

Abstract
Changes in RNA secondary structure play fundamental roles in the cellular functions of a growing number of non-coding RNAs. This chapter describes NMR-based approaches for characterizing microsecond-to-millisecond changes in RNA secondary structure that are directed toward short-lived and low-populated species often referred to as “excited states”. Compared to larger-scale changes in RNA secondary structure, transitions towards excited states do not require assistance from chaperones, are often orders of magnitude faster, and are localized to a small number of nearby base pairs in and around non-canonical motifs. Here we describe a procedure for characterizing RNA excited states using off-resonance R1ρ NMR relaxation dispersion utilizing low-to-high spin-lock fields (25–3000 Hz). R1ρ NMR relaxation dispersion experiments are used to measure carbon and nitrogen chemical shifts in base and sugar moieties of the excited state. The chemical shift data is then interpreted with the aid of secondary structure prediction to infer potential excited states that feature alternative secondary structures. Candidate structures are then tested by using mutations, single-atom substitutions, or by changing physiochemical conditions, such as pH and temperature, to either stabilize or destabilize the candidate excited state. The resulting chemical shifts of the mutants or under different physiochemical conditions are then compared to those of the ground and excited state. Application is illustrated with a focus on the transactivation response element (TAR) from the human immune deficiency virus type 1 (HIV-1), which exists in dynamic equilibrium with at least two distinct excited states.
Keywords: NMR, relaxation dispersion, RNA switches, chemical exchange, RNA dynamics, RNA secondary structure, RNA flexibility, HIV, TAR
1. Introduction
Many regulatory RNAs (Storz, 2002; Serganov et al., 2007; Cech et al., 2014) undergo conformational changes that play essential roles in their biological functions (Mandal et al., 2004; Tucker et al., 2005; Schwalbe et al., 2007; Cruz et al., 2009; Rinnenthal et al., 2011; Dethoff, Chugh, et al., 2012). Among many functionally important motional modes, changes in RNA secondary structure can expose or sequester key regulatory elements, and thereby provide the basis for molecular switches that regulate and control a wide range of biochemical processes (Serganov & Patel, 2007; Breaker, 2011; Dethoff, Chugh, et al., 2012).
For example, riboswitches (Grundy et al., 2002; Winkler et al., 2002) are RNA-based genetic elements typically embedded in the 5′ untranslated region (5′ UTR) of bacterial genes that employ changes in secondary structure in order to regulate expression of metabolic genes in response to changes in cellular metabolite concentration (Schwalbe et al., 2007; Serganov & Patel, 2007). In a prototypical metabolite riboswitch (Tucker & Breaker, 2005), a ligand metabolite, such as adenine, binds the aptamer domain and induces a conformational change, which typically sequesters an RNA element into a helix (Fig 1A). The unavailability of this element in turn changes the folding pathway of a downstream decision-making expression platform, directing it towards structures that turn off (and in some cases, on) gene expression, either by forming a transcription terminating helix or by sequestering the Shine-Dalgarno sequence where the ribosome binds, thereby inhibiting translation (Fig 1A) (Haller et al., 2011). Changes in RNA secondary structure can also affect access to RNA sites and thereby allow regulation of post-transcriptional processing, including splicing (Fig 1B) (Cheah et al., 2007), gene silencing by microRNA (miRNA) (Kedde et al., 2010), and RNA editing (Polson et al., 1996). RNA secondary structural switches are also widely used by the RNA genomes of retroviruses to transition between different roles required by various steps of the viral lifecycle (Huthoff et al., 2001; D’Souza et al., 2004). For example, a pH dependent secondary structural RNA equilibrium was recently shown to regulate translational recoding in the murine leukemia virus (Fig 1C) (Houck-Loomis et al., 2011).
Figure 1.
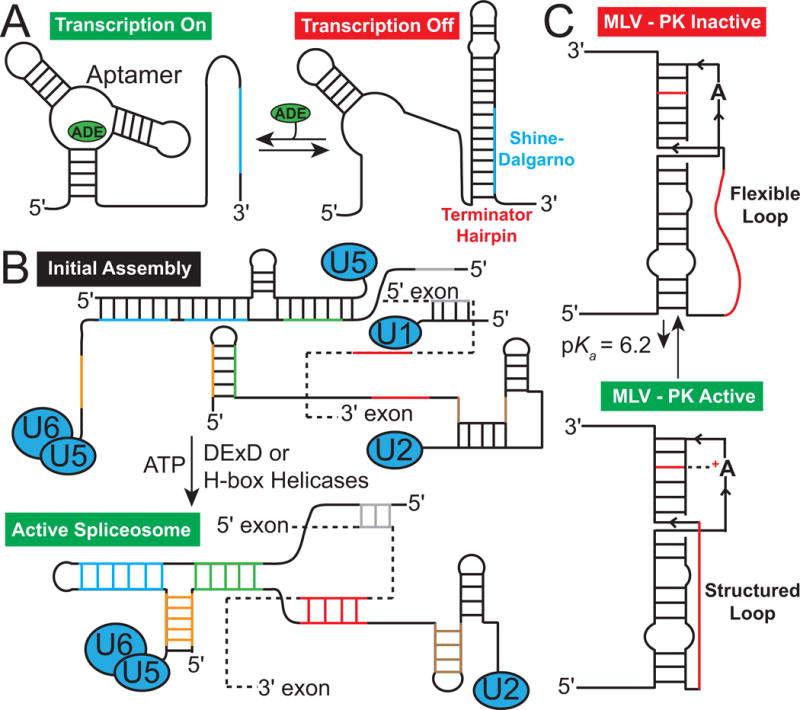
Secondary structural changes in RNA (A) Adenine riboswitch secondary structure with and without the adenine metabolite, which binds to the aptamer domain stabilizing a specific secondary structure turning on transcription. In the absence of metabolite the Shine-Dalgarno sequence (blue) is sequestered turning off transcription (Reining, A. et al., 2013). (B) Secondary structural transitions of mRNA (dashed line) and a spliceosome catalyzed by chaperones (DExD or H-box helicases) that are key for splicing. (C) pH dependent secondary structural equilibrium in the murine leukaemia virus pseudoknot (MLV-PK) mRNA that regulates translational miscoding.
Most RNA secondary structural switches require the melting of several base pairs and result in the creation and disruption of entire helices and hairpins. They can therefore be energetically costly and often require assistance from protein chaperones (Herschlag, 1995; Rajkowitsch et al., 2007) or otherwise must take place during co-transcriptional folding (Garst et al., 2009; Lai et al., 2013). Recently, a new mode of RNA secondary structural transitions has been uncovered through application of solution state NMR spectroscopy (Hoogstraten et al., 2000; Venditti et al., 2009; Dethoff, Petzold, et al., 2012; Lee et al., 2014). These transitions are directed toward low populated (typically <5%) and short-lived (lifetime typically <2 ms) alternative secondary structures that feature local reshuffling of base pairs (bps) in and around non-canonical motifs (Fig 2) (Dethoff, Petzold, et al., 2012; Lee et al., 2014). These transient species are often referred to as “excited states” (ES) (Sekhar et al., 2013) because they represent a higher free energy state (typically destabilized by 1–3 kcal/mol) as compared to the energetically more favorable ground state (GS).
Figure 2.
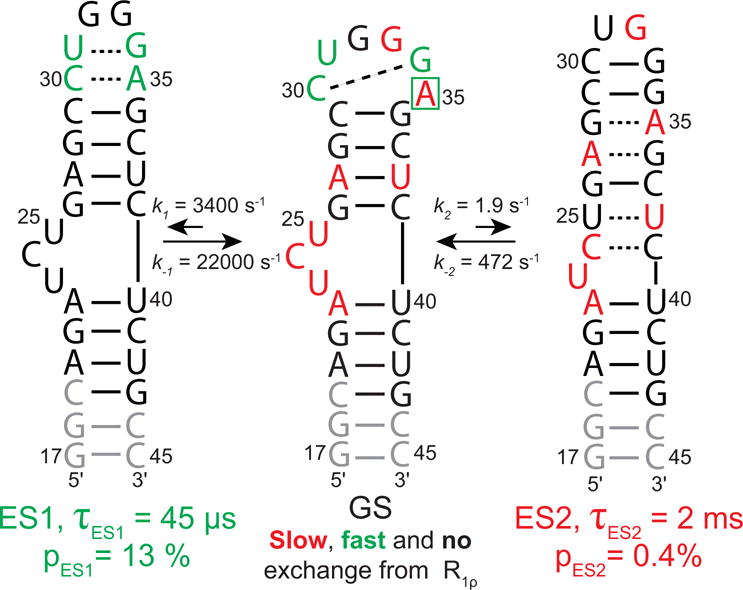
HIV-1 TAR exists in dynamic equilibrium with two excited states (ES1 and ES2). Shown are the ES secondary structure, population (pES), lifetime (τ), as well as the forward (k1/k2) and reverse (k−1/k−2) rate constants. The GS is labeled to show sites with slow (red), fast (green) and no chemical change (black) measured by R1ρ relaxation dispersion. Note that A35 shows a combination of fast (C1′) and slow (C8) exchange. Sites in grey were not measured due to spectral overlap.
An example of such RNA transitions is shown in Figure 2 for the transactivation response element (TAR) RNA from the human immune deficiency virus type 1 (HIV-1) (Weeks et al., 1990; Puglisi et al., 1992; Aboul-ela et al., 1996). TAR has been shown to exist in dynamic equilibrium with two distinct excited states (ES1 and ES2) (Dethoff, Petzold, et al., 2012; Lee et al., 2014). ES1 is 13% populated, has a short lifetime of ~45 μs and features localized changes in base pairing within an apical loop (Fig 2). ES1 sequesters apical loop residues by base pairing interactions that would otherwise be available to interact with proteins (Fig 2) (Dethoff, Petzold, et al., 2012). ES2 is only ~0.4% populated, but has a longer lifetime of ~2 ms (Fig 2) (Lee et al., 2014). It features longer-range reshuffling of base pairs that span the bulge and apical loop that are separated by a four bp helix, remodeling all non-canonical features of the TAR structure (Fig 2) (Lee et al., 2014).
Recent studies suggest that transitions toward ESs are common in many regulatory RNAs (Hoogstraten et al., 2000; Venditti et al., 2009; Zhang et al., 2011; Dethoff, Petzold, et al., 2012; Lee et al., 2014; Tian et al., 2014; Zhao et al., 2014). Compared to conventional secondary structural rearrangements, transitions towards RNA ESs are orders of magnitude faster, occur without assistance from external co-factors, and result in smaller, yet significant, changes in RNA secondary structure, which can include changes in base protonation state (Lee et al., 2014). They can therefore meet unique demands for RNA biological functions (Lee et al., 2014). In addition to playing new roles in RNA-based regulation, the unique features of RNA ESs make them potentially attractive targets for the development of RNA-targeting therapeutics (Lee et al., 2014). Here, we review NMR methods that can be used to characterize structural and energetic properties (population and lifetime) of RNA ESs.
