

Abstract
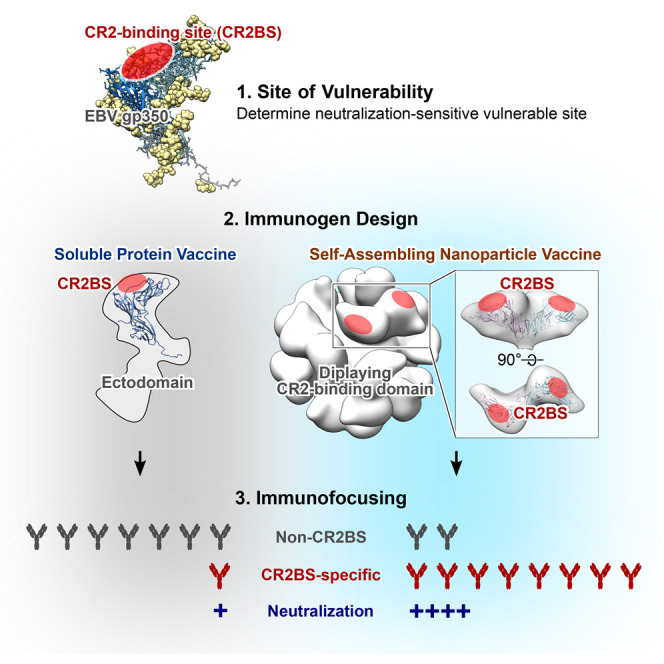
Epstein-Barr virus (EBV) represents a major global health problem. Though it is associated with infectious mononucleosis and ∼200,000 cancers annually worldwide, a vaccine is not available. The major target of immunity is EBV glycoprotein 350/220 (gp350) that mediates attachment to B cells through complement receptor 2 (CR2/CD21). Here, we created self-assembling nanoparticles that displayed different domains of gp350 in a symmetric array. By focusing presentation of the CR2-binding domain on nanoparticles, potent neutralizing antibodies were elicited in mice and non-human primates. The structurally designed nanoparticle vaccine increased neutralization 10- to 100-fold compared to soluble gp350 by targeting a functionally conserved site of vulnerability, improving vaccine-induced protection in a mouse model. This rational approach to EBV vaccine design elicited potent neutralizing antibody responses by arrayed presentation of a conserved viral entry domain, a strategy that can be applied to other viruses.
PaperClip
Graphical Abstract
Highlights
-
•
Self-assembling nanoparticles present the conserved gp350 receptor-binding domain
-
•
The nanoparticles elicit more potent neutralizing antibodies than soluble gp350
-
•
These neutralizing antibodies predominantly target the CR2-binding site on gp350
-
•
The nanoparticles elicit potent neutralizing antibodies in mice and non-human primates
Structurally designed EBV vaccine candidates based on self-assembling nanoparticles elicit potent and durable virus-neutralizing antibodies that target the receptor-binding site on the viral envelope protein gp350, a site of vulnerability, serving as a template to develop an EBV vaccine and providing a basis for immunofocusing through rational vaccine design.
Introduction
Epstein-Barr virus (EBV) infection is associated with multiple human diseases, including infectious mononucleosis (IM) and a variety of malignancies. Burkitt and Hodgkin lymphoma, gastric, and nasopharyngeal carcinoma are among the neoplasms observed after infection, as are lymphoproliferative disorders. The prevalence and the severity of these diseases underscore the potential public health benefit of an EBV vaccine (Cohen et al., 2011).
Despite the morbidity associated with EBV, there are no prophylactic vaccines, though the virus was isolated and identified more than a half century ago (Epstein et al., 1964). Attempts to develop a vaccine have focused on the viral major envelope glycoprotein 350/220 (gp350), because it represents a principal target of neutralizing antibodies in naturally infected individuals (Hoffman et al., 1980, Thorley-Lawson and Geilinger, 1980, Thorley-Lawson and Poodry, 1982). Prototype gp350-based vaccines have induced protective immunity against EBV-mediated lymphomas in a cottontop tamarin challenge model of disease (Epstein et al., 1985) and more recently reduced infection in a rhesus macaque model in which both immunization and challenge were performed using rhesus macaque lymphocryptovirus, a homolog of EBV (Sashihara et al., 2011). Selected candidate prophylactic EBV vaccines have been evaluated in humans (Elliott et al., 2008, Gu et al., 1995, Moutschen et al., 2007, Sokal et al., 2007). The only phase II trial of an EBV prophylactic vaccine used recombinant soluble gp350 with an AS04 adjuvant. That vaccine demonstrated a 78% reduction in the rate of IM in EBV-seronegative vaccine, but it did not protect against acquisition of primary infection (Sokal et al., 2007), limiting enthusiasm for its further development.
EBV infects B cells by engaging viral gp350 to its primary receptor, complement receptor 2 (CR2/CD21) (Fingeroth et al., 1984), or alternatively CR1 (CD35) (Ogembo et al., 2013). The heterotrimeric viral glycoprotein complex, gH/gL/gp42, binds to HLA class II molecules as a co-receptor on B cells, while heterodimeric gH/gL and BMRF2 engage integrins as primary receptors to infect epithelial cells (Connolly et al., 2011, Hutt-Fletcher, 2007, Tugizov et al., 2003). Although inhibition of any of these viral glycoproteins by antibody or gene disruption prevents or severely impairs viral infection of cells, the degree of virus neutralization varies by antibody specificity and the target cell type. A murine monoclonal antibody (mAb) 72A1 potently neutralizes EBV infection of B cells (Hoffman et al., 1980), and its predicted epitope on gp350 largely overlaps the inferred binding site of CR2, suggesting a mechanism of neutralization mediated by this antibody (Tanner et al., 1988). The mAb 72A1 also blocks the interaction of gp350 with CR1 (Ogembo et al., 2013), further indicating that this epitope is functionally important and hence an attractive vaccine target to prevent viral infection of B cells, a principal target cell type of EBV. Here, we aimed to elicit potent neutralizing antibodies to this epitope by immunizing with a vaccine designed based on a rational understanding of structural biology and nanotechnology to optimize presentation and recognition of this site of vulnerability.
Results
Design and Expression of EBV gp350-Based Nanoparticles
To display monomeric antigens on the surface of self-assembling nanoparticles, we identified two potential platforms, ferritin (Cho et al., 2009) and encapsulin (Sutter et al., 2008). Ferritins form an octahedral cage consisting of 24 subunits, while encapsulin forms an icosahedron made of 60 identical subunits (T = 1). Ferritins have been engineered for multiple purposes (Huard et al., 2013, Jääskeläinen et al., 2007, Li et al., 2006, Meldrum et al., 1992). Among these innovations, we have recently developed a platform to mount a viral trimeric glycoprotein at ferritin’s 3-fold axes, providing a basis for structure-based nanoparticulate immunogens (Kanekiyo et al., 2013). Comparison of ferritin structures revealed that certain ferritins, including human light chain (Wang et al., 2006) (PDB: 2FFX) and bullfrog lower subunit (Trikha et al., 1995) (PDB: 1RCC), contained an NH2-terminal extension that was not present in ferritin from Helicobacter pylori (Cho et al., 2009) (PDB: 3EGM). This NH2-terminal extension makes the NH2-terminal residue project radially from the nanoparticle core, and the termini are evenly spaced on the surface. Due to relatively high sequence homology between bullfrog and human ferritins (62%), we were concerned that use of either as a vaccine platform could induce an autoimmune reaction.
