

Abstract
In the heart, excitation-contraction (E-C) coupling is mediated by Ca2+ release from sarcoplasmic reticulum (SR) through the interactions of proteins forming the Ca2+ release unit (CRU). Among them, calsequestrin (CSQ) and histidine-rich Ca2+ binding protein (HRC) are known to bind the charged luminal region of triadin (TRN) and thus directly or indirectly regulate ryanodine receptor 2 (RyR2) activity. However, the mechanisms of CSQ and HRC mediated regulation of RyR2 activity through TRN have remained unclear. We first examined the minimal KEKE motif of TRN involved in the interactions with CSQ2, HRC and RyR2 using TRN deletion mutants and in vitro binding assays. The results showed that CSQ2, HRC and RyR2 share the same KEKE motif region on the distal part of TRN (aa 202–231). Second, in vitro binding assays were conducted to examine the Ca2+ dependence of protein-protein interactions (PPI). The results showed that TRN-HRC interaction had a bell-shaped Ca2+ dependence, which peaked at pCa4, whereas TRN-CSQ2 or TRN-RyR2 interaction did not show such Ca2+ dependence pattern. Third, competitive binding was conducted to examine whether CSQ2, HRC, or RyR2 affects the TRN-HRC or TRN-CSQ2 binding at pCa4. Among them, only CSQ2 or RyR2 competitively inhibited TRN-HRC binding, suggesting that HRC can confer functional refractoriness to CRU, which could be beneficial for reloading of Ca2+ into SR at intermediate Ca2+ concentrations.
Keywords: calsequestrin; histidine rich Ca2+ binding protein, and triadin; junctin; ryanodine receptor
INTRODUCTION
In the heart, pacemaker activities in the form of action potential depolarize the cell membranes leading to activation of dihydropyridine receptors (DHPRs)/L-type Ca2+ channels, resulting in increased cytosolic Ca2+ concentration. The small amount of fluxed Ca2+ could trigger Ca2+-induced calcium release (CICR), the amplified Ca2+ release from the SR through ryanodine receptor 2 (RyR2)/Ca2+ release channel (CRC) located in the SR membranes (Bers, 2002). This transitory elevation of cytosolic Ca2+ is responsible for activation of actin-myosin cross-bridges resulting in the generation of heartbeat.
Ca2+ release through RyR2/CRC is mediated by interactions between multiple proteins in a complex called calcium release unit (CRU) that includes triadin (TRN), calsequestrin (CSQ), junctin (JTN) (Gyorke et al., 2004), and histidine rich Ca2+ binding protein (HRC) (Fan et al., 2004). Both JTN and TRN may act as scaffold proteins anchoring CRU proteins such as CSQ and HRC (Goonasekera et al., 2007; Guo et al., 1996; Jones et al., 1995; Zhang et al., 1997) in the vicinity of RyR2. The SR luminal Ca2+ binding proteins, CSQ and HRC have effects on RyR2 through their Ca2+ sensitivity and interactions with TRN and JTN (Arvanitis et al., 2007; Boncompagni et al., 2012; Fan et al., 2004; Guo and Campbell, 1995; Kim et al., 2003; Park et al., 2012; Picello et al., 1992; Zhang et al., 1997). The interactions of CSQ or HRC with TRN are known to occur at highly conserved luminal domains within the structure of TRN. These domains have been characterized by the presence of alternating positively and negatively charged amino acids known as KEKE motif (Kobayashi et al., 2000; Lee et al., 2001; 2004b; Zhang et al., 1997).
The regulation of Ca2+ cycling by those CRU proteins is considered as an important area of investigation, since desynchronized Ca2+ release from SR (called Ca2+ leak) has been associated with arrhythmogenesis and heart failure (Arvanitis et al., 2007). Furthermore, the dynamically changing luminal Ca2+ levels in the SR could have critical influences on the interactions among the proteins in CRU, since the structural and functional defects of CRU are associated with aberrant Ca2+ homeostasis, as shown in myocardial hypertrophy, heart failure and ventricular arrhythmias (Hasenfuss et al., 1997; Kim et al., 2013; Lehnart et al., 2009; Postma et al., 2002; Priori and Napolitano, 2005). Hence, understanding the nature of protein-protein interactions (PPI) in CRU and its influence on RyR2 activities are critical for elucidating mechanistic insights of Ca2+ cycling in the pathological states of the heart.
