

Abstract
AIM: To address the effect of heat-shock protein 90 (HSP90) inhibitors on the release of the hepatitis C virus (HCV), a cell culture-derived HCV (JFH1/HCVcc) from Huh-7 cells was examined.
METHODS: We quantified both the intracellular and extracellular (culture medium) levels of the components (RNA and core) of JFH-1/HCVcc. The intracellular HCV RNA and core levels were determined after the JFH1/HCVcc-infected Huh-7 cells were treated with radicicol for 36 h. The extracellular HCV RNA and core protein levels were determined from the medium of the last 24 h of radicicol treatment. To determine the possible role of the HSP90 inhibitor in HCV release, we examined the effect of a combined application of low doses of the HSP90 inhibitor radicicol and the RNA replication inhibitors cyclosporin A (CsA) or interferon. Finally, we statistically examined the combined effect of radicicol and CsA using the combination index (CI) and graphical representation proposed by Chou and Talalay.
RESULTS: We found that the HSP90 inhibitors had greater inhibitory effects on the HCV RNA and core protein levels measured in the medium than inside the cells. This inhibitory effect was observed in the presence of a low level of a known RNA replication inhibitor (CsA or interferon-α). Treating the cells with a combination of radicicol and cyclosporin A for 24 h resulted in significant synergy (CI < 1) that affected the release of both the viral RNA and the core protein.
CONCLUSION: In addition to having an inhibitory effect on RNA replication, HSP90 inhibitors may interfere with an HCV replication step that occurs after the synthesis of viral RNA, such as assembly and release.
Keywords: Hepatitis C virus, Inhibition of hepatitis C virus release, Cell culture-derived hepatitis C virus, Heat-shock protein 90 inhibitors, Hepatitis C virus RNA replication
Core tip: Hepatitis C virus (HCV) is a major causative agent of hepatocellular carcinoma. Several non-structural proteins of HCV physically and functionally interact with heat-shock protein 90 (HSP90). Although HSP90 inhibitors, which inhibit the chaperone function of HSP90, have been shown to inhibit HCV replication by several groups, a recent report using a reporter system for HCV RNA replication (replicon) suggests that the effect is nonspecific. Thus, the inhibitory mechanism of HSP90 inhibitors remains controversial. Here, we address the effect of HSP90 inhibitors on the release of JFH1/cell culture-derived HCV from Huh-7 cells, and suggested that, HSP90 inhibitors may also interfere with an HCV replication step that occurs after the synthesis of viral RNA, such as assembly and release.
INTRODUCTION
Chronic infection with hepatitis C virus (HCV) frequently causes liver cirrhosis and hepatocellular carcinoma[1]. Approximately 170 million individuals have been infected by HCV[2] and are at risk for developing liver disease[3]. HCV has a positive-sense single-stranded RNA genome that encodes a 3000 amino acid polyprotein and also contains an internal entry site for translation and a non-coding region for genome replication at the 5’- and 3’- flanking region. Translation of the polyprotein is followed by cleavage by the host and viral proteases, which yields three structural proteins (core, E1 and E2) and seven nonstructural (NS) proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B)[4]. Establishment of the cell culture-derived HCV (HCVcc) system (i.e., human hepatoma Huh-7 cells propagating the highly infectious clone of HCV genotype 2a, JFH-1[5]) provides a native infection-cycle system, which is suitable for determination of the precise function of HCV proteins and thus the mechanism for replication and secretion of viral particles. Accumulating evidence has indicated that NS3, NS4B and NS5A are required for not only genome RNA replication but also virus assembly[6], whereas P7 and NS2 are dispensable for RNA replication but are required for virus production[7].
