

Abstract
In two manuscripts published in Neuron (Beg et al. and Wegmeyer et al.) and one published in Cell (Iwasato et al.), investigators have found that a particular GAP, α-chimaerin, is required in vivo for ephrinB3/EphA4-dependent motor circuit formation.
The diversity of signaling molecules has long fascinated scientists and suggested the possibility that particular proteins might be required for specific cellular events. However, examples of signal transduction proteins acting only during particular cellular events have been few and far between. Now, three papers provide a beautiful in vivo example of a specific function for a particular signaling molecule. Together, the three manuscripts are striking for the extent that they each describe a similar set of results but use independent approaches to do so: one group began by identifying a spontaneous mouse mutant (Iwasato et al., 2007), a second used a yeast two-hybrid screen (Wegmeyer et al., 2007), and a third used a screen for protein-protein interactions (Beg et al., 2007). While each paper offers its own strong evidence for the role of α-chimaerin in mediating ephrinB3/EphA4-dependent motor circuit formation, together these manuscripts provide a compelling and unusually comprehensive picture of an essential role for a particular GAP.
Small GTPases in the Rac, Rho, and Cdc42 families act as molecular switches in signaling pathways, with the GTP-bound “ON” state and GDP-bound “OFF” state. Regulating the cycle between ON and OFF are over 70 small RhoGTPase-activating proteins (GAPs) in the mammalian genome, with 12 known to have specific activity for Rac. Chimaerins are Rho-GAPs with specific activity for Rac that contain a C1 domain that allows them to bind phorbol esters (Yang and Kazanietz, 2007). There are two α-chimaerins and two β-chimaerins made as alternatively spliced products of two genes. α2- and β2-chimaerin also contain SH2 domains that enable them to bind phosphorylated tyrosine residues. The α-chimaerin GAPs are expressed in brain and linked to axon guidance (Diaz et al., 2002), semaphorin signaling (Brown et al., 2004), dendritic spine development (Buttery et al., 2006), and the NMDAR (Van de Ven et al., 2005). These properties made chimaerin an interesting candidate for regulating axonal growth in the nervous system.
Work from a number of laboratories has identified a specific role of ephrinB3/EphA4 forward signaling in the formation of the motor circuit between neurons in layer 5 of motor cortex and the principle motor neurons in the lumbar spinal cord (Dottori et al., 1998; Coonan et al., 2001; Kullander et al., 2001; Kiehn and Butt, 2003). Mice with mutations to either of these proteins exhibit a hopping rabbit-like gait where both hindlimbs move together—arising from the failure of cortical motor axons to innervate only contralateral motor neurons in the spinal cord and from defects in spinal interneuron axonal projections (Figures 1A and 1C). These defects appear to be due to the failure of these axons to respond to an ephrinB3 repulsive guidance cue along the midline of the spinal cord that allows axons to innervate both left and right motor neurons (Kullander et al., 2001, 2003). Moreover, ephrin-Eph signaling induces growth cone collapse that appears to be mediated via regulation of small GTPase activity (Kullander and Klein, 2002; Cowan et al., 2005; Sahin et al., 2005).
Figure 1. Model for α2-Chimaerin Activity in EphrinB3/EphA4 Forward Signaling.
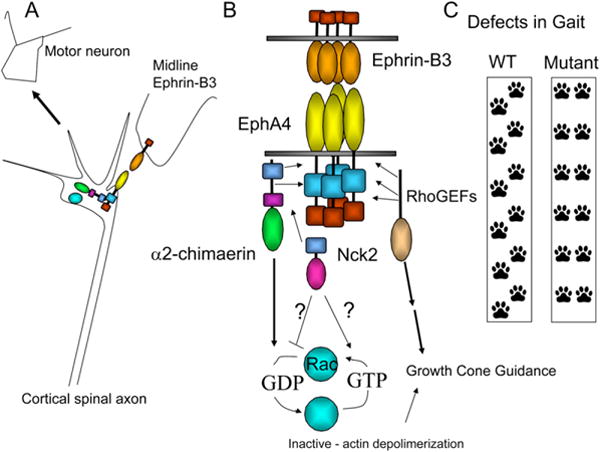
(A) EphrinB3 mediated axon guidance via EphA4 and α2-chimaerin in axonal process of cortical projection neurons.