2. NMR Relaxation Dispersion
2.1 Chemical Exchange
NMR relaxation dispersion (RD) experiments, which probe line-broadening contributions to NMR resonances arising from chemical exchange, can be used to characterize ESs of biomolecules. Several excellent reviews (Palmer et al., 2001; Palmer et al., 2006; Korzhnev et al., 2008; Bothe, Nikolova, et al., 2011; Sekhar & Kay, 2013; Palmer, 2014) describe the theoretical underpinnings of RD NMR techniques. They are briefly reviewed here to make the basic concepts accessible to the general reader. Readers are referred to the above reviews for a more rigorous treatment of these experiments.
The key NMR interaction underlying the RD experiments is the chemical shift. Nuclei behave as tiny magnets and due to quantization of the nuclear spin angular momentum, they align parallel (α state) or anti-parallel (β state) relative to the static NMR magnetic field (B0). Since the parallel alignment is more energetically favorable (for nuclei with a positive gyromagnetic ratio), a net bulk magnetization over an ensemble of spins build up parallel to the magnetic field. In a basic 1D NMR experiment, radiofrequency (RF) pulses are used to realign this bulk magnetization along a direction perpendicular to the B0 field. The bulk magnetization then precesses about the B0 field at a characteristic resonance Larmor frequency and gives rise to a detectable oscillating magnetic field. This time-domain signal is then Fourier transformed to yield the standard frequency-domain NMR spectrum, in which unique signals at characteristic frequencies are observed for various spins for a given nuclei. These frequencies are referenced against a standard frequency (e.g. tetramethylsilane for 1H) at that given B0 field and this field-independent parameter is called the “chemical shift”.
The chemical shift is directly proportional to the energy gap between the a and b states, which in turn is proportional to the magnetic field strength experienced by the nucleus. Because electronic clouds surrounding nuclei “shield” or “deshield” the nucleus from the external magnetic field by variable amounts that are highly dependent on the specific electronic environment, a wide range of chemical shifts are typically observed for different sites in a molecule. This makes the NMR chemical shifts exquisitely sensitive to structure (e.g. torsion angles, sugar pucker, etc.), protonation state, and interactions (hydrogen bonding, electrostatic interactions, etc.). For nucleic acid applications, one is typically interested in the NMR active nuclei 1H, 13C, 15N, 2H, and 31P, with 13C, 15N, and 2H introduced during synthesis, typically by using labeled nucleotide triphosphates (NTPs) in in vitro transcription reactions.
To understand how exchange between a GS and ES gives rise to line-broadening of the NMR resonance, consider an RNA molecule exchanging between two states; a major GS, in which a base is flipped in, and a minor ES, in which the base is flipped out (Fig 3A). Nuclei belonging to this base (along with its immediate neighbors) will experience different electronic environments before and after the flip, therefore will be associated with distinct NMR chemical shifts, ωGS and ωES (Fig 3B). In the absence of exchange between the GS and ES, two NMR peaks are observed centered around ωGS and ωES with integrated volumes reflecting the relative populations of the GS and ES (Fig 3B). However, when the GS and ES exchange at rates (kex = k1 + k−1) comparable to their NMR frequency difference (Δω = ωGS−ωES), the chemical shift of a given nucleus fluctuates back and forth between ωGS and ωES. Because the fluctuation is stochastic, nuclei in different molecules spend varying amounts of time in the GS and ES, and therefore spend differential amounts of times with ωGS and ωES frequencies. This results in the broadening of the NMR signal, reflecting a wide range of apparent frequencies due to variable admixing of ωGS and ωES (Fig 3B). Because of its lower starting population and signal intensity, this broadening often puts the minor ES outside NMR detection limits (Fig 3B).
Figure 3.
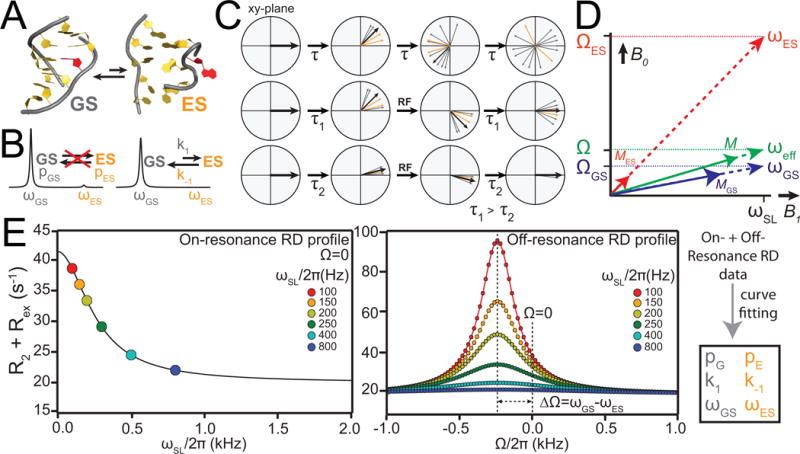
Characterizing chemical exchange using NMR relaxation dispersion. (A) Example of an equilibrium between a GS and ES. (B) Chemical exchange between GS and ES leads to broadening of resonances and disappearance of minor ES signal. (C) Bulk magnetization aligned along the Y-axis of the lab frame, followed by magnetization dephasing due to transverse relaxation. The middle and lower panel show the dephasing of bulk magnetization due to chemical exchange suppressed by application of RF fields for long (τ1) and short (τ2) delays in the CPMG experiment. (D) Resonance offset (Ω), effective field (ω) and magnetization (M) vectors for the GS and ES in R1ρ experiment (denoted by the subscripts GS and ES, respectively). ωeff, Ω and M represent the effective field, average offset and magnetization, (M), respectively. (E) Simulated examples of on-resonance (left) and off-resonance (right) RD profiles showing the dependence R1ρ on spin-lock power ωSL and offset Ω.
It is instructive to conceptualize chemical exchange in terms of magnetization precession (Fig 3C). Here, nuclei in the sample are associated with a bulk “magnetization” vector that is initially aligned along an axis in the laboratory frame by a “preparation” pulse. The individual magnetization vectors from nuclei in different molecules are initially co-aligned along the Y- (or X-) axis via phase coherence of the nuclear spins. The magnetization vector then precesses about the B0 field (Z-axis) with the frequency ω (Fig 3C). In the absence of chemical exchange, two species precess at their respective chemical shift frequencies, ωGS and ωES. However, in the presence of exchange, the precessing frequency of a given magnetization vector varies stochastically between ωGS and ωES. Since nuclei from different molecules spend varying amounts of time precessing with frequencies ωGS and ωES, they no longer precess in synchrony. Rather, they “fan out” and cause phase decoherence or “dephasing” (Fig 3C). This dephasing leads to an additional exchange contribution referred to as Rex to the observed transverse relaxation rate R2,obs = R2+Rex describing the rate at which the Y (or X) component of the magnetization vector decays over time. The NMR line width is directly proportional to R2+Rex, and exchange manifests as a line-broadening contribution.
2.2 Relaxation Dispersion Experiments
RD NMR experiments can be used to characterize properties of the often invisible ES by measuring the exchange broadening contribution to the visible GS resonance (reviewed in (Palmer et al., 2001; Palmer & Massi, 2006; Korzhnev & Kay, 2008; Bothe, Nikolova, et al., 2011; Palmer, 2014)). In particular, experiments are used to measure the chemical exchange contribution to line-broadening (Rex) during a relaxation period (Trelax) during which the sample is subjected to radiofrequency frequency (RF) irradiation. Various types of RD experiments differ in the initial state of the magnetization and the type of RF irradiation that is used.
In the Carr-Purcell-Meiboom-Gill (CPMG) experiment (Carr et al., 1954; Meiboom et al., 1958; Loria et al., 1999a; F. A. Mulder et al., 2001), the initial state generally consists of transverse magnetization (e.g. aligned along the Y-axis) and the RF irradiation consists of a series of equally spaced high-power 180° refocusing pulses. In the case of the spin relaxation in the rotating frame experiment, or R1ρ (Deverell et al., 1970; Akke et al., 1996; F. A. A. Mulder et al., 1998; Korzhnev et al., 2002), the initial magnetization is typically aligned along an effective field direction (which is defined by both the RF irradiation power and offset) and the irradiation consists of a weaker, but continuous RF with a specified power level (ωSL) and frequency or offset (Ω) (Fig 3D).
The RF field employed in this case is called a “spin-lock” (SL), as it locks the magnetization along its effective field. In both the CPMG and R1ρ experiments, the RF irradiation perturbs the precession of magnetization so as to diminish the efficiency with which chemical exchange results in dephasing of the magnetization, and therefore exchange broadening. For example, in the CPMG experiment, the series of 180° pulses effectively “invert” the precession of magnetization at a constant time interval (τCPMG); in this manner, some of the dephasing occurring prior to the 180° pulse is refocused in the period following the pulse, with the degree of refocusing increasing with shorter τCPMG delays (Fig 3C). In the case of the R1ρ experiment, the two effective field directions associated with the GS and ES are brought into closer alignment by application of a continuous RF field (Fig 3D), thereby decreasing the extent of dephasing arising due to precession around the GS and ES effective fields. The dependence of the exchange broadening contribution (Rex) on τCPMG in the case of CPMG and ωSL, as well as Ω in the case of R1ρ (Fig 3E), can be used to extract exchange parameters of interest. For slow (kex << |Δω| to intermediate (kex~|Δω|) exchange, these experiments can be used to determine the population (pES), lifetime (τES = 1/[(1−pES)kex]), and chemical shift of the ES (ωES). For fast exchange (kex>>|Δω|), it becomes more difficult to reliably determine exchange parameters though one can typically accurately measure the factor Φ = pGSpESΔω2 where additional experiments are needed to resolve the ES chemical shift and population (Bothe et al., 2014). It is important to note the chemical shift of the ES carries the desired structural information.
CPMG relaxation dispersion experiments can be used to characterize processes with exchange rates (kex = k1+k−1) in the range of ~10 s−1 < kex < ~6000 s−1 (Palmer et al., 2001; Korzhnev & Kay, 2008). CPMG data is typically measured at multiple magnetic field strengths and combined with additional experiments (e.g. HSQC/HMQC) in order to determine the sign of the excited state chemical shift (Skrynnikov et al., 2002). Although widely used in studies of proteins, the CPMG experiment proves difficult to apply to nucleic acids. This is due to a paucity of ideal imino nitrogens, and because 13C experiments are complicated by extensive C–C interactions that are difficult to suppress due to challenges in achieving selective carbon excitation with hard pulses (Yamazaki et al., 1994; Johnson et al., 2008). The exchange timescale accessible to R1ρ is broader than CPMG (~60 s−1 < kex < ~100,000 s−1) (Palmer & Massi, 2006) and for slow-intermediate exchange, the sign of excited state chemical shift sign can deduced at a single magnetic field strength (Trott et al., 2002). For processes occurring at even slower timescales (~20 s−1 < kex < ~300 s−1) chemical-exchange saturation transfer (CEST) experiments employing weak RF spin-lock fields have recently been shown to be a robust approach to characterize lowly populated conformational states in both proteins and nucleic acids (Fawzi et al., 2011; Vallurupalli et al., 2012; Long et al., 2014; Zhao et al., 2014).