Heat-shock protein 90 (HSP90) functions as a molecular chaperone for various client proteins and interacts with a cohort of co-chaperones that modulate the HSP90 ATPase cycle. The ATPase activity of HSP90 is inhibited by HSP90 inhibitors, which compete with ATP for binding and thereby eliminate HSP90 chaperone activity[8]. HSP90 clients include not only host proteins but also some virus proteins. Thus, some of the replication steps are required for HSP90 activity[9]. In fact, activities of multiple HCV proteins are affected by inhibition of HSP90 by specific inhibitors of HSP90, such as radicicol and geldanamycin and its derivatives. HSP90 inhibitors restrict the activity of NS2/3 protease[10], the stability of NS3[11], and the RNA replication of HCV in cells harboring the HCV replicon by inhibiting a complex consisting of HSP90, NS5A and a human FK506-binding protein (FKBP8)[12]. The host factors that are required for HCV replication are also affected by inhibition of HSP90[13,14]. Apart from these positive results, Beran et al[15] have shown that an HSP90 inhibitor elicits effects similar to those of known cytostatic compounds, abrogating propagation of the mini-genome (subgenomic) replicon of HCV indirectly, through slowing cell growth. HCV subgenomic replicons are generally composed of a selection marker gene, such as G418 resistance, a reporter gene such as luciferase, and a whole HCV genome, except for the regions encoding the structural proteins and NS2 protease, which are deleted[16]. The replicons propagating in Huh-7 cells are widely used for studying RNA replication as well as for screening for anti-HCV drugs. However, it is possible that the replicon system does not fully represent the native function of HCV proteins, because mutations that accumulate in NS3 and NS5A in the replicon during enhancement of the RNA replication interfere with virus assembly when the mutation is introduced in the HCVcc construct[17]. Hence, the HCVcc system may be more suitable than the subgenomic replicon system to evaluate the anti-viral effect of HSP90 inhibitors. In addition, HSP90 is required for NS3 stability[11] and for formation of the complex composing NS4A[12]. Because NS3 and NS4A can act on both RNA replication and HCV assembly, it is possible that HSP90 activity may contribute to post-RNA replication steps, such as virus assembly and release, which is represented by HCVcc but not the replicon system.
In this study, we used JFH1/HCVcc to demonstrate the effect of HSP90 inhibition on HCVcc release from infected Huh-7 cells. Our results showed that the HSP90 inhibitor radicicol preferentially reduced the levels of the core and the HCV RNA released from cells in the medium compared with those in the cells. The HSP90 inhibitor had a more potent effect on viral release in the medium than that of the inhibitor for RNA replication.
MATERIALS AND METHODS
Reagents
Radicicol (Sigma-Aldrich, St. Louis, MO, United States) was dissolved in methanol (1 mg/mL). Cyclosporin A [cyclosporin A (CsA); Wako Pure Chemical, Osaka, Japan], 17-AAG [17-(allylamino)-17-demethoxygeldanamycin, Sigma-Aldrich] and 17-DMAG [17-(dimethylaminoethylamino)-17-demethoxygeldanamycin; BIOMOL, Plymouth Meeting, PA, United States] were dissolved in ethanol (1 mg/mL). Interferon-α (IFN-α; PeproTec EC, London, United Kingdom) was dissolved in water (0.1 mg/mL).
Culture of human hepatoma Huh-7 cells and preparation of JFH1/HCVcc
Human hepatoma Huh-7 cells (purchased from Human Science Resources Bank, Osaka, Japan) were cultured in DMEM (Nissui Pharmaceuticals, Tokyo, Japan) supplemented with 10% fetal calf serum, 0.06% glutamine, 0.35% glucose and 1% penicillin-streptomycin (Life Technologies, Carlsbad, CA, United States). The cells were maintained at 37 °C in an atmosphere of 5% CO2. We synthesized the HCV genomic RNA of genotype 2a (JFH1) in vitro using a MEGAscript™ T7 kit (Ambion, Austin, TX, United States) and introduced the RNA into Huh-7 cells by electroporating the cells with the GenePulser II electroporation system (Bio-Rad, Hercules, CA, United States) as previously described[5]. The cytotoxic effects of the reagents were examined with Alamar Blue cell viability reagent (Serotec, Raleigh, NC, United States), which allows an estimation of the oxidation levels in the cellular electron-transport pathways with a fluorescent indicator. Alamar Blue was used as described by the manufacturer.