Although the previous studies have shown evidence of the regulatory interactions between RyR and its protein partners within CRU, the dynamic interactions of RyR2 in the heart with the key luminal CRU proteins such as CSQ and HRC under different SR luminal Ca2+ levels remain to be investigated. In this study, we determine the minimal motif on the luminal part of TRN, which is critical for PPI of the CRU proteins, the characteristic of HRC-TRN binding and the nature of HRC-TRN binding with CSQ2 or RyR2, to provide new insights into the molecular events involved in the regulation of Ca2+ release from SR.
MATERIALS AND METHODS
Generation and purification of the recombinant proteins used for the present study
GST-protein fusion constructs were generated from mouse cardiac cDNAs using PCR. The PCR products were digested with restriction enzymes BamHI and XhoI, and subcloned into Bam-HI/XhoI cloning sites of pGEX 4T-1 vector (Amersham Biosciences). Clones with desired inserts were confirmed by direct sequencing, and the fusion proteins were subjected to translation as described previously (Zhang et al., 1997). The expression of GST fusion proteins were induced in Escherichia coli (E. coli) BL21 with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 12 h at 18°C. Cells were harvested by centrifugation, resuspended in phosphate buffered saline (PBS), lysed by ultrasonication on ice, and then incubated in 1% Triton X-100 for 30 min. The soluble fraction was obtained by centrifugation at 20,000 × g for 10 min at 4°C. The fusion proteins were immobilized by incubating 1 ml of the soluble E. coli fraction with 50 μl bead volume of glutathione-Sepharose 4B (Pharmacia) for 3 h and washed thrice with PBS. Primer sets used for generation of TRN mutants are as follows: 5′-CGC GGA TCC ATG ACT GAG ATC ACT GCT GAA-3′, 5′-CCG CTC GAG AGG GGA ACT GAA TGT TGT CAC-3′ for the cytoplasmic domain (aa 2-47); 5′-CGC GGA TCC GAT TTA GTG GAT TAT AAA AAC-3′ and 5′-CCG CTC GAG TTA GTG TAT TTC TTT TCT TTT-3′ for proximal TRN (aa 69–146); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG CTC TTT GTC ATC CTT CTT TCT-3′ for TRN Del 1 (aa 147–254); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG TTT CAC TTT CTC CTG TTT TCC-3′ for TRN Del 2 (aa 147–231); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG TTC AGT CTT TTC CTT AGT CTT-3′ for TRN Del 3 (aa 147–215); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG TGC CAT CAT CTT CGT CTC TGG-3′ for TRN Del 4 (aa 147–202); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG GTG AGT TGC TGT CTT CTC AGG-3′ for TRN Del 5 (aa 147–186); 5′-CGC GGA TCC GAA AAG GCT GAA AAA GAG GAG-3′ and 5′-CCG CTC GAG TCT GTG TGA AGC TTT AGT TTG-3′ for TRN Del 6 (aa 147–166).
GST pulldown assay and Western blot
GST pulldown assays was performed as described previously with minor modifications (Lee et al., 2004a). Briefly, the mouse heart homogenates were solubilized for 1 h on ice in buffer containing 2% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 1 mM phenylmethylsulfonyl fluoride, 1.5 mM sodium orthovanadate, 15 mM sodium fluoride, 1 mM DTT and protease inhibitor mixture. Solubilized proteins were obtained by centrifugation and diluted with 50 mM Tris-HCl, pH 7.4, 1 mM phenylmethylsulfonyl fluoride, 1 mM DTT, and protease inhibitor cocktail to reduce high salt and detergent concentration. Solubilized proteins were pre-cleared with 50% slurry of glutathionesepharose 4B beads for 2 h at 4°C. The pre-cleared supernatant was incubated with equivalent amount of GST fusion proteins coupled to glutathione-Sepharose 4B beads for 12 h at 4°C. After the incubation, the fusion protein-Sepharose 4B complexes were collected by centrifugation and washed five times with wash buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.3% CHAPS. The bound proteins were eluted, and boiled in 2X sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and subjected to SDS-PAGE, followed by immunoblotting with HRC, CSQ or RyR antibodies. Antibodies of HRC, CSQ2 and TRN (homemade rabbit polyclonal sera), RyR (ABR, MA3-925), western blot signal was detected by ImageQuant LAS 4000 mini (GE Healthcare Bio-Sciences AB) with a SuperSignal West Pico chemiluminescence kit (Thermo Fisher Scientific, Inc.). Western blot band intensities were measured using ImageJ software. Ca2+ dependent GST pulldown assay was performed after adjusting different concentrations of free Ca2+ as described previously (Zhang et al., 1997). In brief, the heart homogenate was prepared in the presence of 2 mM EGTA. Based on the maxchelator program, specific concentrations of CaCl2 were added to the reaction solutions after homogenates were diluted. GST pulldown assay was performed as mentioned above.