2.3 R1ρ with Low-to-High Spin-Lock Fields
The application of low ωSL is required in order to characterize ESs with exchange rates slower than kex < ~2,000 s−1. However use of lower ωSL (<1000 Hz) in the conventional 2D R1ρ experiment is complicated due to the fact that one has many spins with a broad range of chemical shift frequencies, each with a distinct effective field (Fig 3D) that have to be aligned along their individual effective fields during the preparation phase of the experiment. For relatively high ωSL (>1000 Hz), there is a limited range of effective field orientations for the various spins, and it is possible to align the magnetization efficiently using adiabatic ramps, which are generally 3–4 ms long (Zinn-Justin et al., 1997; F. A. A. Mulder et al., 1998; Kim et al., 2004; Massi et al., 2004; Igumenova et al., 2006; Hansen et al., 2007). However, for low spin-lock fields (<1000 Hz), one has a broader range of orientations, and proper alignment of the magnetization requires longer delays of the adiabatic ramps, resulting in severe sensitivity loss due to relaxation (Kim & Baum, 2004; Massi et al., 2004; Palmer & Massi, 2006). For a spin-lock field of 275 Hz, the alignment of initial magnetization is efficient only for offset values >325 Hz (Kim & Baum, 2004).
Recent advances in the 15N R1ρ experiment introduced by the groups of Palmer and Kay address these limitations and allow use of much lower spin-lock fields (Massi et al., 2004; Korzhnev et al., 2005), on the order of 25–150 Hz, thus extending sensitivity to exchange timescales on the order of tens of milliseconds comparable to those accessible by CPMG. These advances have been integrated into an NMR experiment for measuring 13C (Hansen et al., 2009) and 15N (Nikolova et al., 2012) R1ρ in nucleic acids (Fig 4). These experiments have so far been applied in the characterization of systems in fast to intermediate exchange in both proteins and nucleic acids (kex ~ 300 – 30000Hz, Δω ~ 20 – 800Hz), and will be discussed in this chapter in the context of characterizing RNA ESs.
Figure 4.
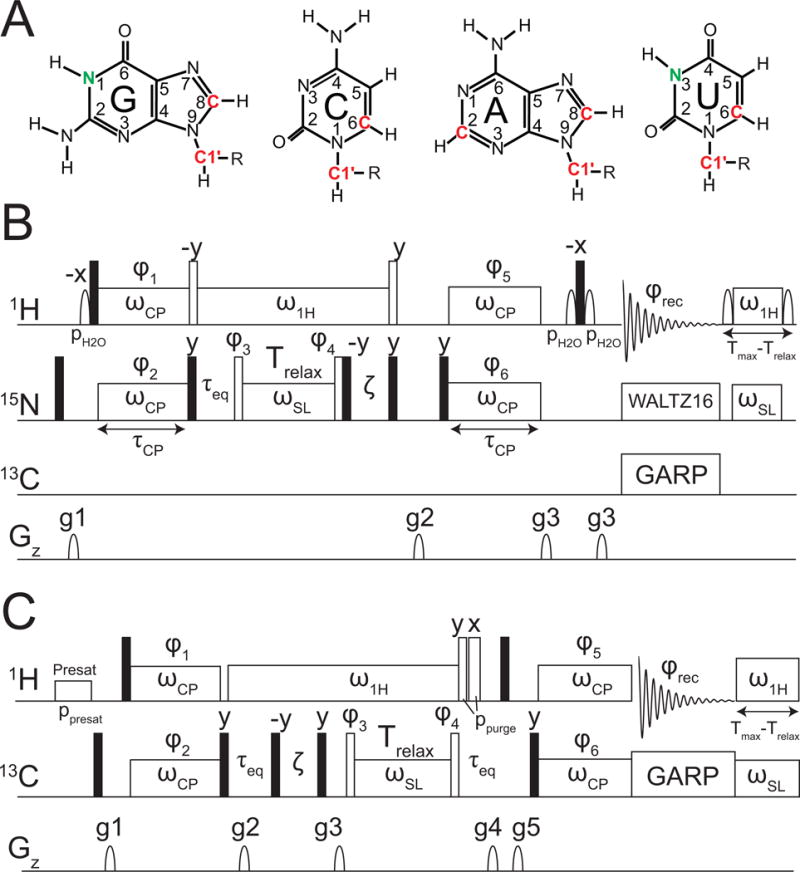
Pulse sequences for the measurement of 15N and 13C R1ρ in uniformly 13C/15N labeled nucleic acids (A) The carbon and nitrogen nuclei that are targeted in RNA for R1ρ measurements are highlighted. Pulse sequences for 1D (B) 15N (Nikolova, E. N., et al., 2012) and (C) 13C (Hansen, A. L., et al., 2009) R1ρ experiments are as shown.
The basic experiment uses selective Hartmann-Hahn polarization transfers developed by Bodenhausen (Pelupessy et al., 1999; Ferrage et al., 2004) to excite specific spins of interest and collect data in a 1D manner. Both the GS and ES magnetization is aligned along the average effective field (ωeff, Fig 3D) through application of a hard 90° pulse (Fig 4B,C) (Bothe et al., 2014). Upon application of the spin-lock, magnetization in states GS and ES will evolve about their respective effective fields ωGS and ωES (Fig 3D). In cases of highly asymmetric exchange (pGS >> pES) the effective fields of the GS and ωeff are essentially the same and it has been shown that the effective field can be considered to be on the GS (Korzhnev et al., 2005; Bothe et al., 2014).
Because only one spin is aligned along its effective field, it is possible to use significantly weaker RF fields, for a full range of 25–3500 Hz (Massi et al., 2004; Korzhnev et al., 2005; Hansen et al., 2009). The lowest spin-lock power attainable is predominately limited by ~3-times the largest homonuclear scalar coupling (JCC/JNN) (Zhao et al., 2014): G-N1 and U-N3 ~50 Hz, A/G-C8 ~45 Hz, C1′ ~150 Hz, and A-C2 ~40 Hz. By using a continuous low spin-lock RF field rather than hard 180° pulses, it is also possible to suppress or eliminate unwanted C-C and N-C interactions in uniformly 13C/15N labeled samples (Hansen et al., 2009). The C-H (or N-H) scalar coupling evolution and cross-correlated relaxation between C-H (or N-H) dipole-dipole and carbon chemical shift anisotropy (CSA) are efficiently suppressed by using a strong 1H continuous-wave field applied on the resonance of interest (Fig 4B,C). Consistent heating throughout a given experiment is maintained by a heat-compensation element (Korzhnev et al., 2005; Hansen et al., 2009) that applies far off-resonance 1H and 13C/15N spin-locks for a given amount of time (Tmax−Trelax), where Tmax is the longest time delay and Trelax is the time delay in a given scan. Water is efficiently suppressed using either low power presaturation during inter-scan delay or with a WATERGATE element (Piotto et al., 1992) before acquisition. The same 15N R1ρ experiment introduced by Kay and co-workers (Korzhnev et al., 2005) can be used to measure RD in imino nitrogens in DNA (Nikolova et al., 2012) and RNA (Lee et al., 2014). Unlike the 13C resonances, it is generally possible to assign the imino N1/3 resonances in much larger RNA systems. This, combined with the fact that the 15N chemical shift is extremely sensitive to RNA secondary structure makes imino 15N R1ρ an ideal approach for characterizing the secondary structure of RNA excited states. These experiments have so far been applied to measure R1ρ data for base (N1, N3, C2, C6, and C8) and sugar (C1′) nuclei in RNA and DNA (see Fig 4A).
3. General Protocol for Characterizing RNA ESs using Low-to-High Spin-Lock Field 13C and 15N Off-resonance NMR R1ρ and Uniformly Labeled Nucleic Acid Samples
3.1 Construct Design
It is common practice for transcription reactions using T7 RNA polymerase (T7 RNAP) to add two G residues at the 5′ end of the RNA in order to maximize transcription yields. RNA secondary structure prediction is used to make sure that such sequence modifications do not interfere with the RNA GS or ES.
3.2 Sample Preparation and Purification
Because the R1ρ experiment is sensitivity demanding, we typically require NMR samples with ≥1 mM concentration of 13C/15N uniformly labeled RNA. This typically yields spectra with ≥40 signal-to-noise ratios (S/N) at 16 scans (the minimum phase-cycling steps involved in the experiment) and with an inter-scan delay (d1) of ~1.4–2.0 sec, allowing acquisition of ~140 R1ρ data points at various ωSL and within ~12 hrs. This requirement for high sensitivity can also make it challenging to record R1ρ data on slowly tumbling systems such as large RNAs (>70 nt) or RNA-protein complexes. TROSY-based R1ρ experiments can be used to improve sensitivity in such large systems (Loria et al., 1999b; Igumenova & Palmer, 2006). Samples can be prepared by in vitro transcription employing commercially available 13C/15N labeled nucleotide triphosphates (NTPs), followed by polyacrylamide gel electrophoresis (PAGE) purification or other purification methods. It is important to ensure that all running buffers, reagents, glassware, and equipment used are RNAse-free. Protocols for preparation of uniformly 13C/15N labeled RNA samples are described elsewhere (Easton et al., 2010; Petrov et al., 2013; Alvarado et al., 2014).
3.3 Protocol for Measuring R1ρ RD in Uniformly Labeled Nucleic Acids
The RNA nuclei that are typically targeted for R1ρ measurements by the methods described here are depicted in Figure 4A. Well established NMR approaches (reviewed in (Furtig et al., 2003)) are initially used to assign resonances in the RNA.
3.3.1 Calibrating Spin-Lock Power
When setting up the R1ρ experiment on a new spectrometer or newly installed probe/console, it is critical to calibrate 13C and 15N spin-lock powers (Palmer et al., 2001). In general, spin-lock power calibration is not necessary when switching samples or changing temperature.
Prepare a modified version of R1ρ pulse sequence by removing the pulses with phase φ3 and φ4 flanking the spin-lock element (Fig 4B,C). This can be done by setting their duration to zero or by omitting the pulses from the pulse program. The following protocol can then be used to calibrate 15N or 13C spin-lock powers.
Select a sharp and well-resolved resonance to perform calibration. This resonance should exhibit minimal to no chemical exchange to avoid complication from exchange processes.
Compile a table containing spin-lock powers ωSL (in Hz) to be calibrated and associated power levels P (in dB). The power levels can be estimated based on the calibrated 15N or 13C rectangular 90° pulse values.