The overall architecture of ferritin subunits from different species is largely similar, although the sequences of H. pylori and human ferritins are diverse (18% and 24% identity to human light and heavy chains, respectively). To avoid the potential for vaccine-induced autoimmunity, we therefore fused the amino terminal extension of bullfrog ferritin to H. pylori ferritin to build a hybrid that has the antigen-attachment sites distributed evenly on the surface (Figure 1A, left). Although encapsulin has not been studied as a scaffold to present heterologous proteins on its surface, we found that the COOH-terminal of encapsulin subunits project outward from the particle core and are located proximal to the 5-fold symmetry axis (Figure 1A, right). This allowed us to create COOH-terminal fusion proteins to present foreign antigen on the nanoparticle surface.
Figure 1.

Molecular Design of gp350-Based Nanoparticles
(A) Assembled hybrid ferritin model (left) and encapsulin (right). Engineered insertion sites for exogenous antigens are indicated as red spheres.
(B) Schematic representation of full-length gp350, its truncated variants, and gp350-based nanoparticles. Domains I, II, and III of gp350 are color coded in dark blue, sky blue, and pale blue, respectively. Structurally undefined region and transmembrane (TM)/cytoplasmic tail (CT) are colored in gray and white, respectively. Hexa-histidine tag (HIS), ferritin, and encapsulin are colored in black, red, and green, respectively. Amino acid (AA) position relative to full-length gp350 is indicated at the bottom.
See also Figure S1.
EBV gp350 is a type I transmembrane protein composed of 907 amino acids, including an ectodomain, a transmembrane domain, and a cytoplasmic tail (Figures 1B and S1 A). We analyzed different gp350 truncation variants for their ability to form nanoparticles and found that the smallest gp350 variant (D12) failed to express on both ferritin and encapsulin platforms, suggesting instability of this variant. The largest variant (ectodomain) with ferritin was expressed; however, a mixture of assembled and misassembled/disassembled species was observed that excluded further use of this construct (Figure S1B). Two truncation variants, termed D2H6O and D123, expressed stably on the ferritin and encapsulin without disturbing self-assembly of the nanoparticles. Although gp350 is extensively posttranslationally modified by both N- and O-linked glycosylations (Machiels et al., 2011, Serafini-Cessi et al., 1989), the shorter D123 truncation eliminates most of the “mucin-like” domain, which may prevent recognition of gp350 by the immune system (Figure S1C), and hence D123 was selected for further studies. The purified soluble gp350 proteins and D123 nanoparticles were homogenous, as shown by size exclusion chromatography (Figure 2 A).
Figure S1.

Design of Truncated gp350 Variants, Related to Figure 1
(A) Crystal structure of gp350 receptor-binding domain (Szakonyi et al., 2006) (PDB: 2H6O) (left). Structure of amino terminal portion of gp350 (residues 4-443) is shown. Structurally defined domains I, II and III are colored in blue, sky blue and pale blue, respectively. Attached N-linked glycans are shown in yellow spheres. Inferred receptor-binding site (CR2BS) is located between domains I and II and is indicated by a dotted circle. Schematic representation of truncated gp350 constructs (right). Amino acid (AA) position of the last residue of each design is denoted. Domains I, II and III are colored as shown in the left panel. (B) Size exclusion chromatography profile of gp350 ectodomain-ferritin purified by GNA lectin resin. Arrow indicates the predicted size of assembled particle. (C) Predicted N- and O-linked glycosylation sites in gp350. N-linked (top) and O-linked (bottom) glycosylation sites in the full-length gp350 (UniProtKB: P03200, spanning residues 1-907) were predicted using NetNGlyc 1.0 and NetOGlyc 3.1 programs (CBS Prediction Servers at http://www.cbs.dtu.dk/services), respectively. Glycosylation potential scores above the dotted line (0.5) were considered as positive.
Figure 2.

Biochemical and Antigenic Characterization of gp350-Based Nanoparticles
(A) Size exclusion chromatography profiles of soluble gp350 ectodomain, D123, and D123 nanoparticles. After affinity purification with Ni-NTA (soluble gp350 proteins) or GNA lectin (D123-ferritin and D123-encapsulin), proteins were separated by size exclusion chromatography using a Superdex 200 10/300 (soluble gp350 proteins) (left) or a Sephacryl S-500 16/60 (D123 nanoparticles) column (right).
(B and C) Binding of anti-gp350 mAbs to purified proteins was assessed by immunoprecipitation (B) and ELISA (C). HC and LC indicate heavy and light chains of antibody, respectively. Each symbol represents mean ± SD.
See also Figure S2.
Antigenicity and Structural Integrity of EBV gp350-Based Nanoparticles
We next examined the antigenicity of gp350 displayed on nanoparticles by using mAbs to gp350. mAb 72A1 (Hoffman et al., 1980) recognizes the CR2-binding site (CR2BS) of gp350 and potently neutralizes virus, whereas mAb 2L10 (Luka et al., 1984) is a non-neutralizing anti-gp350 antibody and does not compete with 72A1. Both D123-ferritin and D123-encapsulin were immunoprecipitated equivalently by 72A1 and 2L10, but not by an irrelevant mAb C179 (Okuno et al., 1993) (Figure 2B). Analogous to soluble gp350 ectodomain, the D123 nanoparticles bound to 72A1 and 2L10; however, soluble D123 showed reduced reactivity to both mAbs by ELISA (Figure 2C), suggesting that the conformation of D123 was preserved on the nanoparticles while that of soluble D123 was altered. This indicated that the soluble form of D123 differed biochemically from either gp350 ectodomain or D123 nanoparticles, although the overall thermostability of these proteins was similar, as measured by differential scanning calorimetry (Figure S2 A). To assess whether the D123 nanoparticles could bind to their cognate ligand, we tested their binding to CR2 on human B cells by flow cytometry. As expected, both soluble gp350 and D123 nanoparticles bound to CR2hi B cells, and the binding was dependent on CR2 expression, as the CR2lo B cells and T cells (CR2−) showed weaker and no binding, respectively (Figure S2B). Preincubation of anti-CR2 antibody diminished the binding of gp350 to CR2, indicating that the interaction was mediated by CR2 (Figure S2B).
Figure S2.

Thermostability and CR2-Binding Properties of Soluble gp350s and gp350-Based Nanoparticles, Related to Figure 2
(A) Thermostability of gp350 ectodomain, D123, D123-ferritin and D123-encapsulin was measured by differential scanning calorimetry (DSC) using a Microcal VP-capillary DSC apparatus (GE Healthcare). Purified proteins were diluted to 0.5 mg ml−1 in PBS and subjected to DSC measurement (30-100°C, 1°C min−1). (B) Binding of gp350-based immunogens to human B cells. Human peripheral blood mononuclear cells were stained with the following monoclonal antibodies: CD3 Cy7-APC, IgG BV421, IgM-Cy5.5-peridinin chlorophyll protein (PerCP), CR2 (CD21) Cy5PE (BD Biosciences), CD14 BV510, CD56 BV510 (Biolegend), CD27-QD605 (Life Technologies), CD19 ECD (Beckman Coulter), CD38 Ax680 (In house: OKT10). Cell viability was assessed using Aqua Blue amine-reactive dye (Life Technologies). Approximately 0.1 ug of EBV gp350 conjugated protein was used for each sample. The gp350 ectodomain, D123, D123-ferritin and D123-encapsulin were biotinylated through primary amine group prior to conjugated with streptavidin-PE. Cells were analyzed on a LSR-II flow cytometer (BD Biosciences) and the data were processed in FlowJo software version 9.8.2 (FlowJo). Cell gating strategy is shown (top). Binding profile of gp350-based immunogens was represented as a histogram for each cell subset (middle). Binding profile of the sample pre-incubated with an anti-CR2 antibody (BD Biosciences, Clone 1048) (bottom).
