

Abstract
Epidemiological studies have shown a correlation between chronic biomass smoke exposure and increased respiratory infection. Pulmonary macrophages are instrumental in both the innate and the adaptive immune responses to respiratory infection. In the present study, in vitro systems were utilized where alveolar macrophages (AM) and bone marrow-derived macrophages (BMdM) were exposed to concentrated wood smoke-derived particulate matter (WS-PM) and mice were exposed in vivo to either concentrated WS-PM or inhaled WS. In vivo studies demonstrated that WS-exposed mice inoculated with Streptococcus pneumoniae had a higher bacterial load 24 h post-exposure, and corresponding AM were found to have decreased lymphocyte activation activity. Additionally, while classic markers of inflammation (cellular infiltration, total protein, neutrophils) were not affected, there were changes in pulmonary macrophages populations, including significant decreases in macrophages expressing markers of activation in WS-exposed mice. The lymphocyte activation activity of WS-PM-exposed AM was significantly suppressed, but the phagocytic activity appeared unchanged. In an effort to determine a pathway for WS-induced suppression, RelB activation, assessed by nuclear translocation, was observed in AM exposed to either inhaled WS or instilled WS-PM. Finally, an analysis of WS-PM fractions determined the presence of 4–5 polycyclic aromatic hydrocarbons (PAHs), and preliminary work suggests a potential role for these PAHs to alter macrophage functions. These studies show a decreased ability of WS-exposed pulmonary macrophages to effectively mount a defense against infection, the effect lasts at least a week post-exposure, and appears to be mediated via RelB activation.
Keywords: Infection, macrophage, mouse, suppression, wood smoke
Introduction
The lung is a major interface with the environment and requires a high level of immune regulation to limit reactivity. The macrophage is an important, if not central, cell in the lung and is primarily responsible for maintaining the delicate immune balance of the lung, including removal of senescent cells and regulation of inflammation and remodeling (Gordon, 1986, 1998). There are two main compartments of pulmonary macrophages: alveolar and interstitial. Alveolar macrophages (AM) constitute more than 90% of the immune cell population of the alveolar spaces (Hocking & Golde, 1979a, Hocking & Golde, 1979b; Lohmann-Matthes et al., 1994), whereas the interstitial macrophage population is very heterogeneous with respect to surface marker expression and constitutes approximately 10–20% of the immune cell population in the interstitial spaces of the lung (Migliaccio et al., 2005). Therefore, due to the important roles of lung macrophages and their proximity to the environment, exposure to bioactive environmental agents, both man-made and natural, has the potential to alter lung macrophage functions resulting in significant consequences on human health (i.e. an increased susceptibility to infectious disease).
Macrophages play a key role in the regulation of immune responses following inhaled exposures. While exposure to toxic airborne agents results in an increased susceptibility to respiratory infections (Akunne et al., 2006; Bautista et al., 2009; Mishra & Retherford 1997; Smith et al., 2000), the exact mechanisms involved still require elucidation. Changes to macrophages that have been observed following wood smoke (WS) exposure in animals include morphology and phagocytic activity (Naeher et al., 2007). Exposure to WS occurs in a variety of ways: residential heating, cooking, forest fires and land management burns. These exposure scenarios can vary greatly with respect to intensity and duration. For example, exposures to wildland fires or agriculture burns can affect ambient air quality in nearby communities for sustained periods depending upon meteorological conditions (Ward et al., 2005). The WS from residential heating can result in moderate, but sustained exposures to indoor PM (Ward et al., 2007). Such indoor exposures can be exceeded by an order of magnitude in developing countries where biomass materials are commonly used for cooking (Naeher et al., 2007; Smith et al., 2000). In cooler seasons when people are more inclined to utilize woodstoves, potential health problems are compounded by the increased time spent indoors; both of which could synergize to increase respiratory illnesses. Furthermore, in the current economy there is an increase in the use of wood as a cheap source of fuel for heating. Recent studies have also shown a dramatic increase in forest fires, especially in areas such as the Northern Rockies, as a result of global climate change (Westerling et al., 2006). Because the snowpack is melting earlier in the spring, the fire season is getting longer and more intense. In addition, land management changes are resulting in fuels not being cleared properly, adding to the intensity of the fires. This increase in exposure to biomass smoke, both acute and chronic, has unknown health consequences.
Infectious disease models using animals have been used extensively in a variety of studies to assess pulmonary immunology following environmental exposures including, but not limited to, studies of diesel exhaust, cigarette smoke, silica and PM. Inhaled diesel exhaust, cigarette smoke and mining particulates have been linked with an increased susceptibility to respiratory infections (Antonini et al., 2000; Arredouani et al., 2004; Castranova et al., 2001; Martin et al., 2006; McDonald et al., 2004). Miners with silicosis have an increased prevalence of human immunodeficiency virus and tuberculosis infections (Ross & Murray, 2004), and cigarette smokers have increased susceptibility to pneumonia, influenza and tuberculosis. Studies using rats found that exposures to WS in the range of 3 h to 2 weeks rendered the animals susceptible to infections with Staphylococcus aureus (Zelikoff et al., 2002). A variety of studies have found increased acute respiratory illnesses in children where biomass burning (i.e. wood) is the method of cooking (Kilabuko et al., 2007; Mishra, 2003; Smith et al., 2000). Although these human studies point to strong links between smoke exposure and respiratory illness, the cellular and molecular mechanisms for impacts of WS are not clear.
In addition to the increased susceptibility to respiratory illnesses, a variety of specific effects of WS on pulmonary immunology have been described. Alterations in macrophages include a decrease in staphylococcus killing, a decrease in Fcγ-mediated phagocytosis, an alteration in morphology and membrane ultrastructure, as well as an increase in recruitment of pulmonary macrophages (Naeher et al., 2007). Changes in secreted factors include a decrease in TNFα and superoxide production, as well as an increase in IFNγ in serum (Loke et al., 1984; Matthew et al., 2001; Naeher et al., 2007). While these data provide additional information indicating that WS exposure can lead to potentially adverse effects, it is still unclear what mechanisms may be involved. In this study, we used both in vivo and in vitro methods to assess the effects of WS on macrophage function and provide evidence on specific mechanisms involved.
Material and methods
Mice
BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) were used for all in vivo studies, as well as the generation of bone marrow-derived macrophages (BMdM) for in vitro studies. DO11.10 mice were used for the isolation of transgenic T cells to use in the in vitro assays described below. Animals were housed in microisolators on a 12:12-h light-dark cycle. The mice were maintained on an OVA-free diet and given deionized water ad libitum. All animal procedures were approved by the University of Montana Institutional Animal Care and Use Committee (AUP 050-06). Euthanasia was performed by intraperitoneal injection of a lethal dose of pentobarbital sodium.