Record 1D experiments for an array of spin-lock durations, Trelax (Fig 4B,C). Typically 33 durations are used in increments of 1/(4ωSL) from 0 to 8/ωSL (Hansen et al., 2009). For example, a 100 Hz spin-lock power would be incremented 33 times from 0–80 ms in increments of 2.5 ms.
Measure the resultant peak intensity. It should be modulated by exp(iωSL,realTrealx) where ωSL,real is the real ωSL.
Calculate the real ωSL by fitting the intensities to the equation I (τ) = I0 exp(−R·Trelax) cos(ωSL,realTrealx + φ), where Trealx is the duration of spin-lock, is the relaxation rate, and I is the peak intensity.
Repeat Step 4–5 for all ωSL in the table, and obtain ωSL,real for all preset P values.
Fit P and ωSL,real to a logarithmic equation P = a log10(ωSL,real) + b. Ideal curve would give an R2 > 0.99. If the data points deviate from linear curve, a polynomial fitting can be adopted instead. An example is shown in Figure 5.
Use the calibration equation given by the fit in 7 to back-calculate P for a desired spin-lock power within the range of powers tested.
Figure 5.
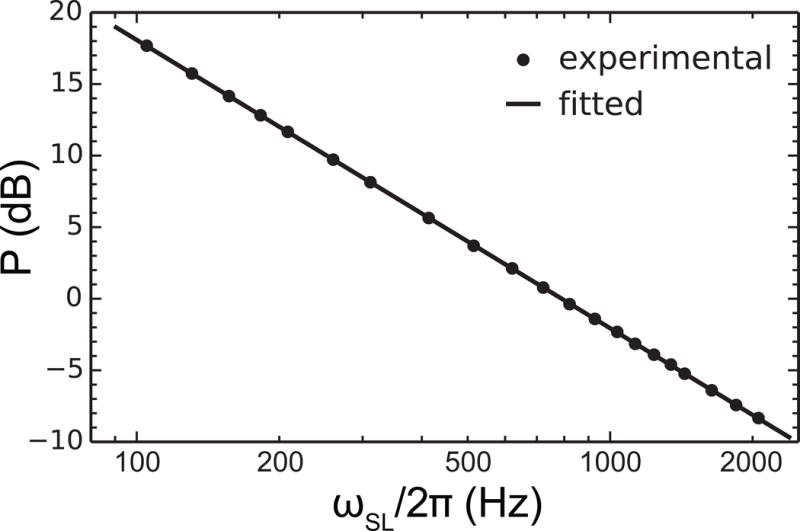
Example of spin-lock power calibration curve. Calibration shown was carried out for 15N on Bruker 600 MHz, fitted with to show a linear regression.
3.3.2 Measurement of 15N R1ρ Data
Figure 4B shows the pulse sequence employed to measure RD for imino 15N nuclei. The RF pulses along with their characteristic power and duration are defined as they appear in the pulse sequence. The key parameters to be optimized are listed below.
pH2O: Sinc shaped pulse for manipulation of water magnetization (for flip-back and WATERGATE). pH2O should be optimized to achieve optimal peak intensity and efficient water suppression. It can be initially set to a value calculated from 1H 90° pulse, and then optimized by arraying 5~7 values of the pulse power centered around the initial guess.
ωCP: strength of 1H/15N RF field for selective Hartman-Hahn polarization transfer pulses. Optimal transfer is achieved when ωCP ≈ 90 Hz for both 1H and 15N pulses. Estimate the power levels for 1H and 15N ωCP pulses corresponding to 90 Hz from the spin-lock power calibration curve of 15N (Fig 5) and from 1H 90° pulse. Fine tune the power levels for both the 1H and 15N pulses around that value to maximize the intensity of the desired peak with Trelax set to 0 ms. Efficient heteronuclear cross polarization can be achieved when ωCP (in Hz) is set to values larger than . (Korzhnev et al., 2005).
τCP: duration of polarization transfer delay is set to ~ 1/|1JNH| and further optimized to obtain the maximum peak intensity of the desired resonance.
ω1H: 1H spin-lock field used to suppress scalar coupling (1JHN), and N-H/N DD/CSA cross-correlated relaxation during the 15N spin-lock period (Trelax). It is set to ~5000 Hz for 15N R1ρ experiment. ω1H should also be chosen to be larger than the effective spin-lock field to avoid polarization transfer during the spin-lock period (Korzhnev et al., 2005). In practice, when setting up 15N R1ρ experiment for the first time, several different ω1H values are tested until dispersion data with minimum artifacts are obtained.
ζ delay (optional calibration): Nearby 15N resonances that have overlapping 1H chemical shifts can be suppressed by using the ζ delay (Fig 4B,C), wherein ζ=π/(2δ) and δ/(2π) is the 15N offset (Hz) of the resonance to be suppressed (Korzhnev et al., 2005; Hansen et al., 2009).
τeq: time delay for equilibration of GS and ES and is set to ~3/kex.
Tmax: the maximum duration of the relaxation time used to measure the R1ρ monoexponential decay. It varies from resonance to resonance, and is typically set to a value that results in decay in the peak intensity to ~30–40% of the intensity measured at Tmin = 0 ms. Care must be taken to ensure the combination of ωSL and Trelax does not exceed the tolerance of the probe.
The following protocol is employed to collect 15N R1ρ
Optimize pulse sequence parameters as described above.
Record on-resonance data by setting Ω = 0 Hz and measure R1ρ decay curves at ~20 powers ranging from 50–2000 Hz. To do this, measure R1ρ monoexponential decay curves at individual power levels and zero offset by acquiring the peak intensity as a function of ~8–10 spin-lock durations (Trelax). Preform duplicate measurements at Tmin and Tmax to help estimate R1ρ uncertainty. Note that the absence of relaxation dispersion – or a dependence for Rex on spin-lock power in the on-resonance profile does not imply absence of chemical exchange. It is possible that the exchange involves a very large Δω such that the carrier offset is not optimally positioned near ωES where the Rex contribution is maximum (Trott & Palmer, 2002). Off-resonance data (see Fig 6 below) must be collected to verify absence of chemical exchange at a given site.
Measure off-resonance R1ρ data by varying both ωSL (typically 3 or more values spanning the limits described above) and Ω (≥20 values per ωSL probed). The maximum value for Ω (hereinafter referred as Ωmax) is generally ~3–4 times the ωSL. Beyond this offset range, R1ρ will be dominated by R1 relaxation contributions, resulting in significant uncertainty when extracting Rex. In general, ≥20 values of Ω are chosen with equal spacing to span ± Ωmax symmetrically about the zero offset.
It may be important to calibrate sample temperature (see note in 3.4.7) when carrying out the R1ρ measurements since small differences (~5 °C) can have significant effect on the observed RD profiles.
Figure 6.
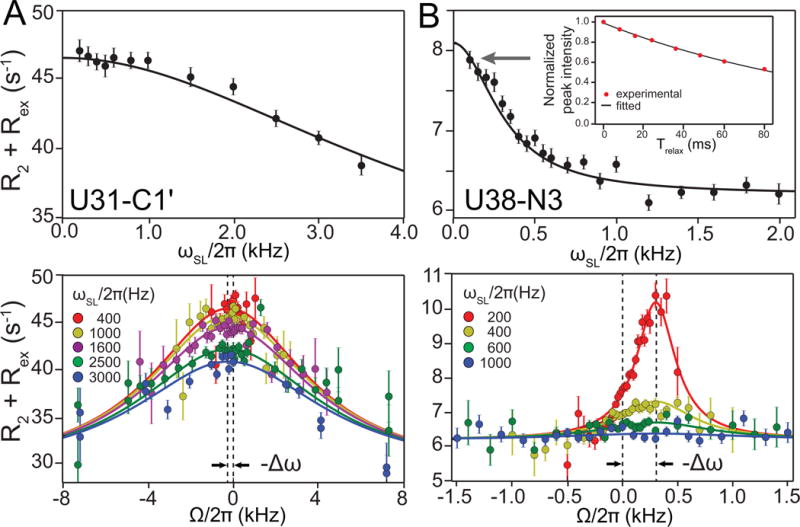
Representative on- (top) and off-resonance (bottom) 1D R1ρ RD profiles for uridine residues from HIV-1 TAR. (A) U31-C1′ exhibiting fast exchange between GS and ES1 measured using 1D 13C R1ρ experiment. (B) U38-N3 exhibiting slow exchange between GS and ES2 measured using 1D 15N R1ρ experiment. The RD data are fitted to Laguerre equation. The inset shows the magnetization decay, as a function of relaxation delay (Trelax), of the first point in the on-resonance profiles, which is fit to a monoexponential to obtain the R1ρ value.
3.3.3 Measurement of 13C R1ρ data
The 13C R1ρ pulse sequence (Fig 4C) can be set up in a similar manner as described for 15N R1ρ (3.3.2) to target the carbon nuclei shown in Figure 4A. A presaturation approach (PRESAT) can be used for water suppression of these non-exchangeable resonances (Hansen et al., 2009). 13C nuclei typically require much shorter relaxation delays (Trelax <50 ms), and a broader range of ωSL can be used (typically 150 – 3500 Hz). Care has to be taken to limit Hartmann-Hahn matching condition with other neighboring carbons that have sizable scalar couplings. See Hansen et al. for an involved discussion (Hansen et al., 2009). Below, we describe the basic elements that differ from those of the 15N R1ρ experiment (3.3.2).
ppresat: presaturation power level. Optimize power level to achieve good water suppression in combination with ppurge (see below). Typically lower power (~100–150 Hz) is used to achieve selective suppression. It is critical to avoid using high power levels for probe safety.
ωCP: strength of 1H/13C RF field of polarization transfer. It is set to ~100 Hz in 13C R1ρ experiment. See 3.3.2.
τCP: duration of polarization transfer. The optimal value is ~ 1/|1JCH|. See 3.3.2.
ω1H: 1H spin-lock field. See 3.3.2. For 13C, ~8–10 kHz ω1H is typically needed to completely suppress unwanted 1JCH and DD/CSA effects (Hansen et al., 2009).
ζ delay (optional calibration): See 3.3.2. It is noted that the effectiveness of the ζ delay is significantly compromised for resonances with strong homonuclear scalar couplings (primarily applicable for 13C resonances such as C6) (Hansen et al., 2009).
ppurge: Water purge element. ppurge need to be optimized by varying their power and duration for every experiment to maximize water suppression.
The protocol for 13C R1ρ measurements is the same as the case of 15N, except for the following points.
On-resonance data are collected at ωSL ranging from 45–3500 Hz or 150–3500 Hz for C2/C8 spins and C1′/C6 spins, respectively.
C-C Hartmann-Hahn transfers need to be computed as described previously (Hansen et al., 2009) in order to avoid spin-lock powers and offset combinations that can result in transfers to neighboring or distant C resonances with significant JCC couplings. For example, lower ωSL are not feasible for C1′ because of sizeable 1JC1′C1′ and need to avoid Hartmann-Hahn transfers (Hansen et al., 2009).