Electron microscopic (EM) analysis confirmed that the D123 and D2H6O nanoparticles formed monodispersed particles with globular protrusions from both ferritin and encapsulin nanoparticles (Figures 3 A and S3 ). Cryo-EM reconstruction further confirmed the symmetry of gp350 D123 on ferritin and encapsulin nanoparticles (Figure 3A). The location of the gp350 protrusions on the ferritin nanoparticles correlated with the 2-fold symmetry and formed a dimer; however, the monomeric gp350 protrusions in the encapsulin nanoparticles are well separated (Figure 3A). Although gp350 D123 is predicted to be highly glycosylated, the dimer interface observed in ferritin cryo-EM contains hydrophobic patches that might be favored to form a dimer. As expected, the inferred CR2BS on the gp350 D123 is exposed on the surface of ferritin and is readily accessible for immune recognition (Figure 3B).
Figure 3.

Structural Characterization of gp350-Based Nanoparticles
(A) Negative-stain transmission EM images (top) and cryo-EM reconstruction model (bottom) of D123 nanoparticles.
(B) Fit model of gp350 D123 protrusion on ferritin nanoparticle. PDB entry 2H6O of gp350 (residues 4–425) was superimposed into cryo-EM density map. Inferred CR2BS is indicated. EM density maps of D123-ferritin and D123-encapsulin were deposited into EMDB under accessions EMDB-3025 and EMDB-6341, respectively.
See also Figure S3.
Figure S3.
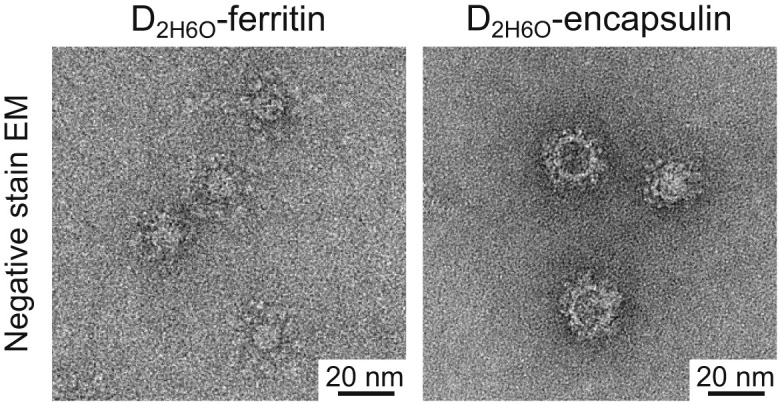
Electron Microscopic Analysis of gp350 D2H6O Nanoparticles, Related to Figure 3
Negative-stain EM images of gp350 D2H6O-ferritin and D2H6O-encapsulin. The D2H6O-nanoparticles were prepared and purified as described for the D123-nanoparticles. The observed protrusions of gp350 D2H6O are slightly larger than that of D123, accounting for the slightly larger overall size of the nanoparticles.
Immunogenicity of EBV gp350-Based Nanoparticles
To characterize the antibody response elicited by soluble gp350 derivatives and D123 nanoparticles, mice were immunized with 5.0 μg of soluble gp350 ectodomain, D123, D123-ferritin, or D123-encapsulin in the presence of Sigma Adjuvant System (SAS) at weeks 0 and 3. After the first dose, antibody binding to gp350 was detected in mice immunized with D123-ferritin and D123-encapsulin (ELISA endpoint titers of 104.6 ± 0.1 and 104.4 ± 0.4, respectively). These titers were >100-fold higher than those found in mice immunized with gp350 ectodomain (Figure 4 A). Antibodies to gp350 were also measured in a luciferase immunoprecipitation system (LIPS) assay, which has been shown to correlate well with EBV neutralization activity, and in a virus neutralization assay using a GFP-reporter virus (Sashihara et al., 2009) (Figure 4A). The second dose of D123-ferritin or D123-encapsulin substantially increased gp350 antibody titers. The binding dissociation rate of anti-gp350 antibodies in sera was also improved by 3- to 5-fold after the second dose, as measured using biolayer interferometry (Figure S4 A), indicating that anti-gp350 antibodies showed greater affinity maturation and bound more tightly to gp350 than antibodies elicited after the first dose, as observed with other vaccines (Bachmann et al., 1997, Khurana et al., 2011, Lee et al., 2012). After the second injection, gp350 ectodomain elicited anti-gp350 antibody titers only ∼2-fold lower than those in mice immunized with D123-ferritin or D123-encapsulin; but the neutralization activity of the sera from mice immunized with gp350 ectodomain was ∼1,000-fold lower than that of sera from D123-ferritin or D123-encapsulin immunization groups (Figure 4A). This discrepancy was also observed in the LIPS assay (Figure 4A). Immunization with soluble D123 elicited a negligible antibody response by ELISA, LIPS, and virus neutralization assays, indicating that the monomeric form of this protein is much less immunogenic. No virus-neutralizing antibody was induced by ferritin alone or ferritin mixed with either soluble gp350 ectodomain or D123 in contrast to immunization with D123-ferritin (Figure 4B). These data document that a physical linkage of antigen to nanoparticle is required for antigen-specific immune induction and show that ferritin does not stimulate a non-specific adjuvant response. Immunization with a 10-fold lower dose (0.5 μg) of D123-ferritin and D123-encapsulin induced virtually the same gp350 ELISA, LIPS, and neutralizing antibody titers in mice (Figure S4B).
Figure 4.

Immunogenicity of Soluble gp350 Monomer and gp350-Based Nanoparticles
(A) Groups of BALB/c mice (n = 5) were immunized with 5.0 μg of soluble gp350 ectodomain, D123, D123-ferritin, or D123-encapsulin in adjuvant at weeks 0 and 3. Immune sera were collected 2 weeks after the first (1) and the second (2) immunizations and analyzed by gp350 ELISA, LIPS, and virus neutralization assays. Endpoint binding titer (left), LIPS relative light units (RLU) (middle), and neutralization IC50 titer (right) were determined. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(B) Neutralization titers from mice 2 weeks after the second immunization with ferritin alone, ferritin mixed with gp350, or D123-ferritin. The data are shown as box-and-whiskers plots (box indicates lower and upper quartiles with line at median, and whiskers span minimum and maximum data points) with individual data points.
(C) Kinetics of serum neutralization titers after immunization with gp350 ectodomain or gp350-based nanoparticles. Groups of BALB/c mice (n = 5) were immunized with 5.0 μg of gp350 ectodomain, D123-ferritin, or D123-encapsulin in adjuvant at weeks 0, 3, and 16. Immune sera were collected periodically after immunization, and serum neutralization IC50 titers were determined and plotted. Each dot represents an individual mouse. The horizontal dotted line represents the detection limit of the assay.
See also Figure S4.
Figure S4.

Affinity Maturation, Low-Dose Immunogenicity, and Anti-Autologous Ferritin Antibody after Immunization with Soluble gp350s and gp350-Based Nanoparticles, Related to Figure 4
(A) Avidity of anti-gp350 antibody after immunization with gp350-based immunogens. Serum antibody binding to gp350 was measured after the first (1) and second (2) immunizations with 5.0 μg of gp350 ectodomain, D123-ferritin or D123-encapsulin with adjuvant by biolayer interferometry (fortéBio Octet HTX, Pall). Dilutions of pooled sera (n = 5) are indicated. Antibody dissociation rates (off-rate, kd) were calculated by Octet analysis software version 8.0 using 1:2 bivalent analyte model. (B) Groups of BALB/c mice (n = 5) were immunized intramuscularly with 0.5 μg of soluble gp350 ectodomain, D123, D123-ferritin or D123-encapsulin mixed with adjuvant at weeks 0 and 3. Immune sera were collected 2 weeks after the first (1) and the second (2) immunizations and analyzed by gp350 ELISA, gp350 LIPS, and virus neutralization assays. Endpoint binding titer (left panels), LIPS relative light units (RLU) (middle panels), and IC50 neutralization titer (right panels) were determined and plotted. The data are shown as box-and-whiskers plots (box indicates lower and upper quartiles with bar at median and whiskers spanning minimum and maximum data points) with individual data points. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The horizontal dotted line represents the detection limit of the assay. (C) Kinetics of serum neutralization titers after immunization with gp350 ectodomain or gp350-based nanoparticles. Groups of BALB/c mice (n = 5) were immunized intramuscularly with 0.5 μg of gp350 ectodomain, D123-ferritin or D123-encapsulin mixed with adjuvant at weeks 0, 3 and 16. Immune sera were collected periodically after immunization and serum neutralization IC50 titers were determined and plotted. Each dot represents an individual mouse. The horizontal dotted line represents the detection limit of the assay. (D) Antibody response to murine ferritin. Serum antibody response to autologous murine ferritin was measured after the second (2) and third (3) immunization with 5.0 μg of gp350 D123-ferritin with adjuvant by ELISA. Rabbit anti-mouse ferritin polyclonal antibody (pAb) was used as positive control in the assay. The horizontal dotted line represents the detection limit of the assay.