Quantification of the HCV core protein and genomic RNA
We washed the JFH1/HCVcc cells with PBS and lysed them in lysis buffer (20 mmol/L Tris-Cl, pH 7.5, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 0.1 mmol/L EDTA, 0.1 mmol/L phenylmethanesulfonyl fluoride, 50 μmol/L N-p-tosyl-L-phenylalanine chloromethyl ketone, 5 μmol/L Nα-tosyl-L-lysine chloromethyl ketone hydrochloride, 5 μg/mL aprotinin and 5 μg/mL leupeptin). We quantified the level of core protein present in the lysates and spent culture medium using the Ortho HCV antigen ELISA test (Ortho Clinical Diagnostics, Rochester, NY, United States). We used a QIAamp™ Viral RNA Mini kit (Qiagen, Hilden, Germany) to isolate the HCV genomic RNA from the medium. We used an RNeasy™ mini kit (Qiagen) to isolate total RNA from the HCV-infected cells. We quantified the HCV genomic RNA using TaqMan™ EZ RT-PCR Core Reagents (Applied Biosystems, Foster City, CA, United States) and the iCycler™ iQ real-time detection system (Bio-Rad) as previously described[18].
Effects of radicicol and CsA
The JFH1/HCVcc cells (5 × 104 cells in one well of a 24-well culture dish) were treated with radicicol, CsA and/or IFN-α for 12 h, at which point the medium was replaced by fresh medium that contained the same level(s) of drug(s). After culturing for another 24 h, the core and the HCV RNA in each cell lysate and culture medium were quantified as described above. The synergism between CsA and radicicol was evaluated by the combination index (CI) equation and the Chou and Talalay method[19,20] using CalcuSyn software (BIOSOFT, Cambridge, United Kingdom). The CI equation is based on the multiple drug-effect equation of Chou et al[20], which is derived from enzyme-kinetic models. Assuming that CsA and radicicol were mutually non-exclusive drugs that have totally independent modes of action, we used the following equation:
Equation 1 dictates that the combination of drug 1 (D)1 and drug 2 (D)2 inhibits a reaction (or phenomenon) by x% in an actual experiment. (Dx)1 and (Dx)2 are the doses of drug 1 and drug 2 alone that inhibit the same reaction by x%.
CI = [(D)1/(Dx)1] + [(D)2/(Dx)2] + [(D)1(D)2/(Dx)2(Dx)2] (1)
Statistical analysis
We used Student’s t-test to examine statistical significance (P < 0.05). All of the experiments were performed with multiple independent replicates, and all of the data are presented as the mean results of three independent experiments with the standard error of the mean. The statistical methods of this study were reviewed by professor Kotaro Tanahashi from Mathematics, Tohoku Pharmaceutical University.
RESULTS
HCV released into the medium is preferentially reduced by HSP90 inhibitors
To examine the effects of HSP90 inhibitor on the release of HCV, we quantified both the intracellular and extracellular (culture medium) levels of the components (RNA and core) of JFH-1/HCVcc. The intracellular HCV RNA and core levels were determined after the cells were treated with radicicol for 36 h. The extracellular HCV RNA and the core were determined from the medium of the last 24 h of radicicol treatment. The radicicol treatment (50-300 nmol/L) exhibited no apparent cytotoxic effect (Figure 1A), reduced both the intracellular and extracellular (medium) levels of the HCV RNA (Figure 1B) and the core (Figure 1C) in a dose-dependent manner. Interestingly, the RNA level in the culture medium relative to the total RNA level was apparently reduced by radicicol even at a low concentration (50 nmol/L) (Figure 1D). Similarly, the core level in the medium relative to the total core level was also significantly decreased (P = 0.029) in the presence of 50 nmol/L radicicol (Figure 1E). Furthermore, two derivatives of the geldanamycin HSP90 inhibitor, 17-AAG and 17-DMAG, also inhibited the release of the HCV RNA and core more effectively than they decreased the intracellular HCV RNA and core levels (Figure 2).
Figure 1.