3.3.4 Trouble Shooting R1ρ
Expected peak is absent. Either the Hartmann-Hahn polarization transfer failed or resulted in negligible signal. Make sure 1H and 13C/15N carrier positions have been set properly, and that a well estimated τCP has been given based on 1JCH or 1JNH. Try increasing the number of scans and array both ωCP pulses around power levels estimated for a 90–100 Hz transfer.
Oscillations are seen in the monoexponential decay of peak intensity. Check that the lowest 13C/15N spin-lock power used is at least 3 times the largest JCC/JNN. For slow exchanging systems with large ES populations (kex <250 s−1, and pES >10%), oscillations can arise due to precession of the ES magnetization. It may be possible to suppress such oscillations by reducing the length of the τeq (see Fig 4) to ~5 ms (Qi Zhang, University of North Carolina, personal communication).
3.4 Data Analysis
3.4.1 Fitting Monoexponentials to Obtain R1ρ Values
The R1ρ data is collected as a series of 1D spectra for each combination of ωSL and Ω. The procedure below is used to extract the R1ρ data from each set of 1D spectra recorded using different spin-lock durations, Trelax.
Spectra are processed by nmrPipe (Delaglio et al., 1995) and then are assembled into a single pseudo-2D spectrum using an nmrPipe script.
The pseudo-2D spectrum is fitted to Gaussian or Lorentzian line-shape using nmrPipe autoFit script. This fitting scheme assumes that all 1D peaks share the same line-width and resonance frequency. We find that this global approach to curve fitting improves accuracy and robustness.
Peak intensities are extracted from the Step 2, and are subsequently fitted against Trelax according to equation I = I0 exp(−R1ρ·Trelax) using a non-linear fitting algorithm. The nonlinear fitting could be sensitive to the initial values of I0 and R1ρ, especially when the signal-to-noise is poor. To address this potential problem, carry out Steps 4 and 5.
Normalize the peak intensities in the 1D arrays by scaling down these intensities so that the intensity at the shortest delay (Tmin) equal to 1.0. The initial estimate for I0 is thus set to 1.0.
The initial estimation for R1ρ is obtained by linearly fitting these intensities to the equation ln I = ln I0 − R1ρTrelax. The non-linear fitting is then performed under these initial guess, and R1ρ is extracted. An example of a fit is shown in Figure 6B (inset).
Monte-Carlo method or Jackknife algorithms can be used to estimate the uncertainty of the fitted R1ρ value. These errors will be used in the fitting of R1ρ data described value. in 3.4.2 and 3.4.3.
3.4.2 Fitting Off-Resonance R1ρ Data using Algebraic Equations
Several algebraic expressions, derived using various approximations, can be used to fit the dependence of R1ρ on ωSL and Ω to extract the desired exchange parameters (reviewed in (Palmer & Massi, 2006)). These equations can be used to extract exchange parameters from the R1ρ data, and also, can provide a powerful tool for exploring the experiment. The most versatile expression uses the Laguerre approximation developed by Palmer and co-workers (Miloushev et al., 2005):
| (1) |
where , , , , , , where R1 and R2 are the longitudinal and transverse relaxation rates, respectively, ΩGS and ΩES are the resonance offsets from the spin-lock carrier for the respective states, ωrf and ωSL are reference frequency and the strength of the spin-lock carrier, kex is the exchange rate, all in units of s−1. The Laguerre equation is valid under conditions of fast exchange, and deviations can arise in slow to intermediate exchange with a large excited state population (pES >10%). These deviations generally increase with slower exchange rates and increasing populations, particularly for low spin-lock fields (ωSL ≤400 Hz) (Miloushev & Palmer, 2005).
For >2-state exchange, there can be a range of exchange topologies (Palmer & Massi, 2006). Typically, the R1ρ data is analyzed assuming ES1⇌GS⇌ES2 with or without minor exchange between ES1 and ES2. It should be noted that under certain exchange regimes, the R1ρ data will be sensitive to the presence of minor exchange between ES1 and ES2, and therefore, under favorable exchange conditions can in principle be used to define the topology of the exchange process (Palmer & Massi, 2006). For 3-state without minor exchange, one can use the following equation (Palmer & Massi, 2006):
| (2) |
where ΔωES1 = ΔΩES1−ΩGS, ΔωES2 = ΔΩES2−ΩGS, k1 and k−1 are the GS-ES1 forward and reverse rate constants, respectively, and k2 and k−2 are the GS-ES2 forward and reverse rate constants, respectively.
A Laguerre approximation has also been used to analyze 3-state exchange (Dethoff, Chugh, et al., 2012), and has shown excellent agreement with numerical solutions, though its range of validity has not been fully examined:
| (3) |
Other expressions for 3-state exchange with minor exchange have been reported (Trott et al., 2004).
When carrying out a fit to the R1ρ data, one starts out with broad estimates of initial exchange parameters and performs a fit using the equations described above. However, starting estimates combined with conventional local-minimum optimization methods (such as the Levenberg-Marquadt algorithm) can lead to fits being trapped in local minima. In such cases, use of simulated annealing and basin-hopping (Wales et al., 1997) schemes can prove very beneficial for finding global minima.
The quality of the fit is assessed using a χ2 analysis, for which reasonable fits typically yield values below 1.5, though care should be taken if a fit gives a χ2 of <1 as this may indicate over-fitting. A poor fit may suggest deviations from a two-state model or from the range of validity of the Laguerre equation or >2-state exchange. Statistical tests (e.g. F-Test and Akaike information criterion) (Akaike, 1974) can provide insights in to the likelihood of one model over the other.
3.4.3 Fitting Off-Resonance R1ρ Data using Bloch McConnell Equations
Alternatively, one can fit the R1ρ data directly by numerical integration of the Bloch-McConnell (B-M) equations (McConnell, 1958). The B-M equation does not rely on any algebraic approximations and makes it possible to compute R1ρ data for arbitrarily complex exchange models consisting of many ESs with distinct exchange topologies. The disadvantage of this method is the high computational cost. Typically, we find that using parameters from the Laguerre fit simplifies the search for the global minimum. More typically, we use the B-M equations to establish the validity of the Laguerre approximation through comparison of computed R1ρ given a set of ωSL and Ω around a given set of speculated exchange parameters (for example see (Bothe, Lowenhaupt, et al., 2011)). B-M simulations can also be used to examine effects arising from >2-state exchange.
3.4.4 Determining the chemical shifts of the ES
Fitting of the R1ρ data yields Δω and pES. This together with the observed chemical shift, , can be used to determine the ES chemical shift, ΩES:
| (3) |
It worth noting that the same unit should be used for ΩES, and Δω either in Hz or in ppm. To convert Δω from Hz to ppm, one can use equation Δω (ppm) = Δω(Hz)×106/ωLamor where ωLamor (in Hz) is Larmor frequency of the spin of interest. For example, if Larmor frequency of a 15N spin is 60.772 MHz, then a Δω=−257 Hz will be translated to −4.23 ppm. The GS chemical shift can be calculated by .
3.4.5 Plotting RD profiles
When displaying RD profiles, we plot R2 + Rex versus spin-lock offset frequency (Ω) for various (typically color coded) spin-lock powers. This omits the R1 contribution to R1ρ, which does not provide any information regarding exchange parameters. Note that Ω is the 13C or 15N resonance offset frequency from the carrier position, and ΔΩ = −Δω. An example plot is shown in Figure 6 for fast (ES1, Fig 6A) and slow (ES2, Fig 6B) exchange processes in HIV-1 TAR. Note that in general, the maximum Rex occurs when the carrier is on resonance with the ES chemical shift, thus simple inspection of the profile can be used to estimate the chemical shift of the ES. For fast exchange, one generally sees a broader line and it becomes more difficult to pinpoint the position of peak maximum and therefore the chemical shift of the ES (Bothe et al., 2014).
3.4.6 Estimating Uncertainties in Exchange Parameters
Bootstrap and Monte Carlo based approaches can be used to estimate uncertainties in the measured exchange parameters (Bothe, et al., 2014). A parent data set of R1ρ data points (Ω, ωSL, R1ρ, R1ρ error) is used for both methods.
In the Bootstrap approach:
1000 child data sets of the same size as the parent data set are generated by randomly choosing data points from the parent data set. Here, each data point can be either excluded or chosen multiple times.
Each child data set and the parent data set are fit to the Laguerre equation to obtain the five exchange parameters: R1, R2 pES, kex and Δω
The uncertainty in the exchange parameters is then determined by calculating the standard deviation of the individual parameters of the child data sets relative to parameters of the parent data set.
In the Monte Carlo approach:
The parent data set is fit to the Laguerre equation to obtain the five exchange parameters.
1000 child data sets are generated using the exchange parameters, the Laguerre equation, and the Ω and ωSL values of the parent data set.
Each child data set is noise corrupted according to the R1ρ error from the parent data set.
The child data sets are fit to the Laguerre equation.
The uncertainty in the exchange parameters is then determined by calculating the standard deviation of the individual parameters of the child data sets relative to parameters of the parent data set.
3.4.7 Kinetic-Thermodynamic Analysis
Valuable kinetic-thermodynamic information can be obtained from the exchange parameters (Korzhnev et al., 2007; Nikolova et al., 2011). This can also provide important insights into the nature of the ES and transition state (TS). For a 2-state system, the free energy difference between the GS and ES at a given temperature can readily be obtained using the following equation:
| (4) |
where ΔGES is the relative free energy difference between the GS (arbitrarily referenced to 0) and the ES, T is the experimental temperature (in Kelvin), and kB is Boltzmann’s constant.
The free energy difference relative to the transition state can be obtained by:
| (5) |
where (i = 1 or −1) are the forward and reverse rate constants, respectively. T is the experimental temperature (in Kelvin), kB is Boltzmann’s constant, κ is the transmission coefficient (assumed to be 1), h is Planck’s constant, ΔGT is the forward or reverse activation barrier, and R is the gas constant.
The enthalpies and entropies can also be obtained through temperature dependent R1ρ measurements.
| (6) |
Where ki (i = 1 or −1) are the forward and reverse rate constants, respectively. T is the experimental temperature (in Kelvin), kB is Boltzmann’s constant, κ is the transmission coefficient (assumed to be 1), h is Planck’s constant, R is the gas constant, ΔGT is the forward or reverse activation barrier, ΔHT is the forward or reverse enthalpy of activation, and Thm is the harmonic mean of the experimental temperatures measured:
| (7) |
Finally, Where ΔST is the forward or reverse entropy of activation.
Note: Here it is critical to ensure the spectrometer temperature is calibrated to match the sample temperature. This can be achieved by recording spectra of 99.8% methanol-d4 (Cambridge Isotope Laboratories, Inc.) at varying spectrometer temperatures. Here, the actual sample temperature is given by T = −16.7467Δδ2 – 52.5130Δδ+ 419.1381, where Δδ is the difference in chemical shift (ppm) between the hydroxyl and methyl proton (Findeisen et al., 2007).