WS exposure
WS emitted from an older model, non-EPA-certified wood-stove (Englander, England Stove Works, Inc., Monroe, VA), was directed by aluminum flex tubing into a modified inhalation chamber. Target PM2.5 concentrations within the chamber were maintained using several in-line valves, with continuous PM2.5 measurement conducted using a TSI DustTrak (TSI, Inc., Minneapolis, MN). Fires were started with 4 g of paper and 20 g of kindling, and maintained by the addition of pre-weighed wood batches (50.00–54.99 g) approximately every 5 min. A mix of local softwoods (Douglas-fir, western larch, ponderosa pine, lodgepole pine) was used for fires and WS generation. Mice were exposed for 2 h each at various target concentrations in the range of 3–15 mg/m3. Temperature and carbon monoxide (CO) readings were constantly measured during exposure studies.
Alternatively, mice were exposed to concentrated WS-PM that was collected from the non-EPA-certified woodstove using a versatile aerosol concentration enrichment system (VACES) particle concentrator. In a controlled simulation, a non-EPA-certified woodstove was loaded with a mixture of locally obtained softwoods, with the smoke pumped into the inhalation chamber to allow the smoke to cool and age (residence time was no more than 2 min). The combustion conditions during the burns ranged from flaming to smoldering. The VACES was then utilized to harvest smoke PM2.5 into a deionized water dropout (Biosampler, SKC, Inc., Eighty Four, PA). At the conclusion of the smoke-PM harvesting trial, there were clearly two fractions that were collected, including a water-soluble fraction (SF) and a water-insoluble fraction (IF) black tar-like material that coated the inside of the impinger. Both fractions were combined (WS-PM) for instillations. Mice were instilled with WS particles using oropharyngeal aspiration. Briefly, mice are anesthetized with isoflurane, suspended on an angled board by hooking the front teeth on a suspended wire, the tongues are gently extended out of the oral cavity and the suspension was instilled in the trachea using a pipette.
Inoculations and bacterial load assessment
Mice were inoculated with Streptococcus pneumoniae (SP) 24 h following exposure to inhaled WS, and then evaluated for bacterial load in the lungs. Animals were inoculated with 107 colony-forming units (CFU) of SP (50 μl, maximum volume) by oropharyngeal aspiration. Briefly, mice were anesthetized with isoflurane, suspended on an angled board, tongues gently extended out of the oral cavity and the inoculum was instilled in the trachea. Mice were sacrificed 2 h post-inoculation, and the lungs harvested. When mice are sacrificed for tissue collection (blood, spleen, bone marrow, lung extraction and/or lavage), they are given a lethal dose of sodium pentobarbital, IP (EuthasolTM, Delmavar Laboratory, Fort Worth, TX; 150 mg/kg, 27 gauge needle). Lungs were homogenized in PBS and frozen with glycerol. Infectious load was then determined using the drop plate method. Briefly, serial dilutions were made (1:10) for enumeration of individual CFU, which were averaged, divided by volume and multiplied by dilution factors to yield the CFU concentration in the initial sample.
Isolation of alveolar cell populations
Cells from the alveoli were isolated by whole lung lavage. Each mouse was lavaged with 3–4 ml of cold PBS and collected fluid was stored on ice. The first draw (1 ml) was centrifuged (500× g for 2 min) and this supernatant was set aside (on ice or at −20 °C) for protein and cytokine analyses. The cell pellet was then added to the remaining 2–3 ml of lavage and centrifuged (500× g for 10 min) to collect all cells. The cell pellet was resuspended in 500-μl PBS, and enumerated using a Coulter Counter. Approximately 30 000 cells were aliquoted for cytocentrifugation and used for cell differentials and immunostaining.
Cytokine and protein analysis
Lavage fluid was analyzed for various markers of inflammation. The first 1 ml of lung lavage fluid collected from the samples was assayed for IL-1β and TNF-α with commercially available kits according to the manufacturer’s protocol (Duo-Set kits, R&D Systems, Minneapolis, MN). Samples were diluted 1:1 in both assays with reagent diluent. Colorimetric analysis was used by analyzing the samples with a Spectra Max 340 plate reader (GE Healthcare, Piscataway, NJ) at 450 nm. Lavage fluid was also assayed for total protein using a BCA Protein kit (Pierce), as well as lactate dehydrogenase (LDH-Cytotoxicity Assay Kit, BioVision, Mountain View, CA).
Phagocytosis assessment
Twenty-four hours following WS exposure, phagocytosis ability of AM was assessed using two techniques. In one assay, Fluorophorex™ Fluorescent Microspheres (Phosphorex, Inc, Fall River, MA) (10 mg/ml stock, 25 μl of stock) were administered via nonsurgical oropharyngeal aspiration. Four hours post-instillation, AM were collected by whole lung lavage as described previously. Cell pellets were resuspended in 50-μl cold PAB (PBS with 0.02% sodium azide and 1% BSA) and cells were stained with F4/80 antibody (PE-conjugated by CalTag Laboratories; Burlingame, CA). Samples were analyzed using a FACSAria system (BD Biosciences, San Jose, CA). AM phagocytosis was also assessed using the Vybrant Phagocytosis Assay Kit (V-6694, Molecular Probes, Eugene, OR). Collected AM were plated in 96-well plates at 1.0 × 105 cells per well. Plates were incubated at 37 °C for 2 h, and then assessed for fluorescent particle uptake using a fluorescence plate reader at 490 nm and 540 nm.
Antigen-presenting cell assay
AM were assayed for antigen-presenting capability using an ex vivo assay previously described (Migliaccio et al., 2005). Following exposure, the AM were isolated by lavage and aliquoted into 96-well plates at a density of 1 × 105 cells/well (100 μl/well) plus 50 μl of media either with or without OVA (Chicken ovalbumin, Grade V, Sigma, St. Louis, MO; 10 mg/ml final concentration). Plates were then incubated (cells + OVA) for 3 h at 37 °C. During this incubation, T cells were prepared from the spleen of DO11.10 mice using a T cell enrichment kit (SpinSep, StemCell Technologies, Vancouver, BC, Canada), where >75% of recovered cells were CD3+ cells expressing the transgenic T cell receptor (data not shown). T cells were added to the 96-well plate following OVA incubation at a concentration of 4 × 105 cells/well (100 μl/well), for a final ratio of 4:1 (T cells:antigen presenting cell (APC)). Plates were incubated at 37 °C for 48 h then centrifuged and supernatants were collected for assay. T cell cytokine (IFNγ) levels were determined by ELISA (Duo-Set kits).
Isolation of interstitial leukocytes
Cells from the interstitial spaces of the lung were isolated from minced tissue treated with collagenase. Minced lung preparations were performed according to the following protocol. Lungs were removed, finely minced and incubated for 2 h in collagenase buffer (~5 ml/lung) at 37 °C. Collagenase buffer consisted of 1 mg/ml collagenase IA (Sigma, St. Louis, MO) in media (RPMI, Mediatech, Herndon, VA; 10% FBS, Gibco, Grand Island, NY; 1% Pen/Strep, Gibco). Post collagenase-treatment, multiple cell populations were isolated by gradient centrifugation (Percoll, GE Healthcare, Piscataway, NJ). The band containing leukocytes (macrophages, dendritic cells, granulocytes and lymphocytes) was collected and washed with PBS. Cells were then enumerated and aliquoted for analysis by flow cytometry.