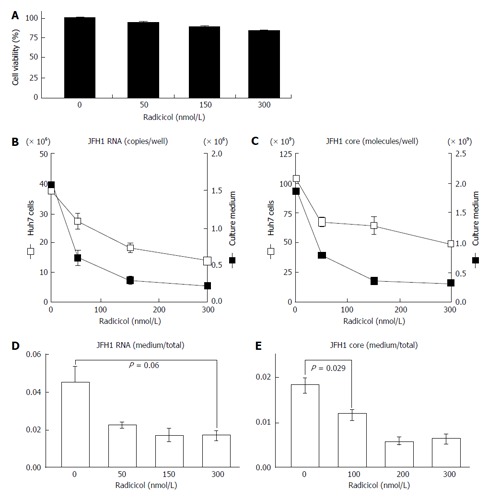
Radicicol affects the relative level of hepatitis C virus (core and hepatitis C virus RNA) produced from the JFH1/cell culture-derived hepatitis C virus system of Huh-7 cells. A: After the cells were treated with radicicol (at final concentration of 0, 50, 150 and 300 nmol/L) for 12 h, the culture medium was replaced with fresh medium containing the same radicicol levels, and the HCV RNA and core levels released into the medium for the next 24 h and produced within cells were determined as described in the text. The cytotoxic effects of the treatment of radicicol on HCVcc Huh-7 cells were examined as described in the text; B and C: The levels of HCV RNA (B) and core (C) in the HCVcc Huh-7 cells (open squares) and the culture medium (filled squares) were quantified. The scales on the left sides (B and C) indicate the scales for the HCV RNA and the core in the Huh-7 cells. The scales on the right sides (B and C) indicate the scales for the HCV RNA and the core in the culture medium; D: The ratios of HCV RNA in the medium to the total HCV RNA (the sum of HCV RNA in the medium and in the cells) are shown; E: The ratios of the core in the medium to the total core (the sum of the core in the medium and in the cells) are shown. The data represent the mean values (± SEM) of the results from three independent experiments. HCV: Hepatitis C virus; HCVcc: Cell culture-derived HCV.
Figure 2.
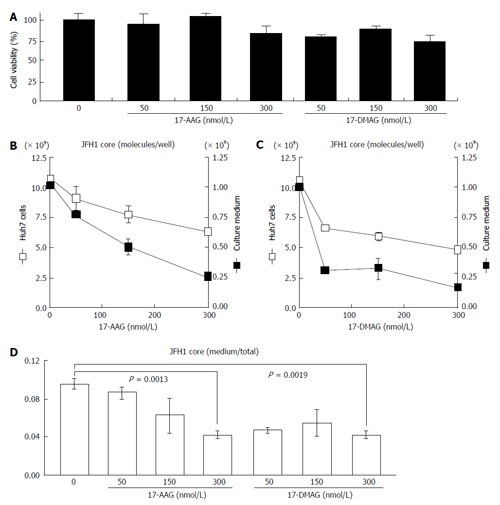
Effects of the geldanamycin derivatives 17-allylamino-17-demethoxygeldanamycin and 17-dimethylaminoethylamino-17-demethoxygeldanamycin on the release of JFH1. A: The cytotoxic effects of 17-AAG and 17-DMAG on Huh-7 cells carrying JFH1/HCVcc were examined as described in the text; B and C: JFH1-infected Huh-7 cells were treated with 17-AAG (B) or 17-DMAG (C) for 24 h and then the protein samples were isolated and quantified. The levels of the HCV core present in the JFH1-infected Huh-7 cells (open squares) and the culture medium (filled squares) were examined as described in the text. The scales on the left (B and C) and right sides (B and C) indicate the scales for the HCV core in the Huh-7 cells and in the culture medium, respectively; D: The ratios of the core in the medium to the total core are shown. The concentrations of 17-AAG and 17-DMAG (0-300 nmol/L) are shown under the histograms. The data represent the mean values (± SEM) of the results from three independent experiments. HCV: Hepatitis C virus; 17-AGG: 17-allylamino-17-demethoxygeldanamycin; 17-DMAG: 17-dimethylaminoethylamino-17-demethoxygeldanamycin; HCVcc: Cell culture-derived HCV.
We next examined whether the integrity of HCV was affected by the radicicol treatment during production of HCV from JFH1/HCVcc. The infectivity of the HCV that had been released into the medium in the presence of radicicol was compared to the infectivity of HCV released in the absence of radicicol. As shown in Figure 3, there was no significant difference (P > 0.3) in the infectivity between the HCV produced in the presence and that produced in the absence of radicicol. These results suggested that even though radicicol preferentially reduced HCV release, radicicol did not affect its infectivity.
Figure 3.