3.5. Inferring Structures of RNA Excited States using NMR Chemical Shifts and Secondary Structure Prediction
3.5.1 13C and 15N Chemical Shift-Structure Relationships in RNA
The 13C and 15N chemical shifts of the ES carry rich structural information. Base carbon chemical shifts (C2/C6/C8) are sensitive to changes in the glycosidic angle χ (anti versus syn) as well as stacking (Dejaegere et al., 1998; Xu et al., 2000; Ebrahimi et al., 2001; Fares et al., 2007; Ohlenschlager et al., 2008). The adenine C2 is sensitive to protonation of N1. Sugar C1′ chemical shifts are sensitive to both sugar pucker and the glycosidic bond angle (Dejaegere & Case, 1998; Xu & Au-Yeung, 2000; Ebrahimi et al., 2001; Fares et al., 2007; Ohlenschlager et al., 2008). The 15N chemical shifts are sensitive to the properties of H-bonding (see below) (Goswami et al., 1993) and also directly report on changes in 15N protonation states (Büchner et al., 1978).
In Figure 7, we show 2D HSQC-style representation that capture key 13C (Fig 7A) and 15N (Fig 7B) chemical-shift-structure relationships in RNA (McBrairty et al., 2015). As can be seen, resonances belonging to Watson-Crick (WC) bps that are surrounded by other WC bps (A-form residues) fall within a narrow region of 13C chemical shifts for both base (C2, C6, C8) and sugar (C1′) resonances (Dejaegere & Case, 1998; Xu & Au-Yeung, 2000; Ebrahimi et al., 2001; Fares et al., 2007; Ohlenschlager et al., 2008). Non-canonical residues have 13C and 15N chemical shifts that deviate from the narrow range defined by the WC A-form conformation. For example, residues in bulges and apical loops have C6/C8 resonances that are downfield shifted in both carbon and proton dimensions. The C1′ chemical shifts in non-A-form regions are also upfield shifted, consistent with changes in sugar pucker from C3′-endo toward C2′-endo. In general, non-canonical bps that resemble the A-form helix geometry will show carbon chemical shifts that fall within the A-form helical distributions, whereas more flexible base-pairing and differences in backbone conformation can lead to differences in both C6/C8 (downfield) and C1′ (upfield) chemical shifts.
Figure 7.
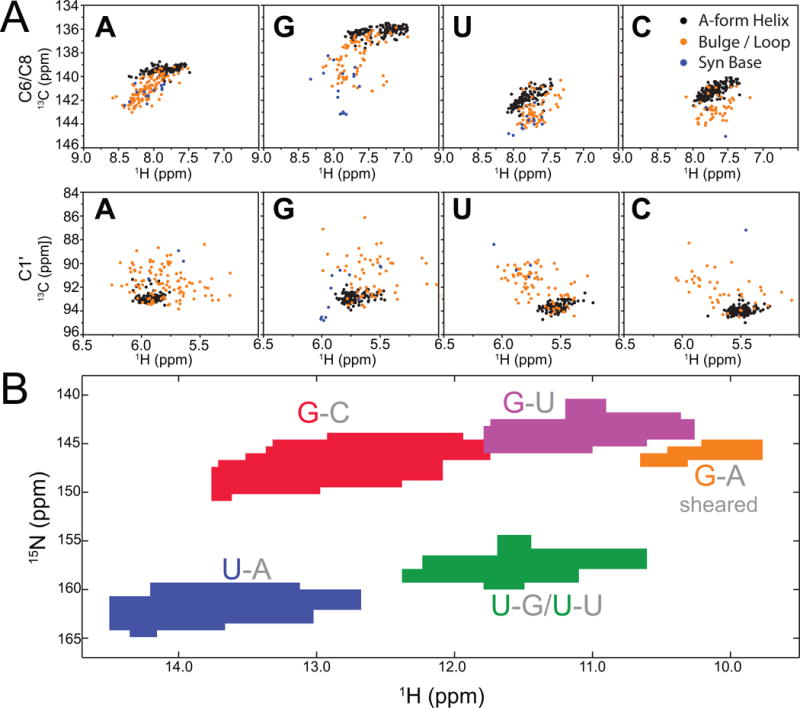
Chemical shift-structure relationships in RNA (A) Distribution of C-H chemical shifts derived from BMRB database (McBrairty, 2015) for different regions of RNAs: A-form helical Watson-Crick base pairs (black); bulge, internal loop or apical loop (gold); residues with a syn base conformation (blue). (B) Distribution of N-H chemical shifts of RNA imino groups for guanine and uracil nucleotides in different base pair contexts: G-C Watson-Crick base pairs (red), G-U wobbles (purple), sheared G-A pairs (gold), U-A Watson-Crick base pairs (blue), and U-G wobbles or U-U mispairs (green).
In addition, the imino (G-N1/U-N3) and amino (A-N6/C-N4/G-N2) 15N chemical shifts are sensitive to changes in base-pairing, including changes in hydrogen bonding partner, as well as the hydrogen bonding distance, which give rise to predictable changes in chemical shift from the standard WC bps to other types of base-pairing (Fig 7B). Guanine N1 in G-C bps resonates in a region downfield (ΔωN1 ~2–5 ppm) from the chemical shifts of the same spin in G-U wobble mispairs. Likewise, uracil N3 chemical shifts of A-U pairs resonate downfield (ΔωN3 ~2–5 ppm) from that of U-N3 in G-U mispairs. In general, G-N1 and U-N3 display upfield shift from the canonical G-C/A-U region upon partial or complete loss of stable nitrogen H-bonding. Therefore, if a base pair involving G/U changes from canonical pair to non-canonical pair or an unpaired base, or vice versa, it is likely to produce a detectable RD profile, providing a clue about the change of base-pairing in the excited state.
It is also possible to capture motif-specific chemical shift signatures. For example, the UUCG apical loop features a syn G that has an unusual C8 resonance that is ~7 ppm downfield shifted from the reference A-form helical chemical shifts (Dethoff, Petzold, et al., 2012). Other common motifs such as GNRA tetraloops can also be readily identified through a combination of the C8, C2, C1′ chemical shifts that remain consistent throughout these motifs.
3.5.2 Secondary Structure Prediction
The information regarding which residues undergo an exchange process and direction and magnitude of the ES change in chemical shift form a “chemical shift fingerprint”, which can be used to infer potential models for the ES structure. The chemical shift fingerprints can be used to infer which residues become more/less helical upon formation of the ES. For example, in the HIV-1 TAR ES1, the G-C8 shows clear signs of a syn G analogous to that seen in the UUCG apical loop, while A35 and many other apical loop residues show signs of moving away from non-helical sugar pucker in the GS (i.e. C2′-endo) toward more helical C3′-endo sugar pucker in ES1. In HIV-1 TAR ES2, the U28-N3 shows clear signs of a weakened hydrogen bond, as the A27-U38 moves towards a non-canonical U25-U38 bp.
RNA ESs often arise due to localized changes in secondary structure (Fig 2). For this reason, it is instructive to test the possibility that the ES represents an alternative secondary structure. A powerful approach for obtaining a list of candidate ES structures is to use commonly available secondary structure prediction programs such as Mfold (Zuker, 2003) or MC-Fold (Parisien et al., 2008) to examine higher energy secondary structures that can be adopted by an RNA (Fig 8A). These alternative secondary structures are examined to see if they can qualitatively explain the observed ES chemical shift fingerprints. For example, in the case of HIV-1 TAR, one finds that the second energetically most favorable structure features a more compact loop that can explain the chemical shift fingerprints measured for ES1 (Fig 8A) (Dethoff, Petzold, et al., 2012). A higher energy secondary structure that features remodeling of bulge, upper helix, and apical loop is also observed that can explain the more broad distribution exchange observed for ES2, and the unique chemical shift fingerprints (Lee et al., 2014). In general, a given secondary structure is interrogated to examine whether it addresses the following questions:
Does the alternative secondary structure result in changes in the environment of all residues showing signs of exchange broadening?
Does the alternative secondary structure preserve the environment of all residues showing no signs of exchange broadening?
Do the specific changes in structure and, specifically, transitions from helical to non-helical residues or inter-conversion of G-C to G-U bps agree with the specific ES chemical shifts?
Figure 8.
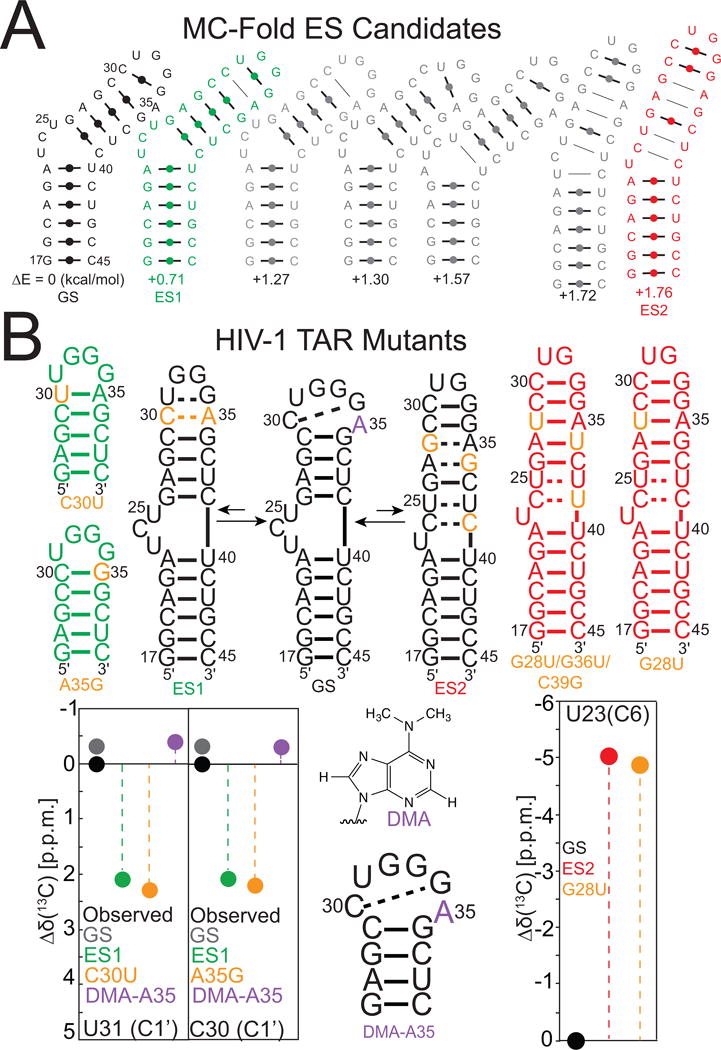
(A) Using secondary structure prediction to generate a list of candidate excited states. Shown are the seven lowest energy secondary structures with estimated energies referenced to the GS of HIV-1 TAR predicted from MC-Fold (Parisien & Major, 2008). The second and the seventh predicted secondary structures are ES1 (green) and ES2 (red), respectively as determined by R1ρ and chemical shift finger prinitng. (B) Mutate and chemical shift fingerprinting analysis of RNA ESs. Shown are TAR mutants designed to stabilize ES1 (green), ES2 (red) or the GS (black) with residue color indicating substitution (orange), chemically modified substitution (purple). Chemical shift fingerprinting is shown as a comparison of chemical shifts determined by HSQC spectra of TAR mutants and R1ρ relative to the chemical shift of HIV-1 TAR.