Durability of Neutralizing Antibodies Elicited by gp350-Based Nanoparticles
We next assessed the kinetics of virus-neutralizing antibody in immunized mice. Two months after the second dose of vaccine (week 11), the neutralizing antibody titers in mice receiving D123-ferritin and D123-encapsulin declined about 10-fold from the peak titers but then remained stable until 3 months after the second dose without further immunization (Figure 4C). Neutralizing antibody titers in mice immunized with gp350 ectodomain were slightly increased over time but still remained at a low level (101.3 ± 0.5) (Figure 4C). Mice were then immunized with a third dose at week 16. The titers were increased by 33- and 26-fold in D123-ferritin and D123-encapsulin, respectively (IC50 titers of 104.3 ± 0.2 and 103.7 ± 0.2, respectively) after boosting, and those titers did not wane as rapidly as after a second dose and remained high even after 3 months (IC50 titers of 103.7 ± 0.3 and 103.6 ± 0.2, respectively) (Figure 4C). The soluble gp350 ectodomain did not substantially enhance the neutralizing antibody titers after three immunizations (Figure 4C). We again observed similar titers and kinetics of neutralizing antibody in the lower-dose (0.5 μg) groups immunized with D123-ferritin or D123-encapsulin (Figure S4C). Importantly, no cross-reactive antibodies to autologous (i.e., murine) ferritin were observed even after three immunizations with D123-ferritin in mice, consistent with previous findings that tolerance to homologous ferritin is preserved (Kanekiyo et al., 2013) (Figure S4D).
Immunogenicity of gp350-Based Nanoparticles in Non-human Primates
To evaluate vaccine-induced immunity in a species more closely related to humans, we immunized cynomolgus macaques (Macaca fascicularis). Monkeys were given 50 μg of gp350 ectodomain or 25 μg of either gp350 D123-ferritin or D123-encapsulin with adjuvant (SAS) at weeks 0, 4, and 12. Because these monkeys are naturally infected with a lymphocryptovirus that shares homology with EBV, cross-neutralizing EBV antibody was found in all monkeys prior to immunization (IC50 titers from 101.1 to 102.0) (Figure 5 A). Nevertheless, the neutralizing antibody titers increased after two doses (week 6) with either D123 nanoparticle, and these titers were further enhanced by 119- and 25-fold from the baseline after a third dose (week 14) of D123-ferritin and D123-encapsulin, respectively (Figure 5A). Neutralization titers in gp350 ectodomain-immunized animals were ∼0.5 to 1.0 log lower, although the overall antibody binding titers were similar to that of D123-ferritin and D123-encapsulin-immunized animals (Figures 5A and 5B). The data confirm the immunogenicity of gp350-based nanoparticles in a second species and indicate that antibody production can be enhanced in the presence of pre-existing immunity.
Figure 5.

Immunogenicity of gp350-Based Nanoparticles in Cynomolgus Macaques
(A) Neutralization titers in monkeys immunized with 50 μg of gp350 ectodomain, 25 μg of gp350 D123-ferritin, or D123-encapsulin in adjuvant at weeks 0, 4, and 12. Plasma was collected prior to immunization and periodically after immunization.
(B) Plasma gp350 binding antibody titer in immunized monkeys after three immunizations. Each symbol represents an individual monkey. The horizontal dotted line represents the detection limit of the assay. The data are shown as box-and-whiskers plot with individual data points.
Protective Immunity against Experimental Challenge with Recombinant Vaccinia Virus Expressing EBV gp350
To test the efficacy of vaccine-induced immunity in a mouse challenge model, we employed a recombinant virus challenge using a vaccinia virus that expressed EBV gp350 (rVV-gp350). We constructed and characterized rVV-gp350 and titrated the dose of virus in mice to establish a model in which weight loss was observed as a result of the infection (Figure S5 ), as has been reported with parental vaccinia virus (Chen et al., 2007). When mice were challenged 2 months after the third immunization, all mice immunized with gp350 ectodomain and control mice, with one exception, lost ≥30% of their weight within 8 days and required euthanization; however, four out of five of the D123-ferritin-immunized mice were protected, showing milder weight loss followed by rapid recovery (Figure 6 A, p = 0.0027 for mice immunized with D123-ferritin versus gp350 ectodomain). We observed similar findings in mice that were challenged even 10 months after the third immunization (Figure 6B, p = 0.0210 for mice immunized with D123-ferritin versus gp350 ectodomain). Protection was not observed in mice immunized with D123-encapsulin in both experiments (Figures 6A and 6B). We note that the majority of the vaccinia virions would not be enveloped by the plasma membrane where the gp350 would be expressed but instead envelope would be derived from the endoplasmic reticulum. Hence, anti-gp350 antibody would likely not completely neutralize the incoming vaccinia virus particles and allow the incoming virus to infect cells for at least one round of replication. In contrast, it is more likely that vaccinia virus-infected cells that express gp350 on the surface are prone to be killed by antibody-dependent cell cytotoxicity, complement-dependent cytotoxicity, or cytotoxic T cells. We think that one or more of these mechanisms mediate viral clearance in the vaccinia challenge model. We observed slightly higher neutralizing activity and increased binding affinity of anti-gp350 antibody in D123-ferritin-immunized groups than in D123-encapsulin groups (Figures 4C, S4A, and S4B), but the difference was not statistically significant, suggesting that other mechanisms may contribute to protection.
Figure S5.

Titration of Recombinant Vaccinia Virus Expressing gp350 in Mice, Related to Figure 6
Groups of BALB/c mice were challenged intranasally with recombinant vaccinia virus expressing gp350 (rVV-gp350) at 105, 106 or 107 PFU. Kaplan-Meier survival curve (left) and body weight change (right) are plotted.
Figure 6.

Protective Immunity against an Experimental Infection with Recombinant Vaccinia Virus Expressing EBV gp350 in Mice
(A and B) Kaplan-Meier survival curve and body weight change after recombinant gp350-vaccinia virus challenge. Mice (n = 5) were immunized with 5.0 μg of gp350 ectodomain, gp350 D123-ferritin, or D123-encapsulin at weeks 0, 3, and 16 and were challenged at 2 months after the final immunization (A) or immunized three times with 0.5 μg of vaccines and challenged at 10 months after the final immunization (B). Summary of p values between groups (generated by log-rank Mantel-Cox test) is shown in each panel (ns, not significant). Mock, irrelevant nanoparticle-immunized mice (n = 5); control, age-matched naive mice (n = 5). See also Figure S5.