Flow cytometry
Analysis of cell surface marker expression was performed using an FACSAria system. Briefly, cells were resuspended in FACS buffer (0.1% BSA + 0.5% sodium azide) and blocked for ≥10 min on ice with anti-CD16/32 (FcBlock, BD Biosciences, San Jose, CA). Cells were then incubated with fluorescently labeled antibodies at previously determined optimal dilutions for ≥30 min on ice. Antibodies against the T cell activation marker (CD25, clone 7D4), as well as the macrophage markers (CD11b, clone M1/70; CD11c, clone HL3; IA/IE, clone 2G9), were obtained from BD Biosciences. Detection of the macrophage marker F4/80 (clone CI:A3-1) was obtained from Caltag Laboratories (Burlingame, CA). Cells were washed with 10% FBS, then transferred to filter-top polypropylene tubes (BD Labware, Franklin Lakes, NJ) for analysis. Data analysis was performed using FACSDiva software (BD Biosciences, San Jose, CA), and histogram overlays generated with FlowJo software (Tree Star Inc., Ashland, OR).
Immunocytochemistry
AM were collected as described above from mice that had been exposed to WS by inhalation or instillation. Cells were cytocentrifuged and air-dried for 15 min then fixed with 4% paraformaldehyde in PBS. Slides were washed for 15 min with sterile PBS changing PBS every 5 min. Cells were blocked and permeabilized with 100 μl solution (2% BSA in PBS + 0.5% Triton X-100) for 1 h at room temperature. Slides were washed three times with sterile PBS, then rabbit anti-p-RelB (Ser 552) antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was added at a dilution of 1:300 in permeability/blocking solution (50 μl/slide) and incubated at 4 °C overnight in the dark humidified chamber. Slides were washed prior to the addition of AlexaFluor488-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR), diluted 1:300 in permeability/blocking solution. Slides were incubated at room temperature for 1 h, washed two times, then cover-slipped using FluorSave mounting media (Calbiochem, Billenica, MA) and ProLong Gold Antifade Reagent (Molecular Probes). Slides were stored in the dark at 4 °C and images taken with either a 40× or 60× objective lens (Nikon Eclipse E800 and a Nuance CRI Multispectral Imaging System, PerkinElmer, Waltham, MA).
Bone marrow-derived macrophages
BMdM were generated for in vitro experiments as previously described (Migliaccio et al., 2008). Briefly, BALB/c mice were euthanized and hind legs removed for bone marrow (BM) isolation. BM was flushed from the femur and tibia with media (RPMI, Mediatech, Herndon, VA; 10% FBS, Gibco, Grand Island, NY; 1% Pen/Strep, Gibco). All cells were incubated in T75 culture flasks with 20-ml media overnight for stromal elimination. Aspirated cells were aliquoted to new T75 flasks (1.5 × 107 cells/flask) in 20 ml of media and 40 μl of M-CSF (R&D Systems, Minneapolis, MN). Cultures continued for 7–10 d with re-feeding every 3–4 d. Generated macrophages were assessed by flow cytometry for expression of CD11b and F4/80 (>90% were positive).
Aryl hydrocarbon receptor and Cyp1A1 activation
BMdM were used in in vitro assays to assess WS-PM activation of the aryl hydrocarbon receptor (AhR) pathway. Cyp1A1 induction was used as the readout of AhR pathway activation. BMdM were aliquoted to 6-well plates at a concentration of 3 × 106 cells/well. Following overnight incubation with WS fractions or the control 2,3,7,8-Tetrachlorodibenzodioxin (TCDD), cell lysates were generated and mRNA was isolated for Cyp1A1 evaluation.
Chemical analysis of WS-PM fractions
WS-PM fractions were analyzed for the presence of polycyclic aromatic hydrocarbons (PAHs) using an Agilent 6890N Gas Chromatograph with an Agilent 5973 Mass Spectrometer (Agilent, Santa Clara, CA). Split injection was used for analysis with an HP-5MS column ((5%-phenyl)-methylpolysiloxane) with dimensions of 0.25 mm ID × 30 m length × 0.25 μm film thickness. A temperature program was used to separate all of the PAHs present in the standard mixture, ramping from 40 °C to 300 °C. An EPA 625 Semivolatile Calibration Mix (Supelco, St. Louis, MO) diluted in methylene chloride was used to identify and quantify each of the PAHs present in the concentrated WS fractions. A separate retene standard was purchased from Chem Service (West Chester, PA) and added to the calibration mixture. Each compound in the standard mixture was identified using the NIST mass spectral library and the list of components in the mixture. Small portions of the water-SF and water-IF of WS-PM were separately dissolved in methylene chloride for analysis. Chromatograms from the standard mixture and the WS samples were overlaid to determine the presence or absence of each PAH in the samples. PAHs present in the concentrated WS-PM samples were quantified using a calibration curve made from dilutions of the EPA Semivolatile Calibration Mix.
PAH-induced RelB activation
BMdM were used in in vitro assays to assess WS-associated PAH activation of the RelB pathway. RelB activation was assessed using cell lysates in the TransAM NFκB Family kit (Active Motif). BMdM were aliquoted to 6-well plates at a concentration of 3 × 106 cells/well. Following overnight incubation with purified PAH, cells were used to generate lysates according to the kit protocol.
Statistical analysis
The values for individual samples/experiments were averaged, and the standard error of the mean (SEM) was calculated. The significance of the differences between the groups was determined by t-test or one-way analysis of variance (ANOVA, >2 groups) followed by Newman–Keuls post hoc test for pair-wise mean comparisons. For histology scoring using subjective scaling the analysis included a Mann–Whitney test to determine differences between median scores. Inter-rater reliability of the scoring was calculated using Cronbach’s alpha. All calculations were performed with Prism software except for Cronbach’s alpha which was calculated with SPSS (SPSS Inc., Chicago, IL). A p value (type I error) of <0.05 was considered statistically significant.
Results
WS exposures
To assess the health effects of WS in our murine model, animals were exposed for 2 h to inhale smoke from the burning of locally obtained softwoods. Exposures were designed for target PM2.5 levels and measured with a DustTrak monitor. The exposure events had an average fuel usage of 930 g (±38.5 SEM) and measured PM2.5 concentrations ranged from 3.62 to 16.2 mg/m3 for the dose studies of bacterial clearance, and averaged 9.27 mg/m3 (±1.34 SEM) for all other exposures. The total fuel (wood) usage was calculated by subtracting the amount of residual ash + wood following the burn from the total amount of wood used during the burn. In the first four exposures, animals were assessed after 24 h, while the remaining seven exposures varied from 2 h to 7 d post-exposure. Each exposure cohort incorporated a minimum of six mice.
Assessment of inflammation
In order to evaluate acute inflammation following exposure to WS, multiple parameters were assessed. Basic cell analysis of primary cells showed no significant increases in leukocyte populations, including macrophages, lymphocytes and granulocytes, in either the alveolar or interstitial compartments (data not shown). Analysis of soluble factors (total protein, cytokines) following WS exposures, in the alveolar spaces (whole lung lavage), showed no significant changes in permeability (total protein) or cytokine (TNFα) levels within the first three days following WS exposure (data not shown). These results indicate a lack of inflammation following acute inhalation of WS.