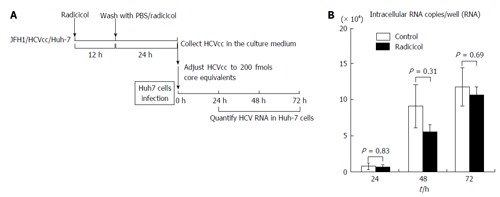
Infectivity of hepatitis C virus produced from Huh-7 cells that were treated with radicicol. A: Experimental design. To examine whether the infectivity of the JFH1/HCVcc released from the Huh-7 cells in the presence of radicicol was altered, we infected fresh Huh-7 cells with JFH1/HCVcc prepared in the presence of radicicol. To prepare a viral stock, the JFH1/HCVcc infected Huh-7 cells with 50 nmol/L radicicol were maintained for an additional 36 h. HCVcc released in the medium during the last 24 h was collected and used as the viral stock. We diluted the viral stock 59.7 times (None) and 28.6 times (Radicicol) to reduce the effects of radicicol carryover and to adjust the HCV levels (core protein level: 200 fmols). The final radicicol concentration was 1.75 nmol/L which did not affect HCV propagation. The HCV samples were added to fresh Huh-7 cells and cultured for 24, 48 and 72 h. The HCV RNA in the infected Huh-7 cells was quantified as described in the text. The multiplicities of infection for HCV were 0.2 copies/cell and 0.16 copies/cell for the HCV infection without and with radicicol, respectively; B: The copies of HCV RNA in the Huh-7 cells of each well after infection (24, 48 and 48 h) were determined as described in the text. The value of the RNA copy number in the Huh-7 cells [which were infected with the viral stock from radicicol-treated cells (values of the closed bars)] was adjusted by multiplying by a factor of 1.25. The data represent the mean values (± SE) of the results from three independent experiments. HCV: Hepatitis C virus; HCVcc: Cell culture-derived HCV.
Radicicol preferentially suppressed the HCV RNA release in the presence of CsA or interferon
NS3 and NS5A, which are known targets of HSP90[11,12], are required for both RNA replication and virus assembly[6,21]. Thus, it is difficult to distinguish whether the inhibitory step of the HSP90 inhibitor affects the RNA replication or the assembly. If the HSP90 inhibitor were to preferentially inhibit the post-RNA replication steps, the HSP90 inhibitor might enhance the RNA-replication inhibitor-dependent inhibitory effect on HCV release. CsA an immunosuppressant, inhibits interaction between a CsA’s target cyclophilin A and NS5A and the interaction of the cyclophilin A with the NS5B polymerase/RNA complex, thereby inhibiting RNA replication[22]. This inhibitory effect is distinct from CsA’s immunosuppressive activity[23]. To determine the possible role of the HSP90 inhibitor on HCV release, we examined the effect of a combined application of low doses of radicicol and CsA. By studying the dose-dependent inhibition of HCV RNA production in Huh-7 cells (data not shown), we determined the doses showing the partial effect of CsA (250 nmol/L) and radicicol (50 nmol/L) on the viral RNA production in cells after 36 h of treatment (Figure 4A, column 1, 2 and 3, open bars). CsA alone, radicicol alone and simultaneous treatment with both drugs did not affect the cytotoxicity in JFH1-infected Huh-7 cells (Figure 4B). When the HCV-infected cells were treated with the low concentration of both drugs simultaneously, the level of HCV RNA released in the medium after 24 h fell significantly (Figure 4A, comparison of the closed bars in columns 3 and 4, P = 0.0015), whereas the level of HCV RNA in the cells was the same as that in the cells treated with CsA alone (Figure 4A, comparison of the open bars in columns 3 and 4). Intriguingly, the RNA level in the culture medium relative to the RNA levels in the infected cells fell in the presence of radicicol (Figure 4C, comparison of columns 1 and 2). However, this medium-to-total ratio was not affected by simultaneous treatment of CsA with radicicol (Figure 4C, column 2 and 4). Thus, radicicol could have more of an inhibitory effect on viral release from infected cells than CsA.
Figure 4.