Induction of CR2BS-Specific Antibodies by D123 Nanoparticle Immunization
To define the specificity of the antibodies elicited by gp350-based immunogens, we used a surface plasmon resonance (SPR)-based antibody competition assay (McLellan, 2013) (Figure S6 ). Strikingly, more than half of the total anti-gp350 antibodies in the sera elicited by either D123-ferritin or D123-encapsulin was competed by CR2BS-directed mAb 72A1 when it was used as a competitor (52% ± 11% or 60% ± 12%, respectively), whereas only 5% ± 4% of antibodies in immune sera from gp350 ectodomain-immunized animals was blocked by this mAb (Figures 7A and 7B). This competition demonstrated that the CR2BS on both D123 nanoparticles was the predominant target of recognition; however, the same site on the gp350 ectodomain was not the major target. In addition, these differences were not due to total levels of gp350 antibody since the ELISA endpoint titers differ only ∼2-fold between the three immunization groups after the second dose (Figure 4A). Interestingly, only a negligible fraction of anti-gp350 antibodies in the same immune sera was competed by non-neutralizing, non-CR2BS-directed mAb 2L10 (6% ± 5%, 5% ± 4%, and 12% ± 16% with gp350 ectodomain, D123-ferritin, and D123-encapsulin, respectively), although 72A1 and 2L10 epitopes were equally available and accessible for immune recognition (Figures 2B, 2C, 7A, and 7B).
Figure S6.

SPR-Based Antibody Cross-Competition Assay, Related to Figure 7
A sensor chip immobilized with purified soluble gp350 ectodomain through amine coupling reaction was used. Either irrelevant mAb (C179) or competing mAb (72A1 or 2L10) was first injected onto the gp350-chip to saturate the mAb’s binding site and then the immune serum was injected onto the mAb-saturated gp350-chip to measure residual binding. In the case of irrelevant mAb-saturated gp350-chip, antibodies in the immune serum can bind to any gp350 surface thus measuring maximum binding (left). In the case of competing mAb-saturated gp350-chip, serum antibodies can only bind to gp350 surface not occupied by the competing mAb thus measuring serum gp350 binding excluding sites blocked by the competing mAb (right).
Figure 7.

Detection of CR2BS-Directed Antibodies in Immune Sera
(A) SPR-based cross-competition assay of immune sera with mAbs. Binding of immune sera (after the second immunization) to the mAb-saturated gp350 was measured by Biacore. Each curve represents an individual serum. All data were normalized with C179-saturated curves and are shown as fraction response. Cross-competition of immune sera by 72A1 (left) and 2L10 (right) are shown for different immunization groups.
(B) Specificity of gp350-binding antibodies in immune sera. Percent inhibition of serum antibodies to bind gp350 by 72A1 or 2L10 was calculated.
(C) Generation of EBV gp350 CR2BS mutants. The gp350 domains I and II are shown in blue and sky blue, respectively. Attached glycans are shown as yellow spheres. Residues 162 and 208 are shown in red, and inferred CR2BS is indicated (PDB: 2H6O) (left). Binding of gp350 WT and CR2BS mutants to mAbs 72A1 and 2L10 was measured by ELISA (right). Each symbol represents mean ± SD.
(D) Serum neutralization titers determined in the absence (closed circle) or presence of gp350 WT (open circle) and glyc162/208 (open square) proteins.
(E) Preferential elicitation of antibody response to CR2BS in mice and monkeys. Serum antibody response to either gp350 WT or glyc162/208 was measured after the second immunization with indicated immunogens. Endpoint ELISA antibody titers for both gp350 WT and glyc162/208 were determined, and ratios of those titers were calculated (WT ⁄ glyc162/208).
The data in (B), (D), and (E) are shown as box-and-whiskers plots with individual data points. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S6.
To confirm the functional relevance of the CR2BS-directed antibodies in immune sera, we designed gp350 ectodomain mutants to abolish CR2 and 72A1 binding (Figure 7C). One mutant had an extra N-linked glycosylation site at residue 162 (glyc162), and another mutant had two extra glycans at residues 162 and 208 (glyc162/208), as both 162 and 208 have been reported as critical residues for CR2 and 72A1 binding (Szakonyi et al., 2006, Young et al., 2008). Expectedly, the glycan mutation at 162 reduced binding to mAb 72A1, and the 162/208 glycans completely diminished 72A1 recognition, whereas binding of mAb 2L10 to gp350 wild-type (WT), glyc162, and glyc162/208 remained constant (Figure 7C). When sera from mice immunized with D123-ferrtin or D123-encapsulin were pre-incubated with gp350 WT to assess virus neutralization, most of neutralizing activity was depleted, as all gp350-specific antibodies in sera were absorbed by excess amount of gp350 WT. In contrast, the 162/208 glycan mutant absorbed only a small fraction of the neutralization activity, indicating that the majority of neutralization activity was targeting the site blocked by this mutant (Figure 7D). These results document the importance of CR2BS-directed antibodies in virus neutralization. Further, we found increased serum antibody binding to the CR2BS relative to full-length gp350 ectodomain after immunization with D123 nanoparticles compared to immunization with gp350 ectodomain (Figure 7E), although the overall antibody binding titers were similar in these immunization groups (Figures 4A and 5B), thus documenting preferential elicitation of CR2BS-directed antibodies in both immunized mice and monkeys.
Discussion
Natural infection with EBV induces a neutralizing antibody response in the majority of individuals, although the titers are generally modest (IC50 ≈101.5) (Sashihara et al., 2009). The quality of the immune response is often a key determinant of vaccine-induced immunity independent of its magnitude. It is noteworthy that the soluble gp350 ectodomain and D123 do not effectively induce neutralizing antibodies in mice, unlike the D123 nanoparticles, despite the fact that both soluble and nanoparticle forms of gp350 have similar antigenic profiles. This observation highlights the importance of antigen presentation, specifically the conformational authenticity of functional domains, in determining the quality of the immune response (McLellan, 2013). Immune focusing through nanoparticle presentation of selective domains of gp350 enabled elicitation of antibody predominantly to CR2BS, providing a mechanism of potent neutralization in sera. The mode of neutralization targeting the receptor-binding site (RBS) has been described in other viruses. The VRC01-class of antibodies that bind the CD4-binding site of the envelope of human immunodeficiency virus 1 (HIV-1) potently neutralize >90% of circulating viral strains regardless of their sequence variability (Wu et al., 2010, Zhou et al., 2010). MAbs CH65 (Whittle et al., 2011), C05 (Ekiert et al., 2012), and some others neutralize influenza A viruses through the conserved sialic acid-binding site on a viral hemagglutinin. Certain mAbs to hepatitis C virus (Krey et al., 2013), poliovirus (Chen et al., 2011, Chen et al., 2013), herpes simplex virus (Lee et al., 2013), and severe acute respiratory syndrome (SARS) coronavirus (van den Brink et al., 2005) also use a similar mechanism to neutralize viruses. Since the RBS must be functionally conserved to engage its host cell receptor, it is one of the most vulnerable targets of the virus. However, isolated RBS-containing domains (or RBDs) are often weakly immunogenic due to steric hindrance from surrounding glycans, hypervariability near the RBS that impairs targeting by the immune system, or altered conformation (Georgiev et al., 2013, Joyce et al., 2013, Julien et al., 2012).
Extensive glycosylation of EBV gp350 is thought to regulate the immune response to the glycoprotein (Machiels et al., 2011, Serafini-Cessi et al., 1989). One of the principles of recombinant vaccine design is to improve immunogenicity of the native protein by modifying and/or altering its function/configuration, otherwise impossible to apply to certain other vaccine platforms (e.g., killed or attenuated virus). Truncation of the COOH-terminal ∼480 residues of gp350 to make the D123 variant almost completely removed the “mucin-like” O-linked glycan-rich domain from the protein; however, this truncation led to protein mis-folding and/or destabilization in a soluble form. Improvement of immunogenicity was observed only when gp350 D123 was displayed on ferritin or encapsulin nanoparticles, suggesting that the D123 was conformationally stabilized on the nanoparticles without the remainder of the gp350 domain(s).
