. The TTR genes from the nine patients were analyzed.
Results:
The onset age varied from 23 to 68 years. The main manifestations were paresthesia, proximal and/or distal weakness, autonomic dysfunction, cardiomyopathy, vitreous opacity, hearing loss, and glossohypertrophia. Nerve biopsy demonstrated severe loss of myelinated fibers in seven cases and amyloid deposits in three. One patient had skin amyloid deposits which were revealed from electron microscopic examination. Genetic analysis showed six kinds of mutations of TTR gene, including Val30Met, Phe33Leu, Ala36Pro, Val30Ala, Phe33Val, and Glu42Gly in exon 2.
Conclusions:
Since the pathological examinations of sural nerve were negative for amyloid deposition in most patients, the screening for TTR mutations should be performed in all the adult patients, who are clinically suspected with hereditary TTR amyloidosis.
Keywords: Amyloidosis, Autonomic Nervous Dysfunction, Cardiomyopathy, Sensory-motor Neuropathy, Transthyretin
INTRODUCTION
Hereditary transthyretin (TTR) amyloidosis is a life-threatening autosomal-dominant disorder characterized by peripheral neuropathy, autonomic dysfunction, heart and kidney failure, and other symptoms leading to death, usually within 10 years of the onset of disease. Peripheral neuropathy is the most predominant feature. It can be classified as early-onset and late-onset types, on the disease onset, before or after 50 years.[1] Misdiagnosis is frequent in the disease with atypical presentations, including chronic inflammatory demyelinating polyradiculoneuropathy[2] or motor neuron disorder.[3] Amyloid deposits containing mutant TTR are the distinctive pathological characteristics. However, the sural nerve biopsies are negative for amyloid deposits in about half of the cases.[4]
At present, over 100 point mutations in the TTR gene have been identified, in which Val30Met is the most common in many countries.[5] The 5-year survival rate of patients who do not have the Val30Met mutation is significantly lower than that of patients with TTR Val30Met. Liver transplantation is the most effective treatment for the disease, especially for patients with Val30Met mutation. Liver transplantation must be done early in the course of hereditary TTR amyloidosis.[5] Hence, the early diagnosis of the disease is significant.
In China, there were only a few reports of hereditary TTR amyloidosis families.[6,7,8] For most of them, amyloid deposits in different tissues were the initial cues for the consideration of the disease. In this article, we will report nine Chinese patients from eight families with hereditary TTR amyloidosis. The diagnosis is on the screening for TTR mutations.
METHODS
Clinical and necessary examination materials were collected from the nine patients of eight families with hereditary TTR amyloidosis at Peking University First Hospital from January 2007 to November 2014. All the research methods and investigational tools in this study were approved by the Ethics Committee of Peking University First Hospital. All the subjects gave a written informed consent.
The sural nerve biopsies were taken for eight patients and skin biopsies were taken in the calf/upper arm for two patients. Parts of the samples were fixed in 4% formaldehyde, paraffin-embedded, 8-μm sections, and stained with hematoxylin and eosin, congo red, as well as luxol fast blue (not for skin tissues). Other parts of the samples were fixed in 3% glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated through serial alcohol baths, and embedded in Epon 812 (Electron Microscopy Sciences, USA). Semithin sections of sural nerve samples for light microscopy examination were stained with toluidine blue. Ultrathin sections of nerve and skin tissues for electron microscope examination were contrasted with uranyl acetate and lead citrate.
The TTR genes from the nine patients were analyzed. We got 5 ml of peripheral blood and extracted genomic DNA by the method of salt precipitation. Polymerase chain reaction (PCR) primers for exons 1–4 of the TTR gene were designed using Primer 3 software. Standard protocols were applied for PCR. The experimental products were purified and then sequenced using the ABI 3730XL automatic sequencing machine (Applied Biosystems, USA).