Respiratory infection
In order to determine if WS exposure alters susceptibility to infection, mice were inoculated with SP following WS inhalation. In addition to a group of sham (air)-exposed mice, mice were exposed to two different levels of WS (Figure 1). In the group exposed to 16.2 mg/m3 of WS, there was a significant increase in the bacterial load in homogenized lungs, as compared to both the 3.62 mg/m3 WS-exposed and sham-exposed mice (Figure 1). These results suggest that acute exposure to WS results in a decrease in the ability of the pulmonary immune system to clear invading bacteria and support the model of respiratory immunity depression.
Figure 1.

Assessment of immunity to respiratory infection following acute WS exposure. Mice were inoculated with SP at 24 h following acute exposure to WS, and whole lung homogenates were cultured and assayed for bacterial load. There was a significant (p<0.05) increase in bacterial load in the “high” WS (16.2 mg/m3) exposed mice as compared to the sham (air) and “low” WS (3.62 mg/m3) groups. Values are averages of 6–8 mice ± SEM.
Assessment of AM phagocytic capacity
In order to assess the phagocytic ability of AM from WS-exposed mice, two different methods were employed. Mice were exposed to a target level of 10 mg/m3 of WS. At the 24-h time point, there was no significant change in AM phagocytosis as measured by the Vybrant assay, and while similar results were obtained using an in vivo fluorescent bead uptake assay, there was a slight, but not significant increase in the percent of AM that took up the fluorescent particles post-WS exposure (data not shown). These results indicate that the phagocytic capacity of WS-exposed AM is unaltered 24h post-inhalation.
AM ex vivo antigen presentation
In order to assess APC activity of macrophages exposed to WS, AM were isolated from WS-exposed mice at various time points and co-cultured with CD4+ T cells. At all time points from 2 h to 7 d, there was a decrease in T cell activation by WS-exposed AM, as determined by the production of IFNγ (Figure 2). While IFNγ levels were only significantly decreased in co-cultures with AM isolated 2h post-exposure, the overall (across the total timecourse) decrease in T cell activation was significant as depicted in the area-under-the-curve (AUC) graph (Figure 2 inset). These results suggest that AM exposed to WS have a compromised ability to activate T cells in an antigen-specific manner, and this supports the model of a depression in respiratory immunity following WS exposure.
Figure 2.

Assessment of AM ability to generate adaptive immunity following exposure to WS. At various time points following smoke exposure, AM were isolated from lung by lavage and assessed in an in vitro assay by co-culturing with transgenic (DO11.10) T cells and measuring IFNγ levels in the supernatants. There was a significant (p<0.05) decrease in IFNγ production at 2 h post–smoke exposure, and the levels remain decreased for up to 7 d post-exposure with a significant effect over the entire timecourse (inset AUC graph). Values are averages of 6–12 mice ± SEM.
Changes in pulmonary leukocyte populations
In order to assess potential WS-induced effects on macrophage constituents of the interstitial space, lungs were homogenized and single cells analyzed by flow cytometry. At 24 h post-exposure (10 mg/m3 target dose), a significant decrease in the percentage of activated (MHC Class II+) macrophages in two distinct populations (based on autofluorescence (AF)) was observed (Figure 3). While the decrease in the Class II+ autofluorescence (AF)+ population was sustained for 48 h, the Class II+ AF− population rebounded by 48 h (data not shown). In addition, there was an initial significant increase in the percentage of CD25+ T cells which was maintained up to one week post-exposure (data not shown). These data indicate changes in specific leukocyte populations in response to WS inhalation that are consistent with decreased pulmonary immunity.
Figure 3.

Flow cytometric analysis of interstitial pulmonary macrophage populations following exposure to WS. Changes in two major populations of the lung were assessed by flow cytometry. Populations were categorized by surface protein expression where F4-80 and CD11b are putative macrophage markers; in addition, pulmonary macrophages were further categorized with AF, a putative marker of phagocytosis, and MHC Class II, a marker of activation. At 24 h post–smoke exposure, there was a significant (*p<0.05, **p<0.01) decrease in the activated (MHC Class II+) macrophages from both the AF+ and AF− populations. Values are averages of 6–12 mice ± SEM.
Assessment of RelB translocation
As RelB (non-canonical NF-κB) has been linked to macrophage suppression, activation of the NF-κB pathway was used to determine the mechanism of macrophage downregulation. AM isolated by lavage from both sham (air)- and WS-exposed mice at 2, 24 and 72 h post-exposure were stained with antibodies against phosphorylated RelB. When RelB becomes activated by phosphorylation, it translocates to the nucleus. RelB positive nuclei were only observed in WS-exposed AM, while AM isolated from sham-exposed mice exhibited RelB staining in the cytoplasm (Figure 4). In addition, WS-PM fractions were instilled in mice and AM isolated by lavage were assessed for RelB translocation. While controls (PBS, DEP and urban PM) and the SF did not activate RelB, both samples containing the IF (IF and WS-PM) did result in RelB translocation (Figure 5). These data suggest that the non-canonical NF-κB pathway is activated in WS-exposed AM, in particular components of the IF may contribute to the decrease in macrophage functions.
Figure 4.

Activation and nuclear translocation of RelB in WS-exposed AM. Following inhalation exposures AM were isolated by lavage and cytocentrifuged for immunocytochemical staining with anti-phosphorylated RelB antibodies. While AM from sham-exposed mice generated cytoplasmic staining patterns in all observed cells (A), a substantial number of AM from smoke-exposed mice presented with a nuclear staining pattern (B). The patterns are at 60× using a confocal laser scanning microscope system (FLUOVIEW FV10i, Olympus, Center Valley, PA) with an overlay of the immunofluorescence staining on phase-contrast scans.
Figure 5.

Activation and nuclear translocation of RelB in particle-exposed AM. Following instillation exposures AM were isolated by lavage and cytocentrifuged for immunocytochemical staining with anti-phosphorylated RelB antibodies. AM from control exposures (PBS (A), DEP (B), PM (C)) generated cytoplasmic staining patterns in all observed cells. While cells exposed to soluble fraction of WS-PM (SF) lacked RelB translocation (D), the AM exposed to either the insoluble fraction (E) or total WS-PM (F) presented with a substantial number of AM with a nuclear staining pattern. The above pictures are at 60× using a confocal laser scanning microscope system (FLUOVIEW FV10i, Olympus, Center Valley, PA) with an overlay of the immunofluorescence staining on phase-contrast scans.
AhR pathway activation
Studies have suggested the activation of RelB occurs via the interaction of chemicals (e.g. PAHS) and the AhR. In an effort to assess the potential role of the AhR pathway in the activation/translocation of RelB, BMdM in vitro cultures were treated with the different fractions of WS-PM. The different fractions of WS-PM were assessed for Cyp1A1 induction as an indicator of AhR activation by using TCDD as a control. While all samples generated some level of Cyp1A1 activation, only the IF-containing (IF and WS-PM) samples were higher than the TCDD control (Figure 6). These results suggest of role of AhR in RelB activation via PAH constituents of one fraction of WS-PM (IF).