(B) EphrinB3 binds to EphA4. α2-Chimaerin binds to EphA4, and, in the presence of ephrinB3, its RAC-inactivating activity is increased. Chimaerin’s RAC-inactivation activity is also modulated with Nck2. EphA4s interact with RhoGEFs that may participate in other aspects of growth cone guidance.
(C) Loss of ephrinB3, EphA4, or α-chimaerin leads to defects in gait resulting from axonal projection defects. Interestingly, only defects in guidance due to ephrinB3/EphA4 forward signaling are phenocopied by the loss of α-chimaerin.
In the manuscript by Iwasato et al. from the RIKEN institute (Iwasato et al., 2007), the authors began by noting a rabbit-like gait in a Chat-Cre line of mice that they generated. Thinking that the insertion of their CRE construct was the cause, they were surprised to learn that the mutation segregated from the CRE, indicating that a spontaneous mutation was causing the phenotype. They determined that the gait defect in the miffy mice was likely due to defects in the motor circuit arising from defects in cortical spinal tract projections (CST). This misprojection resulted in an abnormal rhythmic activity of flexors and tensors reminiscent of that seen in mice lacking EphA4. The authors then isolated the mfy locus using microsatellites and single-nucleotide polymorphisms (SNPs) to a 3.27 Mb region on chromosome 2. Of the 30 genes in that region, ten were ruled out because mice lacking these genes were known not to have defects in gait. Fortunately for the authors, only one of the 20 remaining genes generated different sized transcript when amplified with RT-PCR—α-chimaerin. The region affected in the miffy mutation resulted in a loss of 58 amino acids in both α1- and α2-chimaerin and generated proteins that lacked GAP activity in vitro.
To demonstrate that this mutation was the cause of the defects in motor circuit formation, Iwasato et al. then undertook to rescue the expression of α-chimaerin using a bac-transgenic rescue in the miffy−/− mice and attempted to phenocopy their spontaneous mutation by making a targeted deletion of α-chimaerin. Likely due to its low sub-wild-type level of expression, the bac-transgenic gave a partial rescue, but knockout of the locus phenocopied the spontaneous mutation, indicating that miffy is likely due to a mutation in α-chimaerin.
Iwasato et al. then began to examine whether α-chimaerin and EphA4 might interact. Immunocytochemistry revealed that α-chimaerin and EphA4 proteins colocalize in developing CST axons in vitro. Moreover, Iwasato et al. were able to demonstrate that α2-chimaerin and EphA4 coimmunoprecipitate when coexpressed in HEK293T cells, in neurons, and from brain lysates of mice. By making mutations to α2-chimaerin and EphA4, they found in HEK293T cells that the interaction appears to be EphA4 kinase independent and relies on the C-terminal domain of α2-chimaerin. The interaction between EphA4 and α2-chimaerin also appears to be functional: α2-chimaerin can inactivate Rac downstream of kinase-active EphA4 when these proteins are coexpressed in COS cells and then stimulated with a soluble form of ephrinB3 (ephrinB3-Fc). Finally, the authors use an RNAi knockdown approach to demonstrate that motor cortex axons expressing less α2-chimaerin are less sensitive to ephrinB3-dependent growth cone collapse.
The two papers published in Neuron begin with a more traditional approach, with both groups first demonstrating that α2-chimaerin or domains of this protein interact with Ephs. Using a yeast two-hybrid screen with α2-chimaerin SH2, Wegmeyer et al. identified EphA4 and EphB1 as α2-chimaerin-interacting proteins (Wegmeyer et al., 2007). This group then generated two mice with mutations to the α-chimaerin locus: a conventional knockout and a knockin with a point mutation in the C1 domain. The knockin mutation resulted in a loss of α1-chimaerin expression and reduction of α2-chimaerin transcript. Similar to what Iwasato et al. reported, heterozygous animals had no phenotype, and homozygous mice of both genotypes displayed defects in gait. Examination of the CST in both lines of mutant mice revealed increased axonal recrossing and a decrease in the size of the dorsal funiculus. Recording motor output from lumbar dorsal roots revealed a loss of motor coordination that was similar to that seen in the mice generated by the Iwasato et al. group. Neurons from mice lacking α-chimaerin had impaired growth cone collapse following ephrinB3 or ephrinA1 stimulation. Moreover, consistent with the data from all three papers, while loss of α-chimaerins resulted in defects in CST axonal projections, axons that were dependent on EphA4 reverse signaling were unaffected.