RESULTS
Clinical data
The clinical manifestations of nine patients with hereditary TTR amyloidosis are shown in Table 1. There were five males and four females, ranging in age from 25 to 85 years (mean ± standard deviation, 50.1 ± 20.3 years), with the duration of disease ranging 1–20 years. The initial symptoms were pure paresthesia of lower limbs in two patients, pure weakness of lower limbs in two, paresthesia and weakness of all limbs in one, bilateral dysacusis in one, dizziness resulting from orthostatic hypotension in one, alternating diarrhea and constipation in one, and dysarthria owing to glossohypertrophia in one. At the diagnostic time, all the patients had paresthesia except patient 9, among them seven patients were with distal weakness, six patients were with proximal weakness. Dysautonomic manifestations included diarrhea/constipation in six patients, urinary incontinence in one patient, sexual dysfunction in one patient, and orthostatic hypotension in four patients. Cardiomyopathy occurred in four patients. Vitreous opacity occurred in three patients and pupil abnormality occurred in one patient. Sensorineural hearing loss and macroglossia appeared in one patient, respectively.
Table 1.
Clinical data of nine hereditary TTR amyloidosis patients
| Items | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 |
|---|---|---|---|---|---|---|---|---|---|
| Gender/age (years) | Male/72 | Female/52 | Male/54 | Male/29 | Female/49 | Male/25 | Male/45 | Female/85 | Female/75 |
| Initial symptoms | Paresthesia of lower limbs | Paresthesia and weakness in all limbs | Paresthesia of lower limbs | Weakness of lower limbs | Bilateral dysacusis | Dizziness | Weakness of lower limbs | Constipation and diarrhea | Dysarthria |
| Onset age (years) | 68 | 50 | 52 | 28 | 43 | 23 | 41 | 65 | 67 |
| Paresthesia | + | + | + | + | + | + | + | + | − |
| Weakness | + | + | + | + | − | + | + | + | − |
| Cardiomyopathy | − | − | + | − | + | − | + | − | + |
| Diarrhea/constipation | − | + | + | + | − | + | + | + | − |
| hypotension | + | − | − | − | + | + | − | + | − |
| Vitreous opacity | − | − | − | − | + | + | − | − | + |
| Hearing loss | − | − | − | − | + | − | − | − | − |
| Glossohypertrophia | − | − | − | − | − | − | − | − | + |
| Family history | + | + | − | − | + | − | − | − | + |
+: With the symptom or sign; −: Without the symptom or sign; TTR: Transthyretin.
All the patients took electrophysiological tests except patient 9. Sensory nerve conduction velocity (SCV) tests showed reduced amplitude or disappearance of sensory nerve action potential (SNAP) in four patients, slow SCV combined with reduced SNAP in three patients, and only slow SCV of median nerve in patient 5. Motor nerve conduction velocity (MCV) tests showed reduced amplitude of compound muscle action potential (CMAP) in one patient, slow MCV in one patient and slow MCV combined with reduced CMAP in five patients, and normal MCV in one patient (patient 5).
Four patients had family histories. Patients 1 and 2 came from the same family. Patient 5 had a family history of autosomal-dominant inheritance [Figure 1], in which the clinical phenotypes varied largely in different individuals. The proband suffered from mild peripheral neuropathy. Her mother and brother had severe sensory-motor neuropathy. One sister of patient 8 had numbness of four limbs and alternating constipation/diarrhea.
Figure 1.
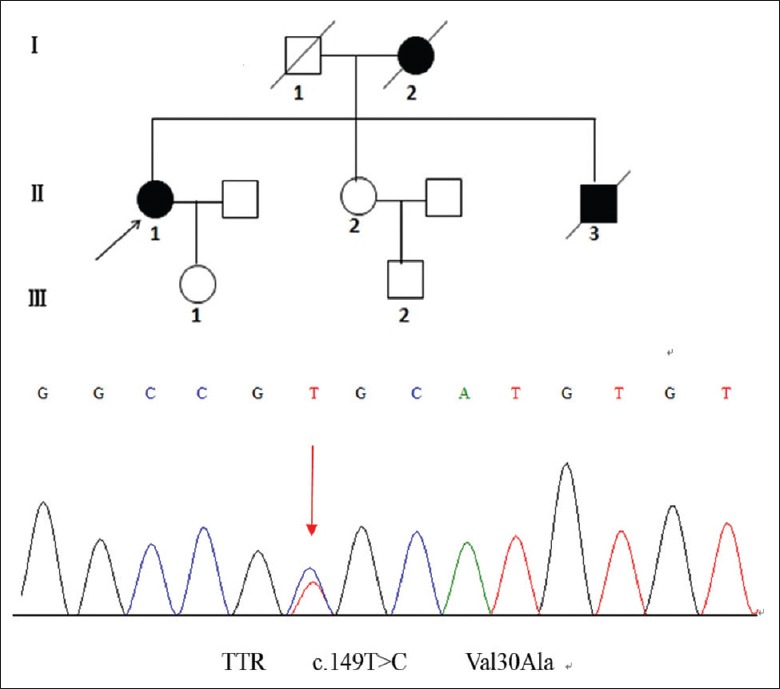
Pedigree of patient 5. The mutation type is Val30Ala. Black arrow indicates the proband. Individual I-2 and II-3 had very severe neuropathy in the early stage of disease and died at ages of 35 and 58, respectively.