Figure 6.

Activation of the AhR pathway in WS-exposed macrophages. BMdM were exposed to a variety of treatments and assessed for Cyp1A1 activation, a marker of the AhR pathway. Under normal culture conditions BMdM do not express Cyp1A1. The known AhR activation control 2,3,7,8-TCDD was used for evaluation of efficient Cyp1A1 activation by measurement of mRNA levels. While all treatments resulted in an increase in Cyp1A1 mRNA levels, only the insoluble fraction (IF)-containing fractions (IF and WS-PM) were above control levels, with the WS-PM fraction levels significantly (*p<0.05) above TCDD. Values are averages of three experiments run in triplicate ± SEM.
Polycyclic aromatic hydrocarbons
PAHs are known activators of the AhR pathway and may be key players in the present model. Therefore, in an effort to evaluate the role of PAHs in WS to activate RelB, PAH chemical analysis was performed and then pure PAHs used in in vitro experiments. Chemical analysis of the fractions showed that PAHs were detected only in the IF portion of WS (Table 1). Consequently, these pure PAHs were utilized in BMdM in vitro cultures to assess activation of RelB. The dilutions used for these experiments encompassed the empirically determined percent composition (~0.1%) for the PAHs in WS. As shown in Figure 7, RelB activation was observed following the treatment with individual PAHs. While the data suggested certain PAHs activate this pathway, especially WS-specific retene which induced levels above WS, there was no significant effect from any single PAH. These data suggest that while PAHs are present in WS, and they may play some role in RelB activation, they apparently do not act alone.
Table 1.
WS-associated PAHs.
| Water soluble fraction | Insoluble fraction | Relative RelB activation | |
|---|---|---|---|
| Acenapthtene | No | No | − |
| Acenaphthylene | No | No | − |
| Anthracene/phenanthrene | No | Yes | − |
| Benz[a]anthracene | No | No | − |
| Benzo[a]pyrene | No | No | − |
| Benzo[ghi]perylene | No | No | − |
| Benzo[k]fluroanthene | No | No | − |
| Benzyl butyl phthalate | No | No | − |
| Bis(2-ethylhexyl)phthalate | No | No | − |
| Carbazole | No | No | − |
| Chrysene | No | No | − |
| Di-n-octyl phthalate | No | No | − |
| Dibenz[a,h]anthracene | No | No | − |
| Dibenzofuran | No | No | − |
| Dibutyl phthalate | No | No | − |
| Diethyl phthalate | No | No | − |
| Fluoranthene | No | Yes | + |
| Fluorene | No | No | − |
| Indeno[1,2,3-cd]pyrene | No | No | − |
| Phenanthrene/anthracene | No | Yes | − |
| Pyrene | No | Yes | − |
| Retene | No | Yes | ++ |
Figure 7.
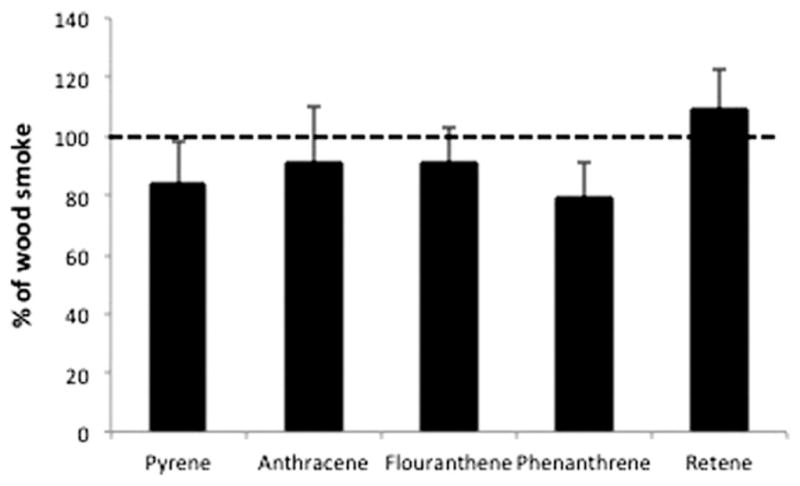
PAH activation of RelB in macrophages. BMdM were generated and cultured with the pure PAHs detected in WS. PAH concentrations were determined using a dilution series focused around the PAH levels detected in WS samples. Cells were incubated with PAHs for 24 h.
Discussion
Exposure to biomass smoke has been epidemiologically associated with pulmonary disease, including asthma and increased susceptibility to respiratory infections (Akunne et al., 2006; Bautista et al., 2009; Kilabuko et al., 2007; Mishra, 2003). While a wide variety of animal models have been utilized in biomass smoke studies including, but not limited to rodents, pigs, dogs and sheep, the mechanism of WS-induced increase in respiratory infection has yet to be defined (Naeher et al., 2007). In addition to constituting the initial line of defense to pathogens, the pulmonary macrophage is a key component in binding and clearing inhaled particulates (Archer et al., 2004; Arredouani et al., 2004; Arredouani et al., 2006; Migliaccio et al., 2005). Therefore, it is highly likely that specific components of WS decrease the antimicrobial activity of pulmonary macrophages.
In the present study, exposures were categorized according to the PM2.5 levels. Exposures ranged from a little over 3 up to nearly 15 mg/m3 with an average of 9.27 mg/m3. This upper range may appear to be well above a “typical” exposure experienced by human populations. However, experience in working with the DustTrak demonstrates that the instrument over-reports PM2.5 concentrations by a factor of 2–3 times when subjected to elevated WS concentrations (Dr. T. Ward, personal communication, 2010). A corrected DustTrak measurement would therefore place the exposures within the range of common, elevated exposures experienced during forest fire, savannah fire or other biomass burning events (Lee et al., 2005; Ward & Lincoln 2006; Zdrahal et al., 2002). In addition, we have previously published (Migliaccio et al., 2009) that comparison of human exposures and those in mouse models requires consideration of multiple parameters. Without compensating for ventilation rates, lung volume and body size (“per kg”), identical PM levels would result in significantly lower amounts of deposition in mice. For example, a 2-h mouse exposure at a concentration of 10 mg/m3 will roughly deposit the same amount as a 2-week human exposure at 0.2 mg/m3. This is a level well within ranges (190–400 μg/m3) reported for communities near wildfires (Brauer & Hisham-Hashim 1998; Naeher et al., 2007; Radzi bin Abas et al., 2004; Ward & Lincoln 2006; Zdrahal et al., 2002).