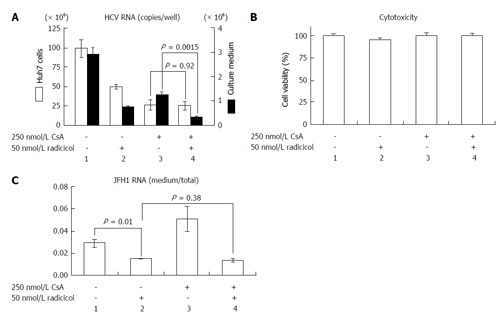
Radicicol and cyclosporin A have a synergistic effect on hepatitis C virus release into the medium. A: JFH1/HCVcc-infected Huh-7 cells were treated with 250 nmol/L CsA alone, 50 nmol/L radicicol alone, or both for 36 h. The total RNA was prepared from the Huh-7 cells, and the HCV RNA was quantified by reverse transcription-polymerase chain reaction (open columns, the scale on the left of the panel indicates the copies/well). The medium was replaced by the fresh medium containing the same levels of the drugs after 12 h, and total RNA was prepared from the medium 24 h later. The copy numbers of the HCV RNA in the medium are indicated by the filled columns (scale on the right of the panel); B: The cytotoxic effects of the drugs used in (A) on the Huh-7 cells carrying JFH1/HCVcc; C: The ratios of the RNA in the medium to the total RNA (as described above) are shown. HCV: Hepatitis C virus; HCVcc: Cell culture-derived HCV; CsA: Cyclosporin A.
Previous results have indicated that the combined effect of IFN and CsA on RNA replication is mostly additive[23] and have suggested that both CsA and IFN target a similar point in HCV replication. However, Robida et al[24] have indicated that CsA resistant mutants maintain their sensitivity to IFN-α. Thus, although both IFN-α and CsA inhibit RNA replication, the inhibitory effects of IFN-α and CsA seems to be different. As shown in Figure 5A, we observed that radicicol efficiently reduced the level of HCV in the medium even in the presence of IFN-α. Although the combined treatment of radicicol and IFN-α and the treatment of IFN-α alone exhibited a similar level of HCV RNA in cells (columns 3 and 4, open bars), HCV RNA in the medium was significantly reduced (columns 3 and 4, closed bars). Again, HCV RNA in the medium was significantly suppressed in the presence of radicicol (Figure 5C). All of these results suggested that HSP90 might be responsible for a post-replication step such as viral release. It should be noted that a similar synergism has been previously reported: A combined administration of an HSP90 inhibitor and polyethylene glycol-conjugated interferon (PEG-IFN) in HCV-infected chimeric mice with humanized livers was more effective at reducing the HCV genomic RNA levels in mouse serum than a single PEG-IFN treatment[25].
Figure 5.
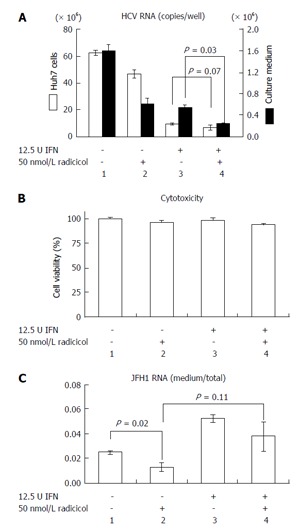
Radicicol and interferon-α have a synergistic effect on hepatitis C virus release into the medium. A: JFH1/HCVcc-infected Huh-7 cells were treated with 12.5 U INF-α alone, 50 nmol/L radicicol alone, or both for 36 h. The total RNA was prepared from the Huh-7 cells, and the HCV RNA was quantified by reverse transcription-polymerase chain reaction (open columns, the scale on the left of the panel indicates the copies/well). The medium was replaced by fresh medium after 12 h, and the total RNA was prepared from the medium 24 h later. The copy numbers of the HCV RNA in the medium are indicated by the filled columns (scale on the right of the panel); B: The cytotoxic effects of the drugs used in (A) on the Huh-7 cells carrying JFH1/HCVcc; C: The ratios of RNA in the medium to RNA in the total RNA (as described above) are shown. HCV: Hepatitis C virus; HCVcc: Cell culture-derived HCV; INF-α: Interferon-α.