Co-immunoprecipitation assay and competitive binding assay
Heart homogenate was used for co-immunoprecipitation (Co-IP). The solubilization step was identical to that of the GST-pull-down assay as described above. The solubilized samples were pre-cleared with protein A sepharose beads (Amersham) and subjected to incubation at 4°C overnight using appropriate antibody. Protein A sepharose beads were added and incubation was continued at 4°C for 2 h. Precipitates were washed four times with washing solution, eluted in SDS sample buffer and analyzed by western blot assay. Competition between RyR2, CSQ2, and HRC for binding to TRN was studied as described previously (Wyszynski et al., 1997). In brief, immunoprecipitation with triadin antibody was performed with increasing concentration of RyR2 peptide IEDPAGDEYEIYRII or with CSQ2 peptide DEDNEDSDDDDDDDE, which have been known to interact with TRN (Lee et al., 2004b; Shin et al., 2000). Bound proteins were eluted and immunoblotted with HRC or CSQ antibody. These experiments were performed at free Ca2+ level of pCa4.
Statistics
All data are shown as mean ± SD. Statistical significance was analyzed by student’s t-test or analysis of variance (ANOVA). A value of P < 0.05 was considered statistically significance.
RESULTS
RyR2, CSQ2 and HRC interact with the charged distal TRN
The KEKE motifs in the luminal region of TRN have been proposed to facilitate PPI with the different CRU proteins (canine cardiac CSQ at aa 210–224 of TRN, Kobayashi et al., 2000; rabbit skeletal RyR1 at aa 200–232 of TRN, Lee et al., 2004b; rabbit skeletal HRC at aa 204–260 of TRN, Lee et al., 2001). Here, we evaluated mouse cardiac TRN interactions with RyR2, CSQ2 and HRC under the same experimental conditions to identify the minimal binding regions of TRN using mouse heart samples. Three GST-tagged deletion mutants, cytosolic region of TRN (aa 2–47), the proximal region of luminal TRN (aa 69–146) and the distal region of luminal TRN (aa 147–277) were generated (Figs. 1A and 1B) and subjected to GST pull-down assay. The results showed that only the distal region of TRN1 containing five KEKE motifs interacted with RyR2, CSQ2 or HRC (Fig. 1C).
Fig. 1.
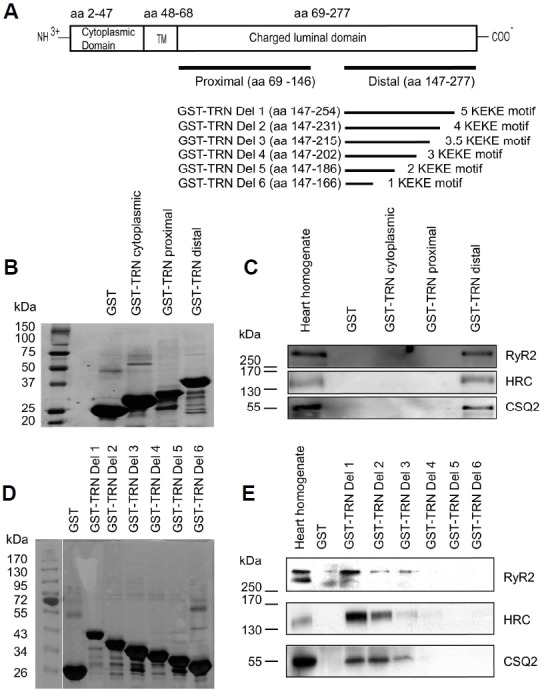
Distal region of the luminal TRN interacts with RyR2, CSQ2 and HRC through the same KEKE motif. (A) Cardiac TRN1 constructs used in the study. Deletion mutants of cardiac TRN1 residues were fused to GST protein. (B) Coomassie-stained gel after protein purification of: GST, GST-TRN cytoplasmic, GST-TRN proximal, and GST-TRN distal regions. GST pull-down assay was performed using mouse heart homogenate. (C) RyR2, HRC and CSQ2 showed interactions with the distal TRN construct containing KEKE motifs. (D) Coomassie stained gel image of the distal TRN mutants containing KEKE motifs after protein purification. (E) The minimal regions for the interactions of RyR2, CSQ2, and HRC needed the presence of KEKE motif (aa 202–231) of TRN.