A variety of inhalation exposures result in pulmonary inflammation in both humans and animal models. The pulmonary macrophage is a key regulator of respiratory immunity and plays an important role in particle-induced inflammation. Studies assessing effects of PM exposures, including urban and diesel exhaust, as well as cigarette smoke and crystalline silica, describe significant pulmonary inflammation (Archer et al., 2004; Das, 2003; Gowdy et al., 2008; Migliaccio et al., 2008; Smith et al., 2002). Typically this inflammation includes an influx of neutrophils as well as increases in soluble markers such as TNFα, IL-1β or total protein (Gowdy et al., 2008; Migliaccio et al., 2005). However, in the present study, following acute exposures to WS, there was no significant or sustained inflammation. There was no increase in total cell numbers, neutrophils, protein or TNFα in lavage. The only indication of inflammation was a significant increase in serum amyloid A at 24 h post-exposure (data not shown). These results show that acute exposure to relatively high levels of WS does not induce a classic inflammatory response, but rather a depression in the normal macrophage immune response.
A significant issue in the field of particle research, which is pertinent to comparison of the present work with other studies, is the concept of standardization. Biomass smoke composition is greatly affected by variables such as fuel source, age of smoke and flaming versus smoldering burning; add to this differences in isolation, concentration and reconstitution of PM in general and one can understand conflicting data between laboratories. While some groups have reported differences with our results (i.e. increase in inflammatory parameters), the lack of standardization for PM acquisition/composition coupled with no unifying models of exposure makes straight comparisons difficult (Danielsen et al., 2008, 2010, 2011; Kocbach et al., 2008; Leonard et al., 2000; Wegesser & Last, 2009; Wegesser et al., 2010). In the present study, in vitro results were compared with in vivo instillation and inhalation models in an effort to validate results. Validation across models and standardization of WS-derived PM would go a long way in increasing the ability of researchers to compare data between studies.
There is ample epidemiological evidence to support the hypothesis that inhalation of biomass smoke can increase susceptibility to respiratory infection (Akunne et al., 2006; Bautista et al., 2009; Kilabuko et al., 2007; Mishra, 2003). In the present study, an increase in SP deposition was observed following WS inhalation (Figure 1). To best determine the role of AM in this infectious model, mice were inoculated 24 h post–WS exposure and processed 2 h later. Preliminary experiments assessed bacterial load 24 h post-inoculation and while there was an increase, it was not significant (data not shown). Because an influx of neutrophils has been observed within 8 h following bacterial inoculations (Clement et al., 2008; Ferreira et al., 2009), it was thought that analysis prior to an influx of neutrophils would be optimal for isolating the affect on the role of macrophages. In addition, recent cigarette smoke studies suggest a more bacterial-specific phagocytic pathway may be involved in an increase in respiratory infection (Phipps et al., 2010). The inoculation studies showed a decrease in the ability of AM to resist bacterial deposition and the APC assay results showed a decreased capacity of AM to induce an adaptive immune response (Figure 2). These results suggest that in the absence of classic inflammation, there is a decreased ability of AM to combat respiratory infection via innate or adaptive immune responses, and that this effect of WS inhalation may contribute to an increased susceptibility to respiratory infection.
Changes in cell populations could also explain and/or contribute to the immune suppression following an acute episode of WS inhalation. The decrease in the activated subset of pulmonary macrophages (Figure 3) is at least a marker of suppression, if not an active component of the process. This observation is opposite of reported increased levels of these subsets in a model of particle (silica) exposure (Migliaccio et al., 2005). Additionally, while the increase of CD25+ T cells in lungs exposed to WS (Figure 4B) appears to be contradictory, it is possible that these lymphocytes are either compensatory T cells (i.e. Th2 T cells down regulating Th1 inflammation via contradictory cytokines) or regulatory T cells. Further studies focusing on this T cell population and their potential cytokines may elucidate this question.
A potential model for the effect of WS on macrophage functions includes PAH components of WS interacting with AhR to activate RelB that then results in a decrease in ability of macrophages to properly respond to pulmonary infections. The AhR is an orphan receptor that binds with dioxin and dioxin-like compounds and can promote alterations in the immune system. The prototypical ligand for AhR is 2,3,7,8-TCDD (i.e. dioxin). Numerous alterations in the immune system have been documented following TCDD exposure (Funatake et al., 2004; Ito et al., 2002; Kerkvliet et al., 2002; Lawrence et al., 2000; Matulka et al., 1997). Multiple types of PAHs and dioxin-like compounds have been described in WS (Ward et al., 2005; Ward & Lincoln 2006). In addition to showing a link between components of cigarette smoke and AhR binding/activation (Baglole et al., 2008; Kitamura & Kasai 2007; Thatcher et al., 2007; Yang et al., 2008), recent studies have described an anti-inflammatory role of AhR in several models (Jensen et al., 2003; Kimura et al., 2009; Lawrence et al., 2008; Negishi et al., 2005). A proposed mechanism for this suppression is via the RelB member of the NF-κB family (Baglole et al., 2008; Thatcher et al., 2007; Vogel et al., 2007). Because RelB has been linked to cigarette smoke-induced suppression of inflammation, we assessed the activation of RelB in mice exposed to WS. AM exposed to inhaled WS or instilled IF-containing PM (IF or WS-PM) stained for nuclear translocation of RelB (Figures 4 and 5). While not every AM from WS-exposed mice presented with RelB nuclear translocation, positive staining was only observed in WS- or IF (IF and WS-PM)-exposed mice and not in controls. In addition, the PM fraction associated with RelB translocation (IF) was found to be a strong activator of the AhR pathway (Figure 6) as well as contain PAHs (Table 1). While the PAHs alone did not fully activate RelB (Figure 7), these data suggest that RelB activation is dependent on the mixture (WS) as a whole, or at least the presence of multiple components including PAHs. These data suggest that suppression of some resident AM via the RelB mechanism is sufficient for a more global suppression of respiratory immunity and an increase susceptibility to infection and may occur via PAH activation of the AhR pathway.
These data support the hypothesis that acute exposure to WS results in an increased susceptibility to respiratory infection. While the phagocytic capacity appears unaffected, the ability of AM to prevent SP deposition is compromised. Besides, the AM ability to mount/induce an adaptive response is also hindered following WS inhalation. Potentially, this is explained by the significant decrease in MHC Class II+ pulmonary macrophages. These detrimental alterations in alveolar immunity are compounded by the decreased ability to mount a normal inflammatory response, which is potentially indicative of further AM functional suppression. In this study these effects were maintained up to one week post-exposure. Therefore, long-term recovery studies, as well as chronic exposures, will need to be assessed using these parameters as a guide.
Acknowledgments
The authors would like to acknowledge Drs Holian and Harmsen, and the Flow Cytometry and Molecular Histology Cores at UM for their aid in these studies, as well as Ray Hamilton for his assistance on statistical analyses.
Footnotes
Declaration of interest
There are no conflicts of interest for any of the authors for the work presented here.
This study was supported by the National Institutes of Health grant R25ES016247 and COBRE grant P20 RR17670. This publication was made possible by grants 4743-RFA04-4/06-4 from HEI, and RR-017670 from the NCRR, a component of NIH, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCRR, NIEHS or NIH.