Analysis of the synergistic effect between radicicol and CsA
Our results suggested that radicicol has a greater inhibitory effect on the release of HCVcc into the medium and that the point of inhibition of HCV production by the HSP90 inhibitor may be different from that of CsA. Thus, we statistically examined the combined effect of radicicol and CsA using the CI and the graphical representation proposed by Chou et al[19,20]. We examined the effects of various CsA (125, 250, 750 and 1500 nmol/L) and radicicol (25, 50, 150 and 300 nmol/L) concentrations on the release of the core into the medium after 24 h (data not shown). Next, we examined the effects of a fixed molar ratio of these drugs (5:1 CsA to radicicol). The results (Figure 6A) indicated that CsA and radicicol had a synergistic effect (CI < 1) above a fractional effect of 0.5 and close to a strong synergism (CI ≤ 0.3) at a fractional effect of 1. The combined effect was additive near a fractional effect of 0.5 and antagonistic at a fractional effect less than 0.4. Figure 6B shows the conservation isobologram (CI = 1) for the different effective doses of the combination treatment that yielded 50% (ED50), 75% (ED75) and 90% (ED90) inhibition of the core release (constructed using actual experimental data). The combined effect at ED50 (CI = 1.05) was additive, whereas the combined effects at ED75 (CI = 0.75) and ED90 (CI = 0.57) were synergistic (Figure 6). The combined use of 408 nmol/L CsA and 82 nmol/L radicicol yielded ED90 (Figure 6B; if a CI = 1 was expected, an estimated 605 nmol/L CsA and 121 nmol/L radicicol would be required to yield ED90). We obtained similar results for the 3:1 and 10:1 molar ratios of CsA to radicicol (Figure 6C). One example, shown in Figure 6D, indicated that treating the cells with both 1000 nmol/L CsA and 100 nmol/L radicicol for 36 h caused the HCV core to be undetectable in the medium for the last 24 h of the combined treatment.
Figure 6.
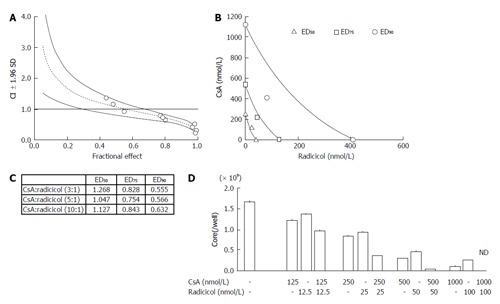
Analysis of the synergistic effect of cyclosporin A and radicicol. A: The graphs were constructed using the Chou and Talalay method. The CI and the fractional effect were derived from the release of the HCV core protein into the culture medium of JFH1-infected Huh-7 cells that were treated with a combination of CsA and radicicol (5:1 molar ratio). The open circles indicate actual experimental data. The CI vs fractional effect plots were generated with CalcSyn software. The dotted line and solid lines represent the mean values and standard deviation (1.96), respectively, of three independent experiments. The CI < 1, CI = 1, and CI > 1 indicate synergy, an additive effect and antagonism, respectively; B: The conservation isobologram (CI = 1) depicting different effective doses that yielded 50% (ED50), 75% (ED75) and 90% (ED90) inhibition of viral release by the combination treatment was graphed with actual experimental data (ED50, open triangles; ED75, open squares; ED90, open circles). The data represent the mean values of the results from three independent experiments; C: The CI values of each ED50, ED75 and ED90 by the combined CsA and radicicol treatment at molar ratios of 3:1, 5:1 and 10:1, respectively; D: The combined effect of CsA and radicicol at a molar ratio of 10:1. The HCV core levels in the medium are shown. The mean values and standard deviation (± SD) of the amounts of the HCV core released into the medium during three independent experiments are shown. The concentrations used in each set of experiments are shown under the histogram. ND: Not detected; CsA: Cyclosporin A; HCV: Hepatitis C virus; CI: Combination index.
DISCUSSION
Collectively, our results suggest that HSP90 inhibitors might affect both the RNA replication and the post-RNA replication stage of viral propagation. Previous reports have indicated that HSP90 is required for NS3 stability[11] and for the formation of a complex consisting of NS5A and FKBP8[12]. Thus, it is possible that HSP90 may affect the post RNA-replication step, such as assembly, through affecting the activity of NS3 and NS5A. Elucidating the precise mechanism for the HSP90 on HCV assembly may provide an alternative drug target for HCV clearance.
COMMENTS
Background
Although heat-shock protein 90 (HSP90) inhibitors, which inhibit the chaperone function of HSP90, have been shown to inhibit hepatitis C virus (HCV) replication by several groups, a recent report using a reporter system for HCV RNA replication (replicon) suggests that the effect is nonspecific. Thus, the inhibitory mechanism of HSP90 inhibitors remains controversial.