The diameter of D123-ferritin and D123-encapsulin particles are within 20–50 nm, and therefore both are likely to traffic through lymph nodes similarly, where they would be taken up by dendritic cells, B cells, and macrophages (Irvine et al., 2013). One potential explanation for the difference in immunogenicity is the orientation of gp350 RBS relative to the surface of the particle and the packing density. These parameters would be quite different between ferritin and encapsulin and could affect the accessibility and angle of approach by immune receptors. On ferritin, the RBS is presented upright and spaced within 50–100 Å. This distance would allow crosslinking of B cell receptors (BCR) and would enhance signal transduction through downstream molecules to stimulate B cell activation (Bachmann and Zinkernagel, 1997). It also remains possible that the dimerization of D123 seen in ferritin effectively limits the solvent-exposed surface of non-RBS sites and focuses the immune response to the RBS more efficiently. Recent advances in nanoparticle technology further accentuate the benefits of symmetrically arrayed antigens to activate B cells through low-affinity BCRs (Jardine et al., 2013, Lingwood et al., 2012) and improve vaccine-induced immunity (Kanekiyo et al., 2013). These technologies allowed us to design immunogens that elicit immune response to targets otherwise difficult to be induced by native protein such as the CR2BS on gp350.
Self-assembling nanoparticle-based EBV vaccine candidates displaying the receptor-binding portion of gp350 elicited potent neutralizing activity in mice and non-human primates by precisely targeting the site of vulnerability of the virus and stimulated neutralizing antibody responses that significantly exceeded the level obtained with soluble gp350 protein. We anticipate that this synthetic approach to design immunogens offers novel opportunities to create and/or redesign vaccines against pathogens for which it has been difficult to induce effective immunity.
Experimental Procedures
Gene Synthesis and Vector Construction
The gene encoding Helicobacter pylori-bullfrog hybrid ferritin was constructed by fusing residues 2–9 of bullfrog (Rana catesbeiana) ferritin lower subunit (UniProt: P07797 with a N8Q mutation to abolish a potential N-glycosylation site) to H. pylori nonheme ferritin (UniProt: Q9ZLI1, residues 3–167) with an I7E mutation to preserve the conserved salt bridge found in human and bullfrog ferritins (6R and 14E in both human light chain and bullfrog lower-subunit ferritins) with 6R of bullfrog ferritin. Mutation N19Q was introduced in H. pylori ferritin to abolish a potential N-glycosylation site. The secreted encapsulin was constructed by fusing a human CD5 signal to Termotoga maritima encapsulin (UniProt: Q9WZP2, residues 1–264). The gene encoding EBV strain B95-8 gp350 (UniProt: P03200, residues 1–907) was synthesized, and the fragments corresponding to ectodomain (residues 2–860), analogous to the region used in the crystal structure (Szakonyi et al., 2006) (D2H6O, residues 2–470), D123 (residues 2–425) and D12 (residues 2–317) were amplified. The fragments were inserted between a bovine prolactin signal (bPRL) and hybrid ferritin or were fused to 3′ end of encapsulin with a (SG3)2 linker to give rise to gp350-ferritin and gp350-encapsulin, respectively. Soluble gp350 ectodomain and D123 were constructed by fusing corresponding fragments with bPRL and COOH-terminal hexa-histidine tag. The CR2BS mutant gp350s were made by introducing N-glycosylation sites at 162 (W162N/N164T) and 208 (D208N/E210T). All genes were then cloned into the CMV/R 8κb (VRC 8405) mammalian expression vector for protein production.
Biosynthesis of Recombinant Proteins and Purification
The expression vectors were transiently transfected into FreeStyle 293F or Expi293F cells using 293fectin or ExpiFectamine 293 transfection reagents, respectively (Life Technologies). The gp350-based nanoparticles were purified by affinity chromatography using Galanthus nivalis agglutinin resins (EY Laboratories) followed by size exclusion chromatography with a HiPrep 16/60 Sephacryl S-500 HR column (GE Healthcare). The soluble gp350 proteins were purified using Ni sepharose excel resin (GE Healthcare) followed by size exclusion chromatography with a Superdex 200 10/300 GL (GE Healthcare).
Electron microscopy and cryo-EM reconstruction
For negative stain electron microscopy (EM), samples of about 50 μg ml−1 were adsorbed to freshly glow-discharged carbon-coated grids and stained with 2% ammonium molybdate. Images were recorded on an FEI T20 microscope with an Eagle CCD camera. For cryo-EM, samples (5 μg μl−1, 3 μl) were applied to holey grids (Quantifoil) and fast-frozen in liquid ethane, as described previously (Meng et al., 2013). Cryo-EM images of the D123-ferritin and D123-encapsulin were acquired on an FEI Titan Krios operated at 300 keV and a CM200 FEG microscope operated at 200 keV, respectively. Image processing and three-dimensional reconstruction were performed using ∼1,000 particles with the EMAN suite of programs (Baker et al., 2010). The final reconstruction was computed and was low-pass filtered to 30 Å in resolution. The fit model was made using the EMFit program (Rossmann et al., 2001).
Immunization
Animal experiments were carried out in accordance with all federal regulations and NIH guidelines and were approved by the Institutional Animal Care and Use Committee. Eight- to 10-week old female BALB/c mice (Charles River Laboratories) were immunized (n = 5) intramuscularly with 5 or 0.5 μg of purified proteins in 100 μl of 50% (v/v) mixture of SAS adjuvant (Sigma) in PBS at weeks 0, 3, and 16. For the non-human primate study, eight cynomolgus macaques (Macaca fascicularis) were immunized with 50 μg (gp350 ectodomain) or 25 μg (D123-ferritin or D123-encapsulin) in SAS adjuvant intramuscularly at weeks 0, 4, and 12.
GFP-Reporter Virus Neutralization Assay
Neutralization of EBV to B cells has been described previously (Sashihara et al., 2009). Briefly, immune sera were serially diluted and incubated with B95-8/F reporter virus (GFP-reporter EBV) for 2 hr. For protein competition neutralization, sera were preincubated with either soluble gp350 WT or glyc162/208 mutant protein (25 μg ml−1) for 30 min. The mixture was added to Raji cells and incubated for 3 days. Cells were fixed and analyzed with an Accuri C6 flow cytometer (BD Biosciences). Neutralization antibody titers were expressed as the concentration of serum antibody needed to inhibit viral entry by 50% (IC50).
Recombinant Vaccinia Virus Challenge
Full-length EBV gp350 was cloned into pRB21, a plasmid encoding vaccinia virus (VV) vp37, which is required for plaque formation (Blasco and Moss, 1995). BSC-1 cells were infected with VV vRB12 (Δvp37) and subsequently transfected with pRB21 containing EBV gp350. Recombinant virus (rVV-gp350) was rescued and purified by plaque purification. Challenge stock was purified on a cushion of 36% sucrose by ultracentrifugation (32,900 × g, 80 min) and titrated in BSC-1 cells. Immunized mice were challenged intranasally with 1 × 106 PFU of virus (10 μl per nostril). Mice were monitored and euthanized when they lost ≥30% of their pre-challenge weight or suffered symptoms.
Surface Plasmon Resonance-Based Antibody Competition
Soluble gp350 WT protein was immobilized on a CM5 sensor chip (GE Healthcare). Fifty microliters (100 μg ml−1) of mAb (72A1, 2L10, or C179) were flowed over the chip at 30 μl min−1 before injecting 50 μl of the serum samples (1/50 dilution) at 30 μl ml−1 on a Biacore 3000 instrument (GE Healthcare). The chip was regenerated using 3M MgCl2 after each run.
Statistical Analysis
p values were generated by one-way ANOVA using the Prism 5 program (GraphPad Software) unless reported otherwise.