To further analyze the specific CRU protein-binding region in TRN, we generated GST-tagged deletion mutants of the KEKE motifs in TRN (Fig. 1D). We prepared different GST fusion proteins such as GST-TRN Del 1 (aa 147-254), GST-TRN Del 2 (aa 147–231), GST-TRN Del 3 (aa 147–215), GST-TRN Del 4 (aa 147–202), GST-TRN Del 5 (aa 147–186), and GST-TRN Del 6 (aa 147–166). The estimated approximate molecular sizes of the proteins were ∼37.77, ∼35.24, ∼33.48, ∼32.05, ∼30.29, and ∼28.09 KDa, respectively. Western blotting of the GST pulldown samples with RyR, CSQ2, and HRC antibodies showed that GST-TRN Del1, GST-TRN Del2, and GST-TRN Del3 interacted with RyR, CSQ2, and HRC, whereas GST-TRN Del 4, GST-TRN Del 5, GST-TRN Del 6, or control GST did not show any interaction with the three proteins (Fig. 1E), suggesting that the KEKE motif region of TRN (aa 202–231) is involved in the interaction with the CRU proteins.
Ca2+-dependent interactions of TRN with RyR2, CSQ2, and HRC
In order to study the optimal Ca2+ concentrations required for binding between TRN-CSQ2, TRN-HRC, and TRN-RyR2 (Arvanitis et al., 2007; Lee et al., 2001), GST pull-down assays were conducted using the mouse cardiac distal TRN under increasing free Ca2+ concentrations followed by Western blot with specific antibodies for RyR, HRC, and CSQ2. The results showed that HRC-TRN had a peak binding at pCa4 (called moderate Ca2+ concentration), but CSQ-TRN binding was maximum at low free Ca2+ level. On the other hand, RyR2-TRN binding showed no apparent Ca2+ dependence (Fig. 2), suggesting that the SR luminal Ca2+ can selectively affect the patterns of PPI depending on the types of CRU proteins.
Fig. 2.
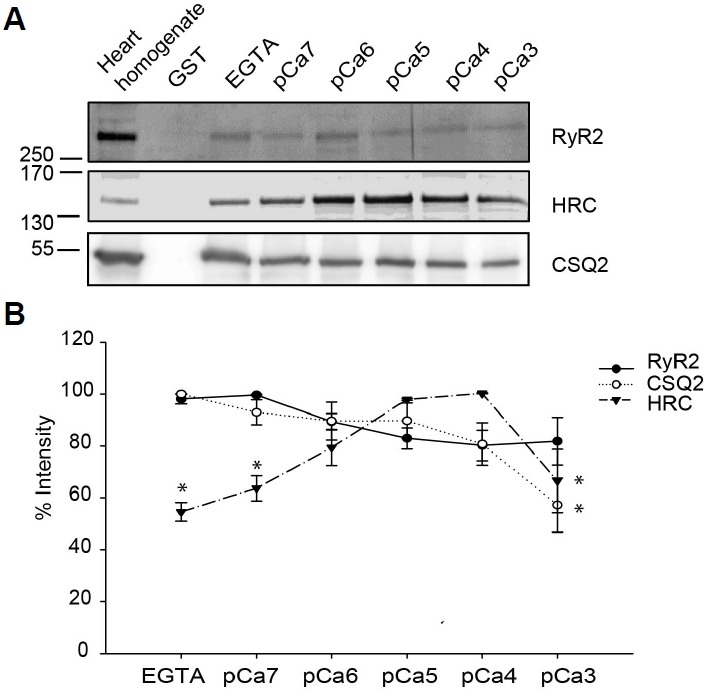
Interaction of TRN with RyR2, HRC, and CSQ2 at increasing Ca2+ concentrations. (A) The distal TRN deletion mutant fused to GST protein was used for pull down assay and its interacting partners in the presence of increasing Ca2+ concentrations were studied. (B) Quantitative representation of the western blot results. (Mean ± SD, *P value < 0.05, n = 3, the statistical significance was calculated with respect to the maximum interaction of the respective protein using one way ANOVA).