Research frontiers
The authors found that the HSP90 inhibitors had greater inhibitory effects on the HCV RNA and core protein levels measured in the medium than inside the cells. This inhibitory effect was observed in the presence of a low level of a known RNA replication inhibitor [cyclosporin A (CsA) or interferon-α].
Innovations and breakthroughs
The authors’ results suggested that, HSP90 inhibitors may also interfere with an HCV replication step that occurs after the synthesis of viral RNA, such as assembly and release.
Applications
This study would benefit the effort to explore new targets for the treatment of HCV infection.
Terminology
HSP90 inhibitors inhibit the chaperone function of HSP90. HSP90 clients include not only host proteins but also multiple HCV proteins including NS2/3 protease, NS3 and the RNA replication complex consisting of NS5A.
Peer-review
The authors suggested that HSP90 inhibitors may interfere with an HCV replication step that occurs after the synthesis of viral RNA such as assembly and release. The results of this study were quite interesting. The data were appropriately presented and interpreted. This manuscript also was well prepared.
Footnotes
Institutional review board statement: No Ethics Committee approval was required.
Informed consent statement: No Ethics Committee approval was required.
Conflict-of-interest statement: All of the authors have no conflict of interest to declare in relationship to this article.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 1, 2015
First decision: November 4, 2015
Article in press: January 19, 2016
P- Reviewer: Chuang WL, Jin B S- Editor: Qiu S L- Editor: A E- Editor: Liu SQ
References
- 1.Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch FX, Ribes J, Borràs J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271–285. doi: 10.1055/s-2007-1007117. [DOI] [PubMed] [Google Scholar]
- 4.Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res. 2004;63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 5.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones DM, McLauchlan J. Hepatitis C virus: assembly and release of virus particles. J Biol Chem. 2010;285:22733–22739. doi: 10.1074/jbc.R110.133017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prodromou C, Pearl LH. Structure and functional relationships of Hsp90. Curr Cancer Drug Targets. 2003;3:301–323. doi: 10.2174/1568009033481877. [DOI] [PubMed] [Google Scholar]
- 9.Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta. 2012;1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci USA. 2001;98:13931–13935. doi: 10.1073/pnas.241510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ujino S, Yamaguchi S, Shimotohno K, Takaku H. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus nonstructural protein NS3. J Biol Chem. 2009;284:6841–6846. doi: 10.1074/jbc.M806452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ujino S, Nishitsuji H, Sugiyama R, Suzuki H, Hishiki T, Sugiyama K, Shimotohno K, Takaku H. The interaction between human initiation factor eIF3 subunit c and heat-shock protein 90: a necessary factor for translation mediated by the hepatitis C virus internal ribosome entry site. Virus Res. 2012;163:390–395. doi: 10.1016/j.virusres.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Kim MG, Moon JS, Kim EJ, Lee SH, Oh JW. Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochem Biophys Res Commun. 2012;421:112–118. doi: 10.1016/j.bbrc.2012.03.126. [DOI] [PubMed] [Google Scholar]
- 15.Beran RK, Sharma R, Corsa AC, Tian Y, Golde J, Lundgaard G, Delaney WE, Zhong W, Greenstein AE. Cellular growth kinetics distinguish a cyclophilin inhibitor from an HSP90 inhibitor as a selective inhibitor of hepatitis C virus. PLoS One. 2012;7:e30286. doi: 10.1371/journal.pone.0030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol. 2013;11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 17.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009;5:e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, Tsukiyama-Kohara K, Kawaguchi R, Tanaka S, Kohara M. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology. 1999;116:636–642. doi: 10.1016/s0016-5085(99)70185-x. [DOI] [PubMed] [Google Scholar]
- 19.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–6442. [PubMed] [Google Scholar]
- 20.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 21.Paul D, Madan V, Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe. 2014;16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F, Robotham JM, Nelson HB, Irsigler A, Kenworthy R, Tang H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J Virol. 2008;82:5269–5278. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 24.Robida JM, Nelson HB, Liu Z, Tang H. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J Virol. 2007;81:5829–5840. doi: 10.1128/JVI.02524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa S, Umehara T, Matsuda C, Kuge S, Sudoh M, Kohara M. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun. 2007;353:882–888. doi: 10.1016/j.bbrc.2006.12.117. [DOI] [PubMed] [Google Scholar]