Having demonstrated that mice lacking α-chimaerin have defects in the CST projection and neuronal growth cones from these animals fail to respond robustly to ephrin treatment, Wegmeyer et al. next examined how EphA4 might interact with and regulate α-chimaerins. Unlike Iwasato et al., Wegmeyer et al. were not able to coimmunoprecipitate α-chimaerin with EphA4; therefore, they turned to a GST-pulldown assay. Using this assay, Wegmeyer et al. determined that both α1- and α2-chimaerin can bind to EphA4 with a complex series experimental results. α1-Chimaerin appears to bind EphA4 strongly, in the kinase domain, but does not require the kinase to be active. α2-Chimaerin, on the other hand, appears to bind to EphA4 both through its SH2 domain and via a second kinase-independent interaction. Because they interact, the authors then asked whether EphA4 might phosphorylate α-chimaerin. Both isoforms of α-chimaerin are phosphorylated in 293FT cells when coexpressed with EphA4, and a mutation of three different tyrosine residues (y202, y303, y333) on α2-chimaerin reduces its phosphorylation, suggesting that EphA4 phosphorylates these sites. Consistent with a role of EphA-dependent regulation of α2-chimaerin, EGFP-tagged α2-chimaerin overexpressed in neurons becomes phosphorylated following soluble ephrinA1 activation of EphA receptors.
The Wegmeyer et al. study implicates the adaptor proteins Nck1 and Nck2 as potentially important in the ephrin/EphA4/α2-chimaerin pathway by showing that these proteins can interact with full-length α2-chimaerin, but not with α1-chimaerin or mutant forms of α2-chimaerin that lack an SH2 domain. The presence of Nck2 negatively regulates the RacGAP activity of only α2-chimaerin in the presence of EphA4. While interpretation of these results was limited because the authors could not control the expression of each protein, the results demonstrate that EphA4, α2-chimaerin, and Nck2 can act in a concerted fashion on Rac activity.
The third paper, by Beg et al. from Columbia University (Beg et al., 2007), uses affinity chromatography with solublized neuronal membrane proteins on an immobilized recombinant α2-chimaerin SH2 domain and GST-pulldown assays in HEK293T cell lysates to identify a number of phospho-tyrosine proteins as potential α2-chimaerin interactors. One of these interacting molecules was EphA4. The SH2 domain of α2-chimaerin fused to GST was able to pull down EphA4 and EphB1, but not TrkA. The SH2 domain interaction required EphA4 kinase activity and likely occurred via interaction with the juxtamembrane tyrosines. While the authors could not demonstrate that EphA4 and α2-chimaerin coimmunoprecipitated, they did note that coexpression of EphA4 and α2-chimaerin led to α2-chimaerin phosphorylation. In contrast to the experiments from Wegmeyer et al., Beg et al. found that the α2- but not the α1-chimaerin interaction with EphA4 required EphA4 kinase activity.
To examine the in vivo significance of α2-chimaerin, Beg et al. used a gene-trap approach that enabled them to substantially reduce α2-chimaerin expression while leaving α1-chimaerin expression at nearly wild-type levels. The authors first asked whether axons from these mutant animals would still respond to ephrinA5-Fc treatment normally and display growth cone collapse. Consistent with a role of α2-chimaerin alone in EphA-mediated growth cone collapse and reports from Iwasato et al. using siRNA, axons from motor cortex explants of mice lacking α2-chimaerin were significantly less responsive to ephrinA5 than controls.
Beg et al. then demonstrated that the loss of α2-chimaerin generated a phenotype strikingly similar to that seen in animals lacking EphA4 and to the reports by the other two groups examining the α1- and α2-chimaerin mutant animals. Gene-trap α2-chimaerin mice showed defective gait, increased CST axon crossing, and increased crossing of spinal interneurons. Then, in an elegant series of experiments, Beg et al. demonstrated a genetic interaction between EphA4 and α2-chimaerin by showing that double-heterozygous mice (α2-chimaerin+/−, EphA4+/−) also have enhanced spinal interneuron axon crossing compared to the single-heterozygous mutants and similar to that seen in the α2-chimaerin mutant. These experiments provide strong genetic evidence for EphA4 and α2-chimaerin interaction.