In this manner, a candidate ES model is generated and tested as described below.
3.6 Testing Model RNA ESs
3.6.1 Stabilizing GS and ES Using Mutations
A candidate ES model with alternative secondary structure is tested using mutations and single atom substitutions that are designed to stabilize the ES and in some cases the GS. Generally, it is possible to design substitution mutations that, for example, convert a non-canonical bp in the ES, such as an A-C bp, into a more stable canonical bp, such as G-C or A-U. This is shown in Figure 8B, where the HIV-1 TAR ES1 is stabilized using two mutations that convert A-C into WC bps. Likewise, mutating the A-G mismatch to a WC A-U can stabilize the TAR ES2. ESs with low energetic stability that have very low populations (<1%) can require more severe mutations in order to completely stabilize the ES. Indeed, complete stabilization of the low populated TAR ES2 (~ 0.4%) required a triple mutant (Fig 8B) (Lee, J., et al., 2014). An ES or GS can also be stabilized through deletion of a residue. For example, in HIV-1 TAR deletion of the three-nucleotide bulge stabilizes the GS of HIV-1 TAR so there is no exchange with ES2, but still allows exchange with ES1. Replacement of the apical loop to a stable UUCG loop stabilizes the GS completely so there is no exchange between either ES1 or ES2 (Fig 8B). All designed mutants are subjected to secondary structure prediction. Ideally, two or more ES-stabilizing mutations should be used independently, each targeting a different region of the RNA (see below). Each mutant is prepared and analyzed by NMR as described below. Because only 2D HSQC spectra are needed for chemical shift fingerprinting, unlabeled RNA samples can be used and obtained by in vitro transcription or by solid-phase chemical synthesis. 2D HSQC NMR measurements at natural abundance can be performed with optimal sensitivity using SOFAST-HMQC (Farjon et al., 2009; Sathyamoorthy et al., 2014) and BEST-TROSY (Ying et al., 2011) that yield higher sensitivity per unit time.
All mutants are subjected to the following analysis:
The secondary structure is determined using 2D NOESY NMR experiments. The goal is to determine whether the mutation stabilizes the proposed ES secondary structure. If the secondary structure does not match that predicted for the ES model, the mutant is not studied further, and the ES model is refined or a new mutant is selected to stabilize it.
The 13C and 15N chemical shifts are assigned and compared (see below for factors that influence this agreement) to the chemical shifts measured for the ES (or GS) in the wild-type construct. Examples of such chemical shift fingerprint comparisons are shown in Figure 8B.
For a concerted exchange directed to a single ES, any ES-stabilizing mutation should affect all of the resonances experiencing exchange, including those that may be distant from the site of mutation (Fig 8B). Moreover, these perturbations should be specifically directed toward the ES chemical shifts fingerprints determined by R1ρ (Fig 8B).
All resonances showing no signs of chemical exchange should experience little chemical shift perturbation upon mutation (typically <1 ppm). At this point, it is possible to examine if a given resonance does not sense a given ES because of too small Δω. For example, the HIV-1 TAR ES2 stabilizing mutant (G28U) shows small Δω values for a number of resonances (U42-N3 and A22-C8) explaining why these residues show little to no sign of chemical exchange in the R1ρ measurements (Lee et al., 2014).
ES-stabilized mutants are not expected to have chemical shifts that perfectly match those of the ES, since the mutation itself is expected to cause changes on the order of 1 ppm (Dethoff, Petzold, et al., 2012; Lee et al., 2014). The ES-stabilized mutant could also be in fast exchange with other higher energy conformations, and this could also impact the observed chemical shifts relative to the ES.
3.6.2 Stabilizing GS and ES Using Single-Atom Substitutions
The GS or ES can be stabilized by chemical modifications, including those that occur naturally. Many chemically modified RNA samples can be obtained commercially (e.g. ChemGenes, Glen Research, Berry and Associates). Typically the sample is lyophilized, to get rid of impurities, and buffer exchanged. Chemical modifications can be used to disrupt base pairing and favor “bulged out” or “flipped out” conformations. For example, N6-N6-dimethyl substituted adenine (DMA) was used to disrupt A-C type H-bonding in the TAR ES1 and to favor the GS in which the adenine is bulged out (Fig 8B) (Dethoff, Chugh, et al., 2012). Indeed, the modification leads to perturbations in the chemical shifts directed toward the GS (Fig 8B). In studies of DNA, ES that feature Hoogsteen (HG) base pairs have been stabilized in G-C and A-T base pairs through methylation of guanine and adenine N1 (Nikolova et al., 2011). Conversely, the single atom substitution converting purine N7 to a carbon (deaza-purine) was used to destabilize the HG bps and trap the WC GS (Nikolova et al., 2012).
3.6.3 Stabilizing GS and ES by Changing pH
In some cases, an ES may be associated with a protonation event. For example, many non-canonical A-C bps are protonated, and therefore their formation can be favored by lowering pH. The HIV-1 TAR ES1 features formation of an A+-C bp, which is absent in the GS, where it is possible lowering the pH will favor the ES over the GS. Indeed, one finds decreasing the pH results in chemical shift perturbations directed toward the ES1, while increase the pH results in perturbations directed toward the GS. In addition, decreasing/increasing the pH results in an increase/decrease in the HIV-1 TAR ES1 population. In general, changing the pH is a powerful approach to obtain insights into any ES that may involve protonation or deprotonation.
Acknowledgments
This work was supported by the US National Institutes of Health (R01 AI066975, NIAID R21Ai096985, and P01 GM0066275) We thank Prof Katja Petzold (Karolinska Institutet Tombtebodavägen) for input on the structure chemical shift relationships.
Footnotes
DISCLOSURE STATEMENT
H.M.A. is an advisor to and holds an ownership interest in Nymirum Inc., which is an RNA-based drug discovery company. The research reported in this article was performed by the University of Michigan faculty and students and was funded by an NIH contract to H.M.A.
References
- Aboul-ela F, et al. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Research. 1996;24(20):3974–3981. doi: 10.1093/nar/24.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike H. A new look at the statistical model identification. Automatic Control, IEEE Transactions on. 1974;19(6):716–723. [Google Scholar]
- Akke M, et al. Monitoring macromolecular motions on microsecond to millisecond time scales by R(1)rho-R(1) constant relaxation time NMR spectroscopy. J Am Chem Soc. 1996;118:911–912. [Google Scholar]
- Alvarado LJ, et al. Chemo-enzymatic synthesis of selectively (13)c/(15)n-labeled RNA for NMR structural and dynamics studies. Methods Enzymol. 2014;549:133–162. doi: 10.1016/B978-0-12-801122-5.00007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe JR, et al. Sequence-specific B-DNA flexibility modulates Z-DNA formation. J Am Chem Soc. 2011;133(7):2016–2018. doi: 10.1021/ja1073068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe JR, et al. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nat Methods. 2011;8(11):919–931. doi: 10.1038/nmeth.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe JR, et al. Evaluating the uncertainty in exchange parameters determined from off-resonance R1rho relaxation dispersion for systems in fast exchange. J Magn Reson. 2014;244:18–29. doi: 10.1016/j.jmr.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR. Prospects for riboswitch discovery and analysis. Mol Cell. 2011;43(6):867–879. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büchner P, et al. Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy of 15N-Labeled Nucleotides. In: Pullman B, editor. Nuclear Magnetic Resonance Spectroscopy in Molecular Biology. Vol. 11. Springer; Netherlands: 1978. pp. 53–70. [Google Scholar]
- Carr HY, et al. EFFECTS OF DIFFUSION ON FREE PRECESSION IN NUCLEAR MAGNETIC RESONANCE EXPERIMENTS. Physical Review. 1954;94(3):630–638. [Google Scholar]
- Cech TR, et al. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Cheah MT, et al. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447(7143):497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- Cruz JA, et al. The dynamic landscapes of RNA architecture. Cell. 2009;136(4):604–609. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- D’Souza V, et al. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature. 2004;431(7008):586–590. doi: 10.1038/nature02944. [DOI] [PubMed] [Google Scholar]
- Dejaegere AP, et al. Density functional study of ribose and deoxyribose chemical shifts. Journal of Physical Chemistry A. 1998;102(27):5280–5289. [Google Scholar]
- Delaglio F, et al. Nmrpipe – a Multidimensional Spectral Processing System Based On Unix Pipes. Journal of Biomolecular Nmr. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dethoff EA, et al. Functional complexity and regulation through RNA dynamics. Nature. 2012;482(7385):322–330. doi: 10.1038/nature10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethoff EA, et al. Visualizing transient low-populated structures of RNA. Nature. 2012;491(7426):724–728. doi: 10.1038/nature11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverell C, et al. Studies of Chemical Exchange by Nuclear Magnetic Relaxation in Rotating Frame. MOLECULAR PHYSICS. 1970;18(4):553–&. [Google Scholar]
- Easton LE, et al. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA. 2010;16(3):647–653. doi: 10.1261/rna.1862210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi M, et al. Dependence of 13C NMR chemical shifts on conformations of rna nucleosides and nucleotides. J Magn Reson. 2001;150(1):1–9. doi: 10.1006/jmre.2001.2314. [DOI] [PubMed] [Google Scholar]
- Fares C, et al. 13C-detection in RNA bases: revealing structure-chemical shift relationships. J Am Chem Soc. 2007;129(51):15814–15823. doi: 10.1021/ja0727417. [DOI] [PubMed] [Google Scholar]
- Farjon J, et al. Longitudinal-relaxation-enhanced NMR experiments for the study of nucleic acids in solution. J Am Chem Soc. 2009;131(24):8571–8577. doi: 10.1021/ja901633y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi NL, et al. Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature. 2011;480(7376):268–272. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrage F, et al. Frequency-switched single-transition cross-polarization: a tool for selective experiments in biomolecular NMR. Chemphyschem. 2004;5(1):76–84. doi: 10.1002/cphc.200300905. [DOI] [PubMed] [Google Scholar]
- Findeisen M, et al. A 1H-NMR thermometer suitable for cryoprobes. Magn Reson Chem. 2007;45(2):175–178. doi: 10.1002/mrc.1941. [DOI] [PubMed] [Google Scholar]
- Furtig B, et al. NMR spectroscopy of RNA. Chembiochem. 2003;4(10):936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- Garst AD, et al. A switch in time: detailing the life of a riboswitch. Biochim Biophys Acta. 2009;1789(9–10):584–591. doi: 10.1016/j.bbagrm.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami B, et al. Nitrogen-15-labeled oligodeoxynucleotides. 5. Use of 15N NMR to probe H-bonding in an O6MeG.cntdot.T base pair. J Am Chem Soc. 1993;115(9):3832–3833. [Google Scholar]