Biopsy
The sural nerve biopsies from the eight patients (except patient 9) showed variable pathological changes [Figures 2 and 3]. Seven patients (1, 2, 3, 4, 6, 7, and 8) had severe loss of myelinated fibers, three (1, 2, and 4) of whom had amyloid deposits, while the other patients had no amyloid deposits from light and electron microscopic examinations. Amyloid deposits were present in the perineurium, endoneurium, and endoneurial arterioles in two patients (1 and 4), and only in a single endoneurial arterioles in one patient (2). One patient (5) had nearly normal sural nerve and skin with no amyloid deposits from light microscopic examinations, but with amyloid deposits among the capillary from skin electron microscopic examinations. One patient (9) had no skin amyloid deposits from light and electron microscopic examinations.
Figure 2.

(Patient 1) Congo red stain demonstrates amyloid deposits at the endoneurium (a), apple green birefringence by polarized light (b). Toluidine blue stained sections show a marked loss of myelinated fibers (c) (patient 3) and nearly normal sural nerve (d) (patient 5).
Figure 3.
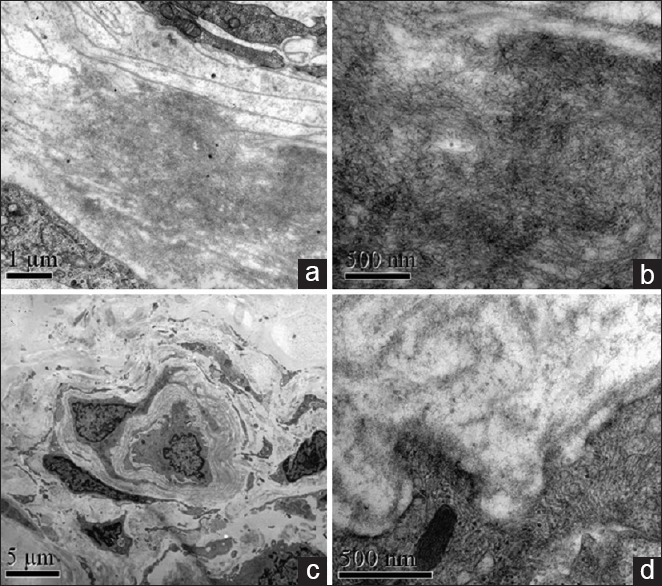
Electron microscopy reveals amyloid deposits around sural nerve capillary (a) and high magnification of amyloid filament (b) in patient 4. Normal control shows no amyloid deposits (c,d).
Genetic analysis
Different missense mutations in the TTR gene were identified in the eight families, including Val30Met (patients 1, 2, 8, and 9), Phe33Leu (patient 3), Ala36Pro (patient 4), Val30Ala (patient 5), Phe33Val (patient 6), and Glu42Gly (patient 7). The most common mutation type Val30Met was found in three families. All the mutations have been reported before.