Molecular Representations
All structural renderings of proteins were generated using the UCSF Chimera package, version 1.8 (http://www.cgl.ucsf.edu/chimera/). The UCSF Chimera is developed by the Resource for Bio-computing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Author Contributions
M.K. and G.J.N. conceptualized and developed the nanoparticulate EBV immunogens; M.K., W.B., J.I.C., and G.J.N. designed research studies; M.K., W.B., M.G.J., G.M., J.R.R.W., U.B., T.Y., S.N., A.B.M., R.A.K., M.G.R., J.I.C., and G.J.N. performed the research and/or analyzed the data; J.-P.T. and S.S.R. assisted the monkey study; M.K., W.B., J.R.M., B.S.G., J.I.C., and G.J.N interpreted and discussed the data; M.K., W.B., J.I.C., and G.J.N. wrote the paper, and all authors participated in manuscript revisions.
Acknowledgments
We thank B. Hartman (VRC) for help with manuscript preparation; S.-Y. Ko, H. Bao, C. Chiedi, M. Dillon, and K. Wuddie (VRC) for help with animal studies; K. Foulds and V. Letukas (VRC) for monkey sample processing; P. Radecki (LID) for help with serum analyses; A. Wheatley, S. Andrews, T. Zhou, and H. Yassine (VRC) for technical advice; Y. Okuno (Osaka University) for providing C179 antibody; and X. Chen (VRC) for help with transfections. This work was supported by the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. M.K., W.B., J.I.C., and G.J.N. are named as inventors on a patent application describing the data presented in this paper, which has been filed by the National Institutes of Health.
Published: August 13, 2015
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.07.043.
Supplemental Information
References
- Bachmann M.F., Zinkernagel R.M. Neutralizing antiviral B cell responses. Annu. Rev. Immunol. 1997;15:235–270. doi: 10.1146/annurev.immunol.15.1.235. [DOI] [PubMed] [Google Scholar]
- Bachmann M.F., Kalinke U., Althage A., Freer G., Burkhart C., Roost H., Aguet M., Hengartner H., Zinkernagel R.M. The role of antibody concentration and avidity in antiviral protection. Science. 1997;276:2024–2027. doi: 10.1126/science.276.5321.2024. [DOI] [PubMed] [Google Scholar]
- Baker M.L., Zhang J., Ludtke S.J., Chiu W. Cryo-EM of macromolecular assemblies at near-atomic resolution. Nat. Protoc. 2010;5:1697–1708. doi: 10.1038/nprot.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R., Moss B. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene. 1995;158:157–162. doi: 10.1016/0378-1119(95)00149-z. [DOI] [PubMed] [Google Scholar]
- Chen Z., Earl P., Americo J., Damon I., Smith S.K., Yu F., Sebrell A., Emerson S., Cohen G., Eisenberg R.J. Characterization of chimpanzee/human monoclonal antibodies to vaccinia virus A33 glycoprotein and its variola virus homolog in vitro and in a vaccinia virus mouse protection model. J. Virol. 2007;81:8989–8995. doi: 10.1128/JVI.00906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Chumakov K., Dragunsky E., Kouiavskaia D., Makiya M., Neverov A., Rezapkin G., Sebrell A., Purcell R. Chimpanzee-human monoclonal antibodies for treatment of chronic poliovirus excretors and emergency postexposure prophylaxis. J. Virol. 2011;85:4354–4362. doi: 10.1128/JVI.02553-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Fischer E.R., Kouiavskaia D., Hansen B.T., Ludtke S.J., Bidzhieva B., Makiya M., Agulto L., Purcell R.H., Chumakov K. Cross-neutralizing human anti-poliovirus antibodies bind the recognition site for cellular receptor. Proc. Natl. Acad. Sci USA. 2013;110:20242–20247. doi: 10.1073/pnas.1320041110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K.J., Shin H.J., Lee J.H., Kim K.J., Park S.S., Lee Y., Lee C., Park S.S., Kim K.H. The crystal structure of ferritin from Helicobacter pylori reveals unusual conformational changes for iron uptake. J. Mol. Biol. 2009;390:83–98. doi: 10.1016/j.jmb.2009.04.078. [DOI] [PubMed] [Google Scholar]
- Cohen J.I., Fauci A.S., Varmus H., Nabel G.J. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci. Transl. Med. 2011;3:107fs7. doi: 10.1126/scitranslmed.3002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S.A., Jackson J.O., Jardetzky T.S., Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011;9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert D.C., Kashyap A.K., Steel J., Rubrum A., Bhabha G., Khayat R., Lee J.H., Dillon M.A., O’Neil R.E., Faynboym A.M. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature. 2012;489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott S.L., Suhrbier A., Miles J.J., Lawrence G., Pye S.J., Le T.T., Rosenstengel A., Nguyen T., Allworth A., Burrows S.R. Phase I trial of a CD8+ T-cell peptide epitope-based vaccine for infectious mononucleosis. J. Virol. 2008;82:1448–1457. doi: 10.1128/JVI.01409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein M.A., Achong B.G., Barr Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet. 1964;1:702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- Epstein M.A., Morgan A.J., Finerty S., Randle B.J., Kirkwood J.K. Protection of cottontop tamarins against Epstein-Barr virus-induced malignant lymphoma by a prototype subunit vaccine. Nature. 1985;318:287–289. doi: 10.1038/318287a0. [DOI] [PubMed] [Google Scholar]
- Fingeroth J.D., Weis J.J., Tedder T.F., Strominger J.L., Biro P.A., Fearon D.T. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. USA. 1984;81:4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev I.S., Gordon Joyce M., Zhou T., Kwong P.D. Elicitation of HIV-1-neutralizing antibodies against the CD4-binding site. Curr. Opin. HIV AIDS. 2013;8:382–392. doi: 10.1097/COH.0b013e328363a90e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S.Y., Huang T.M., Ruan L., Miao Y.H., Lu H., Chu C.M., Motz M., Wolf H. First EBV vaccine trial in humans using recombinant vaccinia virus expressing the major membrane antigen. Dev. Biol. Stand. 1995;84:171–177. [PubMed] [Google Scholar]
- Hoffman G.J., Lazarowitz S.G., Hayward S.D. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc. Natl. Acad. Sci. USA. 1980;77:2979–2983. doi: 10.1073/pnas.77.5.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard D.J., Kane K.M., Tezcan F.A. Re-engineering protein interfaces yields copper-inducible ferritin cage assembly. Nat. Chem. Biol. 2013;9:169–176. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- Hutt-Fletcher L.M. Epstein-Barr virus entry. J. Virol. 2007;81:7825–7832. doi: 10.1128/JVI.00445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D.J., Swartz M.A., Szeto G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jääskeläinen A., Harinen R.R., Lamminmäki U., Korpimäki T., Pelliniemi L.J., Soukka T., Virta M. Production of apoferritin-based bioinorganic hybrid nanoparticles by bacterial fermentation followed by self-assembly. Small. 2007;3:1362–1367. doi: 10.1002/smll.200700011. [DOI] [PubMed] [Google Scholar]