- Grundy FJ, et al. tRNA-mediated transcription antitermination in vitro: codon-anticodon pairing independent of the ribosome. Proc Natl Acad Sci U S A. 2002;99(17):11121–11126. doi: 10.1073/pnas.162366799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller A, et al. The Dynamic Nature of RNA as Key to Understanding Riboswitch Mechanisms. Accounts of Chemical Research. 2011;44(12):1339–1348. doi: 10.1021/ar200035g. [DOI] [PubMed] [Google Scholar]
- Hansen AL, et al. Dynamics of large elongated RNA by NMR carbon relaxation. J Am Chem Soc. 2007;129(51):16072–16082. doi: 10.1021/ja0757982. [DOI] [PubMed] [Google Scholar]
- Hansen AL, et al. Extending the range of microsecond-to-millisecond chemical exchange detected in labeled and unlabeled nucleic acids by selective carbon R(1rho) NMR spectroscopy. J Am Chem Soc. 2009;131(11):3818–3819. doi: 10.1021/ja8091399. [DOI] [PubMed] [Google Scholar]
- Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270(36):20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- Hoogstraten CG, et al. Active site dynamics in the lead-dependent ribozyme. Biochemistry. 2000;39(32):9951–9958. doi: 10.1021/bi0007627. [DOI] [PubMed] [Google Scholar]
- Houck-Loomis B, et al. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature. 2011;480(7378):561–564. doi: 10.1038/nature10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, et al. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7(1):143–157. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igumenova TI, et al. Off-resonance TROSY-selected R 1rho experiment with improved sensitivity for medium- and high-molecular-weight proteins. J Am Chem Soc. 2006;128(25):8110–8111. doi: 10.1021/ja061692f. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Jr, et al. Extensive backbone dynamics in the GCAA RNA tetraloop analyzed using 13C NMR spin relaxation and specific isotope labeling. J Am Chem Soc. 2008;130(49):16757–16769. doi: 10.1021/ja805759z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedde M, et al. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12(10):1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- Kim S, et al. An on/off resonance rotating frame relaxation experiment to monitor millisecond to microsecond timescale dynamics. J Biomol NMR. 2004;30(2):195–204. doi: 10.1023/b:jnmr.0000048941.17540.fb. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, et al. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: an application to protein folding. Acc Chem Res. 2008;41(3):442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, et al. Off-resonance R(1rho) NMR studies of exchange dynamics in proteins with low spin-lock fields: an application to a Fyn SH3 domain. J Am Chem Soc. 2005;127(2):713–721. doi: 10.1021/ja0446855. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, et al. The folding pathway of an FF domain: characterization of an on-pathway intermediate state under folding conditions by (15)N, (13)C(alpha) and (13)C-methyl relaxation dispersion and (1)H/(2)H-exchange NMR spectroscopy. J Mol Biol. 2007;372(2):497–512. doi: 10.1016/j.jmb.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, et al. An NMR experiment for the accurate measurement of heteronuclear spin-lock relaxation rates. J Am Chem Soc. 2002;124(36):10743–10753. doi: 10.1021/ja0204776. [DOI] [PubMed] [Google Scholar]
- Lai D, et al. On the importance of cotranscriptional RNA structure formation. Rna. 2013;19(11):1461–1473. doi: 10.1261/rna.037390.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, et al. Invisible RNA state dynamically couples distant motifs. Proc Natl Acad Sci U S A. 2014;111(26):9485–9490. doi: 10.1073/pnas.1407969111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D, et al. Measuring hydrogen exchange rates in invisible protein excited states. Proc Natl Acad Sci U S A. 2014;111(24):8820–8825. doi: 10.1073/pnas.1405011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria JP, et al. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999a;121(10):2331–2332. [Google Scholar]
- Loria JP, et al. A TROSY CPMG sequence for characterizing chemical exchange in large proteins. Journal of Biomolecular Nmr. 1999b;15(2):151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- Mandal M, et al. Gene regulation by riboswitches. Nat Rev Mol Cell Biol. 2004;5(6):451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- Massi F, et al. NMR R1 rho rotating-frame relaxation with weak radio frequency fields. J Am Chem Soc. 2004;126(7):2247–2256. doi: 10.1021/ja038721w. [DOI] [PubMed] [Google Scholar]
- McBrairty M, et al. The H-factor: A Parameter that Relates RNA Chemical Shifts and Secondary Structure. 2015 In Preparation. [Google Scholar]
- McConnell HM. REACTION RATES BY NUCLEAR MAGNETIC RESONANCE. Journal of Chemical Physics. 1958;28(3):430–431. [Google Scholar]
- Meiboom S, et al. MODIFIED SPIN-ECHO METHOD FOR MEASURING NUCLEAR RELAXATION TIMES. Review of Scientific Instruments. 1958;29(8):688–691. [Google Scholar]
- Miloushev VZ, et al. R(1rho) relaxation for two-site chemical exchange: general approximations and some exact solutions. J Magn Reson. 2005;177(2):221–227. doi: 10.1016/j.jmr.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Mulder FA, et al. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001;123(5):967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- Mulder FAA, et al. An off-resonance rotating frame relaxation experiment for the investigation of macromolecular dynamics using adiabatic rotations. J Magn Reson. 1998;131(2):351–357. doi: 10.1006/jmre.1998.1380. [DOI] [PubMed] [Google Scholar]
- Nikolova EN, et al. Probing transient Hoogsteen hydrogen bonds in canonical duplex DNA using NMR relaxation dispersion and single-atom substitution. J Am Chem Soc. 2012;134(8):3667–3670. doi: 10.1021/ja2117816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova EN, et al. Transient Hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470(7335):498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlenschlager O, et al. Conformational signatures of 13C chemical shifts in RNA ribose. J Biomol NMR. 2008;42(2):139–142. doi: 10.1007/s10858-008-9271-1. [DOI] [PubMed] [Google Scholar]
- Palmer AG., 3rd Chemical exchange in biomacromolecules: Past, present, and future. J Magn Reson. 2014;241:3–17. doi: 10.1016/j.jmr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG, 3rd, et al. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- Palmer AG, 3rd, et al. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106(5):1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- Parisien M, et al. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452(7183):51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Pelupessy P, et al. Excitation of selected proton signals in NMR of isotopically labeled macromolecules. J Magn Reson. 1999;138(1):178–181. doi: 10.1006/jmre.1999.1715. [DOI] [PubMed] [Google Scholar]
- Petrov A, et al. RNA purification by preparative polyacrylamide gel electrophoresis. Methods Enzymol. 2013;530:315–330. doi: 10.1016/B978-0-12-420037-1.00017-8. [DOI] [PubMed] [Google Scholar]
- Piotto M, et al. Gradient -tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2(6):661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- Polson AG, et al. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380(6573):454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- Puglisi JD, et al. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- Rajkowitsch L, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4(3):118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- Rinnenthal J, et al. Mapping the landscape of RNA dynamics with NMR spectroscopy. Acc Chem Res. 2011;44(12):1292–1301. doi: 10.1021/ar200137d. [DOI] [PubMed] [Google Scholar]
- Sathyamoorthy B, et al. Development and application of aromatic [C, H] SOFAST-HMQC NMR experiment for nucleic acids. J Biomol NMR. 2014 doi: 10.1007/s10858-014-9856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbe H, et al. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl. 2007;46(8):1212–1219. doi: 10.1002/anie.200604163. [DOI] [PubMed] [Google Scholar]
- Sekhar A, et al. NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc Natl Acad Sci U S A. 2013;110(32):12867–12874. doi: 10.1073/pnas.1305688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, et al. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat Rev Genet. 2007;8(10):776–790. doi: 10.1038/nrg2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrynnikov NR, et al. Reconstructing NMR spectra of “invisible” excited protein states using HSQC and HMQC experiments. J Am Chem Soc. 2002;124(41):12352–12360. doi: 10.1021/ja0207089. [DOI] [PubMed] [Google Scholar]
- Storz G. An expanding universe of noncoding RNAs. Science. 2002;296(5571):1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- Tian S, et al. High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. Rna. 2014 doi: 10.1261/rna.044321.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, et al. R1rho relaxation outside of the fast-exchange limit. J Magn Reson. 2002;154(1):157–160. doi: 10.1006/jmre.2001.2466. [DOI] [PubMed] [Google Scholar]
- Trott O, et al. Theoretical study of R(1rho) rotating-frame and R2 free-precession relaxation in the presence of n-site chemical exchange. J Magn Reson. 2004;170(1):104–112. doi: 10.1016/j.jmr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Tucker BJ, et al. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15(3):342–348. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P, et al. Studying “invisible” excited protein states in slow exchange with a major state conformation. J Am Chem Soc. 2012;134(19):8148–8161. doi: 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- Venditti V, et al. Minimum-energy path for a u6 RNA conformational change involving protonation, base-pair rearrangement and base flipping. J Mol Biol. 2009;391(5):894–905. doi: 10.1016/j.jmb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales DJ, et al. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. The Journal of Physical Chemistry A. 1997;101(28):5111–5116. [Google Scholar]
- Weeks KM, et al. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 1990;249(4974):1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- Winkler W, et al. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419(6910):952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- Xu XP, et al. Investigation of chemical shift and structure relationships in nucleic acids using NMR and density functional theory methods. Journal of Physical Chemistry B. 2000;104(23):5641–5650. [Google Scholar]
- Yamazaki T, et al. NMR Experiments for the Measurement of Carbon Relaxation Properties in Highly Enriched, Uniformly 13C, 15N-Labeled Proteins: Application to 13Ca Carbons. J Am Chem Soc. 1994;116:8266–8278. [Google Scholar]
- Ying J, et al. Measurement of (1)H-(15)N and (1)H-(13)C residual dipolar couplings in nucleic acids from TROSY intensities. J Biomol NMR. 2011;51(1–2):89–103. doi: 10.1007/s10858-011-9544-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, et al. Comparison of solution and crystal structures of preQ1 riboswitch reveals calcium-induced changes in conformation and dynamics. J Am Chem Soc. 2011;133(14):5190–5193. doi: 10.1021/ja111769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, et al. Characterizing slow chemical exchange in nucleic acids by carbon CEST and low spin-lock field R(1rho) NMR spectroscopy. J Am Chem Soc. 2014;136(1):20–23. doi: 10.1021/ja409835y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn-Justin S, et al. Off-resonance rf fields in heteronuclear NMR: Application to the study of slow motions. J Biomol NMR. 1997;10(4):363–372. doi: 10.1023/A:1018365815186. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]