HRC-TRN binding is competed with RyR2 or CSQ2
We performed a competitive binding assay to examine whether HRC-TRN binding is interfered by RyR or CSQ2. The study also could lead to the answers why the seemingly functionally redundant proteins, HRC and CSQ exist within the SR lumen. Since HRC showed a unique binding pattern with the maximum TRN binding ability at moderate Ca2+ levels, the competitive TRN-HRC binding experiments were conducted at pCa4 by varying concentrations of CSQ2 or RyR2 loop 2 peptide using immuno-precipitation assays with TRN. The results showed that HRC binding to TRN significantly decreased in the presence of RyR2 loop 2 or CSQ peptides in concentration range of 3–30 μM and 1-30 μM, respectively (Fig. 3). Taken together, the results indicate that HRC competes with RyR or CSQ to interact with TRN at the moderate free luminal Ca2+ concentrations.
Fig. 3.
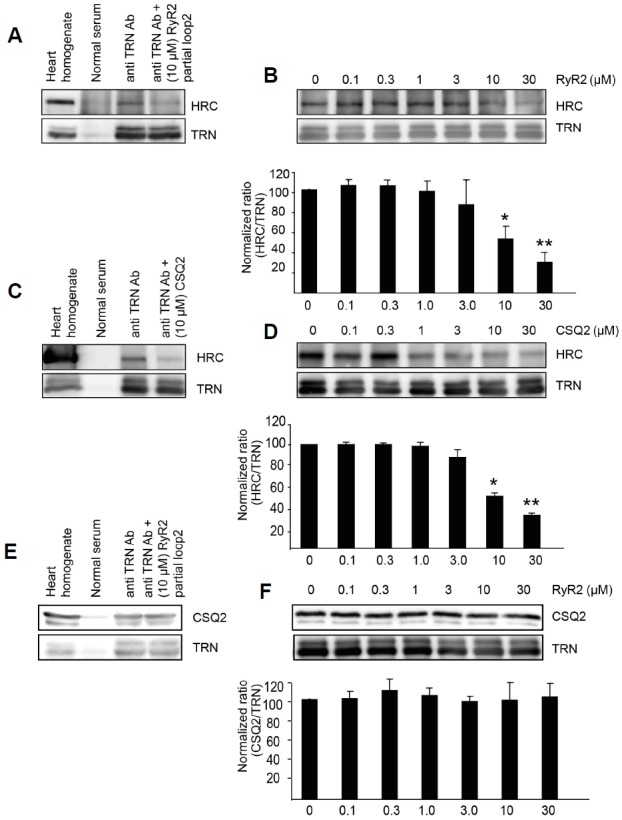
HRC competes with RyR2 and CSQ2 to interact with TRN, whereas RyR2 did not show any competition with CSQ2 to interact with TRN at pCa4 (A) On immunoprecipitation with TRN antibody at pCa4 in the presence of synthesized RyR2 loop 2 (10 μM) peptide, HRC showed significantly reduced interaction with TRN. (B) HRC shows reduced interaction with TRN as the concentration of RyR2 loop 2 increased (0–30 μM). Quantitative representation of the Western blot results of HRC plotted as a ratio to total TRN as the RyR peptide concentration increased. (C) Immuno-precipitation with TRN antibody at pCa4 in the presence of synthesized CSQ2 peptide (10 μM). (D) HRC shows reduced interaction with TRN as the concentration of CSQ2 increased (0–30 μM). Quantitative representation of the Western blot results of HRC plotted as ratio to total TRN as CSQ2 peptide concentration increased. (E) Immunoprecipitation with TRN antibody at pCa4 in the presence of synthesized RyR2 loop 2 peptide (10 μM). CSQ2 showed no change in interaction to TRN. (F) No significant change was observed in the interaction of CSQ2 and TRN as the concentration of RyR2 increased (0–30 μM). Quantitative representation of the Western blot results of CSQ2 plotted as the ratio to total TRN as RyR2 peptide concentration increased. (Mean ± SD, *P value < 0.05, **P value < 0.01, n = 4).