- Jardine J., Julien J.P., Menis S., Ota T., Kalyuzhniy O., McGuire A., Sok D., Huang P.S., MacPherson S., Jones M. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce M.G., Kanekiyo M., Xu L., Biertümpfel C., Boyington J.C., Moquin S., Shi W., Wu X., Yang Y., Yang Z.Y. Outer domain of HIV-1 gp120: antigenic optimization, structural malleability, and crystal structure with antibody VRC-PG04. J. Virol. 2013;87:2294–2306. doi: 10.1128/JVI.02717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien J.P., Lee P.S., Wilson I.A. Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol. Rev. 2012;250:180–198. doi: 10.1111/imr.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo M., Wei C.J., Yassine H.M., McTamney P.M., Boyington J.C., Whittle J.R., Rao S.S., Kong W.P., Wang L., Nabel G.J. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499:102–106. doi: 10.1038/nature12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S., Verma N., Yewdell J.W., Hilbert A.K., Castellino F., Lattanzi M., Del Giudice G., Rappuoli R., Golding H. MF59 adjuvant enhances diversity and affinity of antibody-mediated immune response to pandemic influenza vaccines. Sci. Transl. Med. 2011;3:85ra48. doi: 10.1126/scitranslmed.3002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey T., Meola A., Keck Z.Y., Damier-Piolle L., Foung S.K., Rey F.A. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013;9:e1003364. doi: 10.1371/journal.ppat.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P.S., Yoshida R., Ekiert D.C., Sakai N., Suzuki Y., Takada A., Wilson I.A. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. USA. 2012;109:17040–17045. doi: 10.1073/pnas.1212371109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.C., Lin L.L., Chan W.E., Ko T.P., Lai J.S., Wang A.H. Structural basis for the antibody neutralization of herpes simplex virus. Acta Crystallogr. D Biol. Crystallogr. 2013;69:1935–1945. doi: 10.1107/S0907444913016776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.Q., Soistman E., Carter D.C. Ferritin nanoparticle technology. A new platform for antigen presentation and vaccine development. Ind. Biotechnol. 2006;2:143–147. [Google Scholar]
- Lingwood D., McTamney P.M., Yassine H.M., Whittle J.R., Guo X., Boyington J.C., Wei C.J., Nabel G.J. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 2012;489:566–570. doi: 10.1038/nature11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luka J., Chase R.C., Pearson G.R. A sensitive enzyme-linked immunosorbent assay (ELISA) against the major EBV-associated antigens. I. Correlation between ELISA and immunofluorescence titers using purified antigens. J. Immunol. Methods. 1984;67:145–156. doi: 10.1016/0022-1759(84)90093-0. [DOI] [PubMed] [Google Scholar]
- Machiels B., Lété C., Guillaume A., Mast J., Stevenson P.G., Vanderplasschen A., Gillet L. Antibody evasion by a gammaherpesvirus O-glycan shield. PLoS Pathog. 2011;7:e1002387. doi: 10.1371/journal.ppat.1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan A. Mid Staffordshire inquiry. Too long, too late but Francis can still help make the NHS better. Health Serv. J. 2013;123:3. [PubMed] [Google Scholar]
- Meldrum F.C., Heywood B.R., Mann S. Magnetoferritin: in vitro synthesis of a novel magnetic protein. Science. 1992;257:522–523. doi: 10.1126/science.1636086. [DOI] [PubMed] [Google Scholar]
- Meng G., Zhang X., Plevka P., Yu Q., Tijssen P., Rossmann M.G. The structure and host entry of an invertebrate parvovirus. J. Virol. 2013;87:12523–12530. doi: 10.1128/JVI.01822-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutschen M., Léonard P., Sokal E.M., Smets F., Haumont M., Mazzu P., Bollen A., Denamur F., Peeters P., Dubin G., Denis M. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine. 2007;25:4697–4705. doi: 10.1016/j.vaccine.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Ogembo J.G., Kannan L., Ghiran I., Nicholson-Weller A., Finberg R.W., Tsokos G.C., Fingeroth J.D. Human complement receptor type 1/CD35 is an Epstein-Barr Virus receptor. Cell Rep. 2013;3:371–385. doi: 10.1016/j.celrep.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y., Isegawa Y., Sasao F., Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 1993;67:2552–2558. doi: 10.1128/jvi.67.5.2552-2558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M.G., Bernal R., Pletnev S.V. Combining electron microscopic with x-ray crystallographic structures. J. Struct. Biol. 2001;136:190–200. doi: 10.1006/jsbi.2002.4435. [DOI] [PubMed] [Google Scholar]
- Sashihara J., Burbelo P.D., Savoldo B., Pierson T.C., Cohen J.I. Human antibody titers to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology. 2009;391:249–256. doi: 10.1016/j.virol.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashihara J., Hoshino Y., Bowman J.J., Krogmann T., Burbelo P.D., Coffield V.M., Kamrud K., Cohen J.I. Soluble rhesus lymphocryptovirus gp350 protects against infection and reduces viral loads in animals that become infected with virus after challenge. PLoS Pathog. 2011;7:e1002308. doi: 10.1371/journal.ppat.1002308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini-Cessi F., Malagolini N., Nanni M., Dall’Olio F., Campadelli-Fiume G., Tanner J., Kieff E. Characterization of N- and O-linked oligosaccharides of glycoprotein 350 from Epstein-Barr virus. Virology. 1989;170:1–10. doi: 10.1016/0042-6822(89)90345-0. [DOI] [PubMed] [Google Scholar]
- Sokal E.M., Hoppenbrouwers K., Vandermeulen C., Moutschen M., Léonard P., Moreels A., Haumont M., Bollen A., Smets F., Denis M. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007;196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- Sutter M., Boehringer D., Gutmann S., Günther S., Prangishvili D., Loessner M.J., Stetter K.O., Weber-Ban E., Ban N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008;15:939–947. doi: 10.1038/nsmb.1473. [DOI] [PubMed] [Google Scholar]
- Szakonyi G., Klein M.G., Hannan J.P., Young K.A., Ma R.Z., Asokan R., Holers V.M., Chen X.S. Structure of the Epstein-Barr virus major envelope glycoprotein. Nat. Struct. Mol. Biol. 2006;13:996–1001. doi: 10.1038/nsmb1161. [DOI] [PubMed] [Google Scholar]
- Tanner J., Whang Y., Sample J., Sears A., Kieff E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J. Virol. 1988;62:4452–4464. doi: 10.1128/jvi.62.12.4452-4464.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley-Lawson D.A., Geilinger K. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc. Natl. Acad. Sci. USA. 1980;77:5307–5311. doi: 10.1073/pnas.77.9.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley-Lawson D.A., Poodry C.A. Identification and isolation of the main component (gp350-gp220) of Epstein-Barr virus responsible for generating neutralizing antibodies in vivo. J. Virol. 1982;43:730–736. doi: 10.1128/jvi.43.2.730-736.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha J., Theil E.C., Allewell N.M. High resolution crystal structures of amphibian red-cell L ferritin: potential roles for structural plasticity and solvation in function. J. Mol. Biol. 1995;248:949–967. doi: 10.1006/jmbi.1995.0274. [DOI] [PubMed] [Google Scholar]
- Tugizov S.M., Berline J.W., Palefsky J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003;9:307–314. doi: 10.1038/nm830. [DOI] [PubMed] [Google Scholar]
- van den Brink E.N., Ter Meulen J., Cox F., Jongeneelen M.A., Thijsse A., Throsby M., Marissen W.E., Rood P.M., Bakker A.B., Gelderblom H.R. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J. Virol. 2005;79:1635–1644. doi: 10.1128/JVI.79.3.1635-1644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Li C., Ellenburg M., Soistman E., Ruble J., Wright B., Ho J.X., Carter D.C. Structure of human ferritin L chain. Acta Crystallogr. D Biol. Crystallogr. 2006;62:800–806. doi: 10.1107/S0907444906018294. [DOI] [PubMed] [Google Scholar]
- Whittle J.R., Zhang R., Khurana S., King L.R., Manischewitz J., Golding H., Dormitzer P.R., Haynes B.F., Walter E.B., Moody M.A. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 2011;108:14216–14221. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Yang Z.Y., Li Y., Hogerkorp C.M., Schief W.R., Seaman M.S., Zhou T., Schmidt S.D., Wu L., Xu L. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young K.A., Herbert A.P., Barlow P.N., Holers V.M., Hannan J.P. Molecular basis of the interaction between complement receptor type 2 (CR2/CD21) and Epstein-Barr virus glycoprotein gp350. J. Virol. 2008;82:11217–11227. doi: 10.1128/JVI.01673-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Georgiev I., Wu X., Yang Z.Y., Dai K., Finzi A., Kwon Y.D., Scheid J.F., Shi W., Xu L. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.