References
- Akunne AF, Louis VR, Sanon M, Sauerborn R. Biomass solid fuel and acute respiratory infections: the ventilation factor. Int J Hyg Environ Health. 2006;209:445–50. doi: 10.1016/j.ijheh.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Antonini JM, Roberts JR, Yang HM, et al. Effect of silica inhalation on the pulmonary clearance of a bacterial pathogen in Fischer 344 rats. Lung. 2000;178:341–50. doi: 10.1007/s004080000038. [DOI] [PubMed] [Google Scholar]
- Archer AJ, Cramton JL, Pfau JC, et al. Airway responsiveness after acute exposure to urban particulate matter 1648 in a DO11.10 murine model. Am J Physiol Lung Cell Mol Physiol. 2004;286:L337–43. doi: 10.1152/ajplung.00202.2003. [DOI] [PubMed] [Google Scholar]
- Arredouani MS, Yang Z, Imrich A, et al. The macrophage scavenger receptor SR-AI/II and lung defense against pneumococci and particles. Am J Respir Cell Mol Biol. 2006;35:474–8. doi: 10.1165/rcmb.2006-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arredouani M, Yang Z, Ning Y, et al. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med. 2004;200:267–72. doi: 10.1084/jem.20040731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglole CJ, Maggirwar SB, Gasiewicz TA, et al. The aryl hydrocarbon receptor attenuates tobacco smoke induced cyclooxy-genase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappa B family member RELB. J Biol Chem. 2008;283:28944–57. doi: 10.1074/jbc.M800685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista LE, Correa A, Baumgartner J, et al. Indoor charcoal smoke and acute respiratory infections in young children in the Dominican Republic. Am J Epidemiol. 2009;169:572–80. doi: 10.1093/aje/kwn372. [DOI] [PubMed] [Google Scholar]
- Brauer M, Hisham-Hashim J. Peer reviewed: fires in Indonesia: crisis and reaction. Environ Sci Technol. 1998;32:404A–7A. doi: 10.1021/es983677j. [DOI] [PubMed] [Google Scholar]
- Castranova V, Ma JY, Yang HM, et al. Effect of exposure to diesel exhaust particles on the susceptibility of the lung to infection. Environ Health Perspect. 2001;109(Suppl 4):609–12. doi: 10.1289/ehp.01109s4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement CG, Evans SE, Evans CM, et al. Stimulation of lung innate immunity protects against lethal pneumococcal pneumonia in mice. Am J Respir Crit Care Med. 2008;177:1322–30. doi: 10.1164/rccm.200607-1038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsen PH, Brauner EV, Barregard L, et al. Oxidatively damaged DNA and its repair after experimental exposure to wood smoke in healthy humans. Mutat Res. 2008;642:37–42. doi: 10.1016/j.mrfmmm.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Danielsen PH, Loft S, Jacobsen NR, et al. Oxidative stress, inflammation, and DNA damage in rats after intratracheal instillation or oral exposure to ambient air and wood smoke particulate matter. Toxicol Sci. 2010;118:574–85. doi: 10.1093/toxsci/kfq290. [DOI] [PubMed] [Google Scholar]
- Danielsen PH, Moller P, Jensen KA, et al. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem Res Toxicol. 2011;24:168–84. doi: 10.1021/tx100407m. [DOI] [PubMed] [Google Scholar]
- Das SK. Harmful health effects of cigarette smoking. Mol Cell Biochem. 2003;253:159–65. doi: 10.1023/a:1026024829294. [DOI] [PubMed] [Google Scholar]
- Ferreira DM, Moreno AT, Cianciarullo AM, et al. Comparison of the pulmonary response against lethal and non-lethal intranasal challenges with two different pneumococcal strains. Microb Pathog. 2009;47:157–63. doi: 10.1016/j.micpath.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Funatake CJ, Dearstyne EA, Steppan LB, et al. Early consequences of 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on the activation and survival of antigen-specific T cells. Toxicol Sci. 2004;82:129–42. doi: 10.1093/toxsci/kfh245. [DOI] [PubMed] [Google Scholar]
- Gordon S. Biology of the macrophage. J Cell Sci Suppl. 1986;4:267–86. doi: 10.1242/jcs.1986.supplement_4.16. [DOI] [PubMed] [Google Scholar]
- Gordon S. The role of the macrophage in immune regulation. Res Immunol. 1998;149:685–8. doi: 10.1016/s0923-2494(99)80039-x. [DOI] [PubMed] [Google Scholar]
- Gowdy K, Krantz QT, Daniels M, et al. Modulation of pulmonary inflammatory responses and antimicrobial defenses in mice exposed to diesel exhaust. Toxicol Appl Pharmacol. 2008;229:310–9. doi: 10.1016/j.taap.2008.01.040. [DOI] [PubMed] [Google Scholar]
- Hocking WG, Golde DW. The pulmonary-alveolar macrophage (first of two parts) N Engl J Med. 1979a;301:580–7. doi: 10.1056/NEJM197909133011104. [DOI] [PubMed] [Google Scholar]
- Hocking WG, Golde DW. The pulmonary-alveolar macrophage (second of two parts) N Engl J Med. 1979b;301:639–45. doi: 10.1056/NEJM197909203011205. [DOI] [PubMed] [Google Scholar]
- Ito T, Inouye K, Fujimaki H, et al. Mechanism of TCDD-induced suppression of antibody production: effect on T cell-derived cytokine production in the primary immune reaction of mice. Toxicol Sci. 2002;70:46–54. doi: 10.1093/toxsci/70.1.46. [DOI] [PubMed] [Google Scholar]
- Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health. 2003;2:16. doi: 10.1186/1476-069X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet NI, Shepherd DM, Baecher-Steppan L. T lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lymphocyte response by TCDD. Toxicol Appl Pharmacol. 2002;185:146–52. doi: 10.1006/taap.2002.9537. [DOI] [PubMed] [Google Scholar]
- Kilabuko JH, Matsuki H, Nakai S. Air quality and acute respiratory illness in biomass fuel using homes in Bagamoyo, Tanzania. Int J Environ Res Public Health. 2007;4:39–44. doi: 10.3390/ijerph2007010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nakahama T, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206:2027–35. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M, Kasai A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007;252:184–94. doi: 10.1016/j.canlet.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Kocbach A, Namork E, Schwarze PE. Pro-inflammatory potential of wood smoke and traffic-derived particles in a monocytic cell line. Toxicology. 2008;247:123–32. doi: 10.1016/j.tox.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Lawrence BP, Denison MS, Novak H, et al. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low-molecular-weight compound. Blood. 2008;112:1158–65. doi: 10.1182/blood-2007-08-109645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence BP, Warren TK, Luong H. Fewer T lymphocytes and decreased pulmonary influenza virus burden in mice exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) J Toxicol Environ Health A. 2000;61:39–53. doi: 10.1080/00984100050116771. [DOI] [PubMed] [Google Scholar]