CSQ2 does not compete with RyR2 to interact with TRN
Similar to the competitive binding assay as shown above, we performed a competitive binding assay to examine whether CSQ-TRN binding was interfered by RyR2 at moderate Ca2+ concentration (pCa4) (Figs. 3E and 3F). The result showed no obvious changes of CSQ-TRN binding by increasing concentrations of RyR2 loop 2, suggesting that there is no competition between CSQ2 and RyR2 to interact with TRN at moderate free luminal Ca2+ levels.
DISCUSSION
In the present study, we examined the binding properties of CRU proteins in the heart using deletion mutants and in vitro binding assays. The new findings of this study are: (1) CSQ, HRC, and RyR2 share the same binding sites on the KEKE motif region of the distal part of TRN (aa 202–231, Fig. 1); (2) HRC-TRN binding has a bell-shaped Ca2+-dependence peak at pCa4 (moderate Ca2+) (Fig. 2); (3) HRC-TRN binding was competed by CSQ2 or RyR2 at pCa4, but CSQ2-TRN was not competed by RyR2 (Fig. 3); (4) Therefore, HRC, can confer functional refractoriness to CRU at pCa4, which is considered to be beneficial for reloading of Ca2+ into SR during the filling phase (Fig. 4).
Fig. 4.
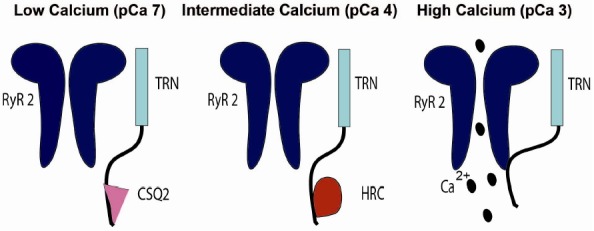
A schematic diagram describing the effect of TRN, CSQ2, and HRC interaction with RyR2 and their response to luminal Ca2+. At low luminal Ca2+ concentration, CSQ2 bound to TRN might help in building Ca2+ concentration near the junctional membrane as a necessary condition for release of Ca2+ ions through RyR2 upon activation; RyR2 Ca2+ leak is prevented by CSQ2. As the luminal Ca2+ concentration reaches moderate levels, CSQ2/TRN/RyR2 complex interaction starts to attenuate. RyR2 inhibition by CSQ2 is gradually relieved enabling RyR2 leakage, but HRC bound to TRN stabilizes RyR2 from being leaky. At higher luminal Ca2+ concentrations HRC is detached from TRN allowing RyR2-TRN interaction resulting in increased RyR2 activity and Ca2+ release.
Previous studies have reported that interactions of RyR/CRC with the luminal auxillary proteins such as CSQ, HRC, TRN, and JTN are involved in the regulation of CRC in cardiac muscle (Knollmann, 2009; Lee et al., 2001). TRN has conserved domains such as KEKE motif, which could act as a nodal point for interactions with RyR, CSQ, and HRC (Guo and Campbell, 1995; Guo et al., 1996; Kobayashi et al., 2000; Lee et al., 2004b). Furthermore, a synthesized KEKE motif of TRN (TRN-KEKE motif) was found to enhance RyR1 channel activity (Wium et al., 2012), suggesting that the TRN-KEKE motif is an important regulator of RyR activity and also serves as the scaffold domain for interactions with the luminal auxillary proteins. Here, we further investigated the presence of a critical interacting region on TRN for binding RyR2, CSQ2, and HRC in the heart. We found that the distal half of TRN (aa 147–277) containing KEKE motif was involved in PPI (Fig.1C), as was partially evidenced in previous reports (Kobayashi et al., 2000; Lee et al., 2004b). We further identified the specific KEKE motif(s) in the distal region that binds the three SR proteins (Fig.1E).
It has been reported that both TRN and junctin (JTN) are capable of interacting with CSQ and RyR, but only JTN has the ability to bind RyR and CSQ simultaneously, thus possessing a unique role in stabilization of RyR in a closed state at low luminal Ca2+ levels (Franzini-Armstrong et al., 2005; Jones et al., 1995; Zhang et al., 1997).We found that similar to JTN, significant fractions of CSQ and RyR2 are bound to TRN at low and intermediate luminal Ca2+ levels (Figs. 2–4) (Beard et al., 2005). Hence, the functional significance of TRN binding of CSQ in the luminal regions may be to anchor CSQ and facilitate building of Ca2+ micro-domain in the vicinity of RyR2 channels (Boncompagni et al., 2012; Gyorke et al., 2004).