DISCUSSION
The present series confirmed polyneuropathy with different severity was the common symptoms of hereditary TTR amyloidosis. Both early-onset and late-onset types of the disease can be found in our patients. The initial symptoms were different among them, including paresthesia or weakness of limbs, dizziness, dysacusis, alternating diarrhea and constipation, and dysarthria. Most of them presented with the clinical features of polyneuropathy. The severity varied from mild sensory neuropathy to severe sensory-motor neuropathy. Similar to other reports,[9] autonomic nervous dysfunction occurred in most patients, especially in early-onset hereditary TTR amyloidosis. Hearing loss is a rare manifestation documented occasionally. Local amyloid deposition or nerve compression, ischemia and siderosis due to leptomeningeal, and vascular amyloidosis explain acoustic dysfunction in such cases.[10] The cardiac involvement appeared in 4/9 of our patients. Echocardiography revealed cardiomyopathy in 72% of patients with Val30Met TTR amyloidosis.[11] We found three patients with vitreous opacity. Ocular abnormalities appeared in about 10% of patients with the disease.[5] Some patients showed ophthalmologic symptoms without the signs of polyneuropathy.[4] We also confirmed that macroglossia resulting in dysarthria may occur in the patients with hereditary TTR amyloidosis, though that is more common in patients with light-chain amyloidosis.[12]
Low amplitudes of CMAP and SNAP in presented series indicated extensive axonal impairment, which are very common in hereditary TTR amyloidosis.[13,14] We found a slowing of conduction velocity suggesting demyelination in some patients. The demyelinating feature can not exclude the possibility of hereditary TTR amyloidosis.[15] In our patient with predominant ophthalmologic symptoms and hearing loss, the NCV showed only mild median nerve involvement, indicating mild peripheral neuropathy for some patients with the disease.
The sural biopsy demonstrated severe axonal neuropathy for most patients. About half of our patients did not show amyloid deposits, similar to other reports.[4,13] Negative congo red stain often leads to diagnostic delay or false.[3] We have proved skin biopsy can show amyloid deposits among the capillary in sural nerve negative pathology for amyloidosis. In addition, salivary gland biopsy, abdominal fat aspiration, and muscle biopsy can be used for the detection of amyloid deposits.[16,17]
TTR should be tested in a wide clinical spectrum of progressive neuropathies even for patients with negative pathological findings. In keeping with other reports, all of our patients showed missense point mutations. Endemic areas of hereditary TTR amyloidosis are Portugal, Japan, Sweden, and Brazil.[1] We found six different kinds of missense mutations in the TTR gene in present series, including Val30Met in three families in which all the patients were late-onset ones. In our patients, Val30Met was the most common genotype which was consistent with foreign reports.[5] Val30Ala, Ala97Ser, and Gly83Arg are recognized as the most common amyloidogenic forms in Chinese patients,[18] but in our patients only Val30Ala appeared in one family. The reasons explaining the discrepancy may be that patients with TTR Gly83Arg mutant, indicating solitary ocular vitreous amyloidosis, without the evidence of systemic involvement, often came from the Department of Ophthalmology,[18] while Ala97Ser may be common in Chinese patients.[19] Some mutations indicating special clinical phenotypes such as familial carpal tunnel syndrome (Tyr114His) and predominant cardiomyopathy (Ala45Ser) proved the genotype-phenotype correlation,[20,21] while sometimes the same genotypes show different phenotypes just like the family of patient 5. The fact that there are large differences in penetrance in different populations may explain different family histories of our patients.[5]
Since there are many patients with negative pathological findings of amyloid deposits in the sural biopsy, TTR gene should be tested in a wide clinical spectrum of chronic progressive neuropathies in adulthood, especially with extra-neural features.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Shao Guo
REFERENCES
- 1.Adams D, Lozeron P, Lacroix C. Amyloid neuropathies. Curr Opin Neurol. 2012;25:564–72. doi: 10.1097/WCO.0b013e328357bdf6. [DOI] [PubMed] [Google Scholar]
- 2.Mathis S, Magy L, Diallo L, Boukhris S, Vallat JM. Amyloid neuropathy mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2012;45:26–31. doi: 10.1002/mus.22229. [DOI] [PubMed] [Google Scholar]