- Lee S, Baumann K, Schauer JJ, et al. Gaseous and particulate emissions from prescribed burning in Georgia. Environ Sci Technol. 2005;39:9049–56. doi: 10.1021/es051583l. [DOI] [PubMed] [Google Scholar]
- Leonard SS, Wang S, Shi X, et al. Wood smoke particles generate free radicals and cause lipid peroxidation, DNA damage, NFkappaB activation and TNF-alpha release in macrophages. Toxicology. 2000;150:147–57. doi: 10.1016/s0300-483x(00)00256-0. [DOI] [PubMed] [Google Scholar]
- Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J. 1994;7:1678–89. [PubMed] [Google Scholar]
- Loke J, Paul E, Virgulto JA, Smith GJ. Rabbit lung after acute smoke inhalation. Cellular responses and scanning electron microscopy. Arch Surg. 1984;119:956–9. doi: 10.1001/archsurg.1984.01390200074017. [DOI] [PubMed] [Google Scholar]
- Martin RJ, Wexler RB, Day BJ, et al. Interaction between cigarette smoke and mycoplasma infection: a murine model. COPD. 2006;3:3–8. doi: 10.1080/15412550500493162. [DOI] [PubMed] [Google Scholar]
- Matthew E, Warden G, Dedman J. A murine model of smoke inhalation. Am J Physiol Lung Cell Mol Physiol. 2001;280:L716–23. doi: 10.1152/ajplung.2001.280.4.L716. [DOI] [PubMed] [Google Scholar]
- Matulka RA, Morris DL, Wood SW, et al. Characterization of the role played by antigen challenge in the suppression of in vivo humoral immunity by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Arch Toxicol. 1997;72:45–51. doi: 10.1007/s002040050467. [DOI] [PubMed] [Google Scholar]
- Mcdonald JD, Harrod KS, Seagrave J, et al. Effects of low sulfur fuel and a catalyzed particle trap on the composition and toxicity of diesel emissions. Environ Health Perspect. 2004;112:1307–12. doi: 10.1289/ehp.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio CT, Bergauff MA, Palmer CP, et al. Urinary levoglucosan as a biomarker of wood smoke exposure: observations in a mouse model and in children. Environ Health Perspect. 2009;117:74–9. doi: 10.1289/ehp.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio CT, Buford MC, Jessop F, Holian A. The IL-4Ralpha pathway in macrophages and its potential role in silica-induced pulmonary fibrosis. J Leukoc Biol. 2008;83:630–9. doi: 10.1189/jlb.0807533. [DOI] [PubMed] [Google Scholar]
- Migliaccio CT, Hamilton RF, Jr, Holian A. Increase in a distinct pulmonary macrophage subset possessing an antigen-presenting cell phenotype and in vitro APC activity following silica exposure. Toxicol Appl Pharmacol. 2005;205:168–76. doi: 10.1016/j.taap.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Mishra V. Indoor air pollution from biomass combustion and acute respiratory illness in preschool age children in Zimbabwe. Int J Epidemiol. 2003;32:847–53. doi: 10.1093/ije/dyg240. [DOI] [PubMed] [Google Scholar]
- Mishra V, Retherford RD. Cooking smoke increases the risk of acute respiratory infection in children. Natl Fam Health Surv Bull. 1997;8:1–4. [PubMed] [Google Scholar]
- Naeher LP, Brauer M, Lipsett M, et al. Woodsmoke health effects: a review. Inhal Toxicol. 2007;19:67–106. doi: 10.1080/08958370600985875. [DOI] [PubMed] [Google Scholar]
- Negishi T, Kato Y, Ooneda O, et al. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J Immunol. 2005;175:7348–56. doi: 10.4049/jimmunol.175.11.7348. [DOI] [PubMed] [Google Scholar]
- Phipps JC, Aronoff DM, Curtis JL, et al. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun. 2010;78:1214–20. doi: 10.1128/IAI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzi Bin Abas M, Oros DR, Simoneit BR. Biomass burning as the main source of organic aerosol particulate matter in Malaysia during haze episodes. Chemosphere. 2004;55:1089–95. doi: 10.1016/j.chemosphere.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Ross MH, Murray J. Occupational respiratory disease in mining. Occup Med (Lond) 2004;54:304–10. doi: 10.1093/occmed/kqh073. [DOI] [PubMed] [Google Scholar]
- Smith KR, Samet JM, Romieu I, Bruce N. Indoor air pollution in developing countries and acute lower respiratory infections in children. Thorax. 2000;55:518–32. doi: 10.1136/thorax.55.6.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Uyeminami DL, Kodavanti UP, et al. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med. 2002;33:1106–14. doi: 10.1016/s0891-5849(02)01003-1. [DOI] [PubMed] [Google Scholar]
- Thatcher TH, Maggirwar SB, Baglole CJ, et al. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–64. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun. 2007;363:722–6. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TJ, Hamilton RF, Jr, Smith GC. The Missoula Valley semivolatile and volatile organic compound study: seasonal average concentrations. J Air Waste Manag Assoc. 2005;55:1007–13. doi: 10.1080/10473289.2005.10464698. [DOI] [PubMed] [Google Scholar]
- Ward TJ, Lincoln E. Concentrations of PM(2.5)-associated OC, EC, and PCDD/Fs measured during the 2003 wildfire season in Missoula, Montana. Environ Monit Assess. 2006;115:39–50. doi: 10.1007/s10661-006-5252-6. [DOI] [PubMed] [Google Scholar]
- Ward TJ, Noonan CW, Hooper K. Results of an indoor size fractionated PM school sampling program in Libby, Montana. Environ Monit Assess. 2007;130:163–71. doi: 10.1007/s10661-006-9386-3. [DOI] [PubMed] [Google Scholar]
- Wegesser TC, Last JA. Mouse lung inflammation after instillation of particulate matter collected from a working dairy barn. Toxicol Appl Pharmacol. 2009;236:348–57. doi: 10.1016/j.taap.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegesser TC, Franzi LM, Mitloehner FM, et al. Lung antioxidant and cytokine responses to coarse and fine particulate matter from the great California wildfires of 2008. Inhal Toxicol. 2010;22:561–70. doi: 10.3109/08958370903571849. [DOI] [PubMed] [Google Scholar]
- Westerling AL, Hidalgo HG, Cayan DR, Swetnam TW. Warming and earlier spring increase western U.S. forest wildfire activity. Science. 2006;313:940–3. doi: 10.1126/science.1128834. [DOI] [PubMed] [Google Scholar]
- Yang SR, Yao H, Rajendrasozhan S, et al. RelB is differentially regulated by IκB-Kinase-α (IKK-α) in B cells and mouse lung by cigarette smoke. Am J Respir Cell Mol Biol. 2008;40:147–58. doi: 10.1165/rcmb.2008-0207OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdrahal Z, Oliveira J, Vermeylen R, et al. Improved method for quantifying levoglucosan and related monosaccharide anhydrides in atmospheric aerosols and application to samples from urban and tropical locations. Environ Sci Technol. 2002;36:747–53. doi: 10.1021/es015619v. [DOI] [PubMed] [Google Scholar]
- Zelikoff JT, Chen LC, Cohen MD, Schlesinger RB. The toxicology of inhaled woodsmoke. J Toxicol Environ Health B Crit Rev. 2002;5:269–82. doi: 10.1080/10937400290070062. [DOI] [PubMed] [Google Scholar]