HRC is a Ca2+ binding protein located in the lumen of SR, which may serve as a Ca2+ buffer, and interacts with TRN and SERCA2 in the lumen of SR. HRC is a bi-functional protein that can regulate activities of both SERCA2 and RyR2. The affinity of HRC to SERCA2 could decrease, when the luminal Ca2+ concentration increases from pCa7 to pCa3 (Arvanitis et al., 2007). As shown in Fig. 2A, HRC binding to TRN showed a bell-shaped Ca2+ dependence, which peaked at pCa4, similar to the previous report (Sacchetto et al., 2001) (Fig. 2A). Recent animal- and cell-level studies using genetic manipulation techniques have shown that deletion or knockdown of HRC gene could increase the leakiness of Ca2+ through RyR2, suggesting that RyR2 activities are inhibited by HRC (Fan et al., 2004; Park et al., 2012). We predicted that HRC might have a regulatory role on RyR stability through its interaction with TRN. A recent CSQ2/HRC double knockout (KO) study suggests an interesting hypothesis that HRC may also act as a primer to enhance RyR2 activity at higher luminal Ca2+ concentrations [Liu. et al., 2015 (in press)], which could support our data.
The model shown in Fig. 4 was constructed based on the results obtained in this study and some of previously published data (For simplicity, the roles of SERCA and JNT are not included).
At low Ca2+: pCa7: CSQ-TRN binding leads to maximum inhibition of RYR2 preventing Ca2+ leak. At intermediate Ca2+: pCa4: CSQ/TRN/RyR2 complex starts to dissociate and RyR2 inhibition by CSQ is gradually relieved as Ca2+ binding sites on CSQ become increasingly occupied with Ca2+ and CSQ-TRN bindings weaken. This event may be further facilitated by HRC binding to TRN conferring the refractoriness to RyR2. At high Ca2+: pCa3: HRC is dissociated from TRN-HRC complex, allowing RyR-TRN interaction, which increases RyR2 activity and facilitates Ca2+ release from SR.
Acknowledgments
This work was supported by the 2015 GIST Systems Biology Infrastructure Establishment Grant and the NRF grant funded by the Korean Ministry of Science, ICT & Future Planning (NRF-2013M3A9A7046297).
REFERENCES
- Arvanitis D.A., Vafiadaki E., Fan G.C., Mitton B.A., Gregory K.N., Del Monte F., Kontrogianni-Konstantopoulos A., Sanoudou D., Kranias E.G. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H1581–1589. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- Beard N.A., Casarotto M.G., Wei L., Varsanyi M., Laver D.R., Dulhunty A.F. Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys. J. 2005;88:3444–3454. doi: 10.1529/biophysj.104.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Boncompagni S., Thomas M., Lopez J.R., Allen P.D., Yuan Q., Kranias E.G., Franzini-Armstrong C., Perez C.F. Triadin/Junctin double null mouse reveals a differential role for Triadin and Junctin in anchoring CASQ to the jSR and regulating Ca(2+) homeostasis. PLoS One. 2012;7:e39962. doi: 10.1371/journal.pone.0039962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G.C., Gregory K.N., Zhao W., Park W.J., Kranias E.G. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1705–1711. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Protasi F., Tijskens P. The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- Goonasekera S.A., Beard N.A., Groom L., Kimura T., Lyfenko A.D., Rosenfeld A., Marty I., Dulhunty A.F., Dirksen R.T. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J. Gen. Physiol. 2007;130:365–378. doi: 10.1085/jgp.200709790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Campbell K.P. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J. Biol. Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- Guo W., Jorgensen A.O., Jones L.R., Campbell K.P. Biochemical characterization and molecular cloning of cardiac triadin. J. Biol. Chem. 1996;271:458–465. doi: 10.1074/jbc.271.1.458. [DOI] [PubMed] [Google Scholar]
- Gyorke I., Hester N., Jones L.R., Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G., Meyer M., Schillinger W., Preuss M., Pieske B., Just H. Calcium handling proteins in the failing human heart. Basic Res. Cardiol. 1997;92(Suppl 1):87–93. doi: 10.1007/BF00794072. [DOI] [PubMed] [Google Scholar]