- 3.Cappellari M, Cavallaro T, Ferrarini M, Cabrini I, Taioli F, Ferrari S, et al. Variable presentations of TTR-related familial amyloid polyneuropathy in seventeen patients. J Peripher Nerv Syst. 2011;16:119–29. doi: 10.1111/j.1529-8027.2011.00331.x. [DOI] [PubMed] [Google Scholar]
- 4.Luigetti M, Conte A, Del Grande A, Bisogni G, Madia F, Lo Monaco M, et al. TTR-related amyloid neuropathy: Clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol Sci. 2013;34:1057–63. doi: 10.1007/s10072-012-1105-y. [DOI] [PubMed] [Google Scholar]
- 5.Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97. doi: 10.1016/S1474-4422(11)70246-0. [DOI] [PubMed] [Google Scholar]
- 6.Li YF, Ng H, Sun IU, Leong W. Clinical and genetic analysis of three families with familiar amyloid polyneuropathy. Chin Med Sci J. 2008;23:230–3. doi: 10.1016/s1001-9294(09)60044-4. [DOI] [PubMed] [Google Scholar]
- 7.Liu T, Zhang B, Jin X, Wang W, Lee J, Li J, et al. Ophthalmic manifestations in a Chinese family with familial amyloid polyneuropathy due to a TTR Gly83Arg mutation. Eye (Lond) 2014;28:26–33. doi: 10.1038/eye.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang AM, Wang H, Sun P, Hu QX, He Y, Yao YG. Mutation p.G83R in the transthyretin gene is associated with hereditary vitreous amyloidosis in Han Chinese families. Mol Vis. 2013;19:1631–8. [PMC free article] [PubMed] [Google Scholar]
- 9.Shin SC, Robinson-Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79:733–48. doi: 10.1002/msj.21352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lossos A, Soffer D, Steiner-Birmanns B, Hassin-Baer S, Sadeh M, Sagi M, et al. Extended phenotype in the transthyretin Tyr77 familial amyloid polyneuropathy. Eur Neurol. 2005;53:55–9. doi: 10.1159/000084299. [DOI] [PubMed] [Google Scholar]
- 11.Koike H, Tanaka F, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: Analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152–8. doi: 10.1136/jnnp-2011-301299. [DOI] [PubMed] [Google Scholar]
- 12.Elsaman AM, Radwan AR, Akmatov MK, Della Beffa C, Walker A, Mayer CT, et al. Amyloid arthropathy associated with multiple myeloma: A systematic analysis of 101 reported cases. Semin Arthritis Rheum. 2013;43:405–12. doi: 10.1016/j.semarthrit.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Leibou L, Frand J, Sadeh M, Lossos A, Kremer E, Livneh A, et al. Clinical and genetic findings in eight Israeli patients with transthyretin-associated familial amyloid polyneuropathy. Isr Med Assoc J. 2012;14:662–5. [PubMed] [Google Scholar]
- 14.Levy J, Hawkins PN, Rowczenio D, Godfrey T, Stawell R, Zamir E. Familial amyloid polyneuropathy associated with the novel transthyretin variant Arg34Gly. Amyloid. 2012;19:201–3. doi: 10.3109/13506129.2012.724035. [DOI] [PubMed] [Google Scholar]
- 15.Koike H, Kawagashira Y, Iijima M, Yamamoto M, Hattori N, Tanaka F, et al. Electrophysiological features of late-onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J Neurol. 2008;255:1526–33. doi: 10.1007/s00415-008-0962-z. [DOI] [PubMed] [Google Scholar]
- 16.Do Amaral B, Coelho T, Sousa A, Guimarães A. Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid. 2009;16:232–8. doi: 10.3109/13506120903421850. [DOI] [PubMed] [Google Scholar]
- 17.van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54:2015–21. doi: 10.1002/art.21902. [DOI] [PubMed] [Google Scholar]
- 18.Yin J, Xia X, Shi Y, Lu Y, Zhao C, Huang Z, et al. Chinese familial transthyretin amyloidosis with vitreous involvement is associated with the transthyretin mutation Gly83Arg: A case report and literature review. Amyloid. 2014;21:140–2. doi: 10.3109/13506129.2014.892871. [DOI] [PubMed] [Google Scholar]
- 19.Liu YT, Lee YC, Yang CC, Chen ML, Lin KP. Transthyretin Ala97Ser in Chinese-Taiwanese patients with familial amyloid polyneuropathy: Genetic studies and phenotype expression. J Neurol Sci. 2008;267:91–9. doi: 10.1016/j.jns.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Murakami T, Tachibana S, Endo Y, Kawai R, Hara M, Tanase S, et al. Familial carpal tunnel syndrome due to amyloidogenic transthyretin His 114 variant. Neurology. 1994;44:315–8. doi: 10.1212/wnl.44.2.315. [DOI] [PubMed] [Google Scholar]
- 21.Janunger T, Anan I, Holmgren G, Lövheim O, Ohlsson PI, Suhr OB, et al. Heart failure caused by a novel amyloidogenic mutation of the transthyretin gene: ATTR Ala45Ser. Amyloid. 2000;7:137–40. doi: 10.3109/13506120009146252. [DOI] [PubMed] [Google Scholar]