Each group conducted a similar series of experiments to demonstrate that the loss of α-chimaerin generates a phenotype similar to that seen with loss or disruption of EphA4. In the end, these three papers paint a compelling picture of a molecule that is essential for a particular aspect of EphA4 receptor signaling (Figure 1). Overall, the data from these three papers fit together nicely, though there are a few places, largely in the biochemical analysis, where there is some disagreement. In most cases, these differences are most likely due to differences in methodology. In any case, the major findings are in agreement. Whether other GAPs and GEFs act so specifically remains to be determined; meanwhile, these papers provide a comprehensive and exciting picture of how a widely expressed signaling molecule can be essential for particular events. There are a number of new questions that are raised by this work. Is axon guidance generated by a balance between GAP and GEF activity? How often do specific GAPs act in particular cells? Do they act sequentially within a signaling cascade? If high levels of specificity do exist, rather than expecting that loss of signaling proteins will generate dramatic phenotypes, we will have to re-examine previously generated models for more specific and subtle phenotypes that point to the essential function of other widely expressed signaling proteins.
References
- Beg AA, Sommer JE, Martin JH, Scheiffele P. Neuron. 2007;55:768–778. doi: 10.1016/j.neuron.2007.07.036. this issue. [DOI] [PubMed] [Google Scholar]
- Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. J Neurosci. 2004;24:8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery P, Beg AA, Chih B, Broder A, Mason CA, Scheiffele P. Proc Natl Acad Sci USA. 2006;103:1924–1929. doi: 10.1073/pnas.0510655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonan JR, Greferath U, Messenger J, Hartley L, Murphy M, Boyd AW, Dottori M, Galea MP, Bartlett PF. J Comp Neurol. 2001;436:248–262. [PubMed] [Google Scholar]
- Cowan CW, Shao YR, Sahin M, Shamah SM, Lin MZ, Greer PL, Gao S, Griffith EC, Brugge JS, Greenberg ME. Neuron. 2005;46:205–217. doi: 10.1016/j.neuron.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Diaz E, Ge Y, Yang YH, Loh KC, Serafini TA, Okazaki Y, Hayashizaki Y, Speed TP, Ngai J, Scheiffele P. Neuron. 2002;36:417–434. doi: 10.1016/s0896-6273(02)01016-4. [DOI] [PubMed] [Google Scholar]
- Dottori M, Hartley L, Galea M, Paxinos G, Polizzotto M, Kilpatrick T, Bartlett PF, Murphy M, Kontgen F, Boyd AW. Proc Natl Acad Sci USA. 1998;95:13248–13253. doi: 10.1073/pnas.95.22.13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Katoh H, Nishimaru H, Ishikawa Y, Inoue H, Saito YM, Ando R, Iwama M, Takahashi R, Negishi M, Itohara S. Cell. 2007;130:742–753. doi: 10.1016/j.cell.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Kiehn O, Butt SJ. Prog Neurobiol. 2003;70:347–361. doi: 10.1016/s0301-0082(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Kullander K, Klein R. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- Kullander K, Croll SD, Zimmer M, Pan L, McClain J, Hughes V, Zabski S, DeChiara TM, Klein R, Yancopoulos GD, Gale NW. Genes Dev. 2001;15:877–888. doi: 10.1101/gad.868901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullander K, Butt SJ, Lebret JM, Lundfald L, Restrepo CE, Rydstrom A, Klein R, Kiehn O. Science. 2003;299:1889–1892. doi: 10.1126/science.1079641. [DOI] [PubMed] [Google Scholar]
- Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O’Connell S, Cowan CW, et al. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Van de Ven TJ, VanDongen HM, Van-Dongen AM. J Neurosci. 2005;25:9488–9496. doi: 10.1523/JNEUROSCI.2450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmeyer H, Egea J, Rabe N, Gezelius H, Filosa A, Enjin A, Varoqueaux F, Deininger K, Schnütgen F, Brose N, et al. Neuron. 2007;55:756–767. doi: 10.1016/j.neuron.2007.07.038. this issue. [DOI] [PubMed] [Google Scholar]
- Yang C, Kazanietz MG. Biochem J. 2007;403:1–12. doi: 10.1042/BJ20061750. [DOI] [PubMed] [Google Scholar]