- Jones L.R., Zhang L., Sanborn K., Jorgensen A.O., Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J. Biol. Chem. 1995;270:30787–30796. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- Kim E., Shin D.W., Hong C.S., Jeong D., Kim D.H., Park W.J. Increased Ca2+ storage capacity in the sarcoplasmic reticulum by overexpression of HRC (histidine-rich Ca2+ binding protein) Biochem. Biophys. Res. Commun. 2003;300:192–196. doi: 10.1016/s0006-291x(02)02829-2. [DOI] [PubMed] [Google Scholar]
- Kim T., Kahng Y.H., Lee T., Lee K., Kim D.H. Graphene films show stable cell attachment and biocompatibility with electrogenic primary cardiac cells. Mol. Cells. 2013;36:577–582. doi: 10.1007/s10059-013-0277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann B.C. New roles of calsequestrin and triadin in cardiac muscle. J. Physiol. 2009;587:3081–3087. doi: 10.1113/jphysiol.2009.172098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y.M., Alseikhan B.A., Jones L.R. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein-protein interaction. J. Biol. Chem. 2000;275:17639–17646. doi: 10.1074/jbc.M002091200. [DOI] [PubMed] [Google Scholar]
- Lee H.G., Kang H., Kim D.H., Park W.J. Interaction of HRC (histidine-rich Ca2+-binding protein) and triadin in the lumen of sarcoplasmic reticulum. J. Biol. Chem. 2001;276:39533–39538. doi: 10.1074/jbc.M010664200. [DOI] [PubMed] [Google Scholar]
- Lee E.H., Rho S.H., Kwon S.J., Eom S.H., Allen P.D., Kim D.H. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. J. Biol. Chem. 2004a;279:26481–26488. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- Lee J.M., Rho S.H., Shin D.W., Cho C., Park W.J., Eom S.H., Ma J., Kim D.H. Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J. Biol. Chem. 2004b;279:6994–7000. doi: 10.1074/jbc.M312446200. [DOI] [PubMed] [Google Scholar]
- Lehnart S.E., Maier L.S., Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail. Rev. 2009;14:213–224. doi: 10.1007/s10741-009-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu. B., Ho. H.T., Brunello. L., Unudurthi. S.D., Lou. Q., Belevych. A.E., Qian. L., Kim D.H., Cho. C., Janssen. P.M.L., et al. Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc. Res. 2015 doi: 10.1093/cvr/cvv222. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.S., Cha H., Kwon E.J., Jeong D., Hajjar R.J., Kranias E.G., Cho C., Park W.J., Kim D.H. AAV-mediated knock-down of HRC exacerbates transverse aorta constriction-induced heart failure. PLoS One. 2012;7:e43282. doi: 10.1371/journal.pone.0043282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picello E., Damiani E., Margreth A. Low-affinity Ca2+-binding sites versus Zn(2+)-binding sites in histidine-rich Ca2+-binding protein of skeletal muscle sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 1992;186:659–667. doi: 10.1016/0006-291x(92)90797-o. [DOI] [PubMed] [Google Scholar]
- Postma A.V., Denjoy I., Hoorntje T.M., Lupoglazoff J.M., Da Costa A., Sebillon P., Mannens M.M., Wilde A.A., Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002;91:e21–26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- Priori S.G., Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J. Clin. Invest. 2005;115:2033–2038. doi: 10.1172/JCI25664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetto R., Damiani E., Turcato F., Nori A., Margreth A. Ca2+-dependent interaction of triadin with histidine-rich Ca2+-binding protein carboxyl-terminal region. Biochem. Biophys. Res. Commun. 2001;289:1125–1134. doi: 10.1006/bbrc.2001.6126. [DOI] [PubMed] [Google Scholar]
- Shin D.W., Ma J., Kim D.H. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 2000;486:178–182. doi: 10.1016/s0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- Wium E., Dulhunty A.F., Beard N.A. A skeletal muscle ryanodine receptor interaction domain in triadin. PLoS One. 2012;7:e43817. doi: 10.1371/journal.pone.0043817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M., Lin J., Rao A., Nigh E., Beggs A.H., Craig A.M., Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Zhang L., Kelley J., Schmeisser G., Kobayashi Y.M., Jones